

Abstract
The response kinetics of rat C6 glioma cells to heat shock was investigated by means of flow cytometric DNA measurements and western blot analysis of HSP levels. The results showed that the effects on cell cycle progression are dependent on the cell cycle phase at which heat shock is applied, leading to either G1 or G2/M arrest in randomly proliferating cells. When synchronous cultures were stressed during G0 they were arrested with G1 DNA content and showed prolongation of S and G2 phases after release from the block. In proliferating cells, HSC70 and HSP68 were induced during the recovery and reached maximum levels just before cells were released from the cell cycle blocks. Hyperthermic pretreatment induced thermotolerance both in asynchronous and synchronous cultures as evidenced by the reduced arrest of cell cycle progression after the second heat shock. Thermotolerance development was independent of the cell cycle phase. Pre‐treated cells already had high HSP levels and did not further increase the amount of HSP after the second treatment. However, as in unprimed cells, HSP reduction coincided with the release from the cell cycle blocks. These results imply that the cell cycle machinery can be rendered thermotolerant by heat shock pretreatment and supports the assumption that HSP70 family members might be involved in thermotolerance development.
INTRODUCTION
Stress‐induced perturbations of the normal sequence of cell cycle processes often lead to transient cell cycle arrests. Heat shock is the most commonly applied stressor and affects various cell cycle processes depending on the cell type, the phase of the cell cycle and the severity of the stress (Roti Roti, Mackey & Higashikubo, 1992; Higashikubo, White & Roti Roti, 1993; Wong et al. 1993; Yonezawa et al. 1996; Van der Waal et al. 1997). Most cell types become arrested or delayed in certain cell cycle phases when exposed to a mild or medium heat shock. In CHO and C6 cells synchronized by serum starvation or mitotic shake‐off, heat shock caused a prolongation of G1 phase and delayed entry into S phase (Martin & Regan 1991; Wong et al. 1993). A similar heat‐induced G1 block and an additional G2/M block was observed in asynchronously proliferating mammalian cells (Read, Fox and Bedford, 1984; Higashikubo et al. 1993; Sugano et al. 1995; Nitta et al. 1997). In other studies, HeLa and CHO cells exhibited a heat shock‐induced prolongation of S phase followed by an arrest in, or a prolongation, of G2/M phase, as revealed by flow cytometry (Read et al. 1984; Fox, Read & Bedford, 1985; Mackey, Anolik & Roti Roti,. 1992; Higashikubo et al. 1993; Hang & Fox 1995).
Severe heat shock treatments ‐ above a cell type‐specific lethality threshold ‐ lead to cell death via cell type specific pathways, e.g. apoptosis, necrosis or loss of clonogenicity (Wong et al. 1993; Yonezawa et al. 1996; Van der Waal et al. 1997). It is not clear why some cell lines are constitutively more resistant to heat stress than other lines, but the constitutive levels of heat shock proteins may play an important role. In agreement with this role, enhanced cell survival after an otherwise lethal heat shock treatment can be induced by a pretreatment of cells with a mild, non‐lethal but HSP‐inducing heat shock followed by recovery at physiological temperatures. This phenomenon was termed ‘acquired thermotolerance’ (Read et al. 1984; Fox et al. 1985; Li, Wong & Dewey, 1990; Mackey et al. 1992; Roti Roti et al. 1992; De Maio 1999).
In order to investigate the heat‐induced HSP expression in rat C6 glioma cells we used a 30 min heat shock treatment at 44°C. This stress condition leads to maximal expression of HSC70 (70 kDa heat shock cognate, constitutively expressed homologue of HSP73 in HeLa cells) and HSP68 (68 kDa heat shock protein, expressed only after stress in rodent cells and homologous to HeLa HSP72) (Subjeck & Shyy 1986; Venetianer et al. 1994). The heat shock also delays the cell doubling time considerably, but does not result in a dramatic increase in cell death in C6 glioma cultures. Considering the putative relationship between thermotolerance and HSP expression mentioned above, we asked whether thermotolerance with respect to the cell cycle can also be induced in our system even though this type of thermotolerance has always been defined and measured in terms of enhanced cell survival.
In order to first thoroughly investigate the heat shock effects on the cell cycle of C6 glioma cells, we analysed a series of flow cytometric DNA‐measurements covering at least one complete division cycle. After determining heat shock‐induced G1 and G2/M phase arrests in asynchronously proliferating cultures, we further characterized these heat shock effects in cultures synchronized by serum starvation and subsequent serum stimulation. In these cultures, heat shock applied in G0 caused a G0 or G1 arrest and considerably delayed the subsequent phases. Thereafter, the same experiments were performed with heat shock pretreated (primed) cells to elucidate possible changes in the arrests and delays. The results clearly demonstrate the phenomenon of ‘acquired thermotolerance’ in asynchronous and synchronous cells. The duration of cell cycle arrests was drastically reduced in heat shock pretreated (primed) cells. Moreover, the delays of S and G2/M phase progression detected in synchronized cultures after a single heat shock were abolished in thermotolerant cells. Therefore, in addition to enhancing cell survival, a priming heat shock renders cells less sensitive to stress‐induced delays in cell cycle progression.
Determination of the heat‐induced HSP70 expression revealed that the cell cycle redistribution of C6 cells, i.e. a first resumption of cell cycle progression occurred while HSP was induced. The later release from both cell cycle blocks coincided with a decline in the HSP level. Primed C6 cells already expressed high levels of HSP before the second treatment, and the second heat shock further increased the levels. However, as in unprimed cells, the release from the heat‐induced cell cycle blocks coincided with a decline of both HSP isoforms. Therefore, high levels of HSP70 isoforms might not only render cells more resistent to lethal heat shock treatments (Venetianer et al. 1994; Thayer & Mirkes 1997; De Maio 1999) but also protect the cell cycle machinery from damage leading to extended cell cycle delays.
MATERIALS AND METHODS
Cell culture
Experiments were performed with asynchronously proliferating and serum‐stimulated synchronous rat C6 glioma cells. This cell line was originally derived from a nitrosomethylurea‐induced astrocytoma (Benda et al. 1968) and is often used either as an undifferentiated glial precursor or as a model system for reactive astrogliosis. Stock cultures were maintained in Dulbecco's modified Eagle's Medium (DMEM, containing 100 U/ml penicillin and 100 µg/ml streptomycin) supplemented with 10% newborn calf serum (NCS) in a humidified 37 °C incubator with a 10% CO2 atmosphere. For all experiments a culture dish with a 70% confluent monolayer was trypsinized, and equal numbers of cells were reseeded in 10 cm culture dishes. Proliferating cells were then cultured for three days in growth medium in order to recover from trypsinisation and to reach exponential growth. Eight hours before heat shock the medium was exchanged to make sure that the pH buffering capacity was maximal. Cultures were synchronized by means of serum restimulation of serum‐deprived cells (Zeise et al. 1998). Cells were initially seeded and kept for 24 h in DMEM supplemented with 10% NCS. This growth phase was followed by six days of cultivation in medium containing only 0.5% NCS. This leads to reversible morphological differentiation and growth arrest in this cell line. Cells were triggered to re‐enter the cell cycle synchronously by addition of CO2‐equilibrated DMEM containing 20% NCS.
Heat shock procedure
All heat shocks (HS) were applied for 30 min in a 44 °C water bath. Culture dishes were sealed with two layers of parafilm to avoid CO2 leakage. Synchronous cultures were heated immediately after serum stimulation. Thereafter, cells were further cultivated for 2–48 h at 37 °C and harvested at the times indicated. Pre‐shocked (primed) cells were derived by heating either randomly proliferating or synchronous cultures for 30 min (44 °C) followed by 24 h recovery at 37 °C before the second heat exposure.
Flow cytometry
Cells were harvested by trypsination at defined times between 0 and 48 h after heat shock, fixed in 70% ethanol and stored overnight at −20 °C. For DNA measurements, cells were centrifuged for 10 min at 300 g, and the pellet was resuspended in phosphate buffered saline (PBS). Big cell aggregates and some cell doublets were excluded by filtering the solution through a 30‐m mesh nylon net. After a second centrifugation, cells were resuspended in PBS containing 50 µg/ml propidium iodide (PI; Sigma, Deisenhofen, Germany) and 10 mg/ml RNase (Sigma) and stained for at least 45 min. DNA analysis was performed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) using the Lysis II and Cellfit software. Cell doublets were completely excluded by software gating tools, and 10 000 single cells were analysed for each DNA‐histogram shown.
Western blot analysis
For all samples the protein concentration (g/l) was determined, and 10% SDS‐PAGE gels were loaded with equal amounts of protein per lane (25 µg). Proteins were separated by electrophoresis and transferred to a nitrocellulose membrane. Membranes were blocked with PBS containing 0.2% Tween20 for 30 min. Antibodies were added for 60 min at room temperature. The antibodies used were: SPA‐820 (clone N27F3–4, monoclonal, 1 : 1000; Biomol, Hamburg, Germany) which recognizes HSC as well as HSP70; SPA‐810AP (clone C92F3 A‐5, monoclonal, 1 : 1000; Biomol) detecting only HSP70 and goat antimouse conjugated to alkaline phosphatase (1 : 1000; Sigma). The relative intensities of the bands were determined using a video scanner and CREAM software.
RESULTS
Heat shock effects on randomly proliferating cells
Heat shock was previously shown to differentially affect cells in G1 and S phase (Higashikubo et al. 1993; Wong et al. 1993). In order to determine the kinetics of these reactions, we used asynchronously proliferating cultures and investigated heat shock effects in all cell cycle phases by flow cytometric DNA‐measurements. Subconfluent C6 cells (Fig. 1a (unprimed), top panel) were heat‐shocked for 30 min at 44 °C and harvested after 2–22 h of recovery at 37 °C. Figure 1a and 1b show that hyperthermia induced two blocks in proliferating cultures ‐ one in G1 and the other in G2/M ‐ probably depending on the individual cell cycle phase in which the cells were exposed to the stress.
Figure 1.
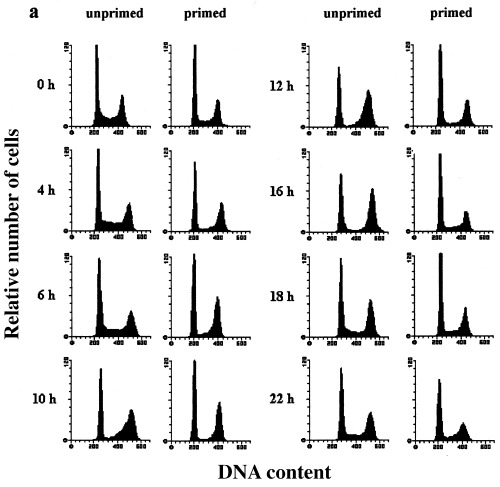
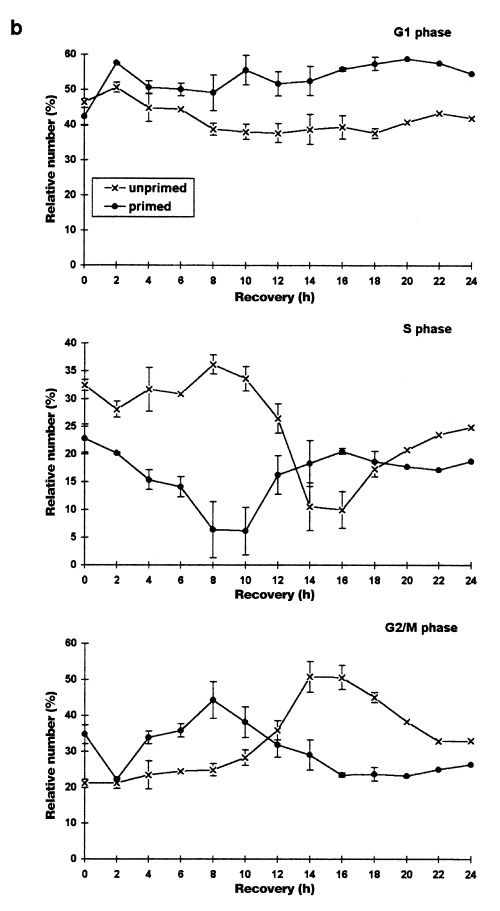
Heat shock effects on the cell cycle distribution of asynchronously proliferating rat C6 cells analysed by flow cytometry. Asynchronously proliferating normal and primed C6 cells were heat‐shocked at 44 °C for 30 min and harvested after the indicated recovery times.(a) Representative DNA‐histograms of C6 cells analysed by flow cytometry. DNA‐content based on PI‐fluorescence (x axis) is plotted against relative number of cells (10 000 cells per histogram; y axis)(b) Quantitative analysis of DNA‐histograms using the Lysis II and Cellfit research programs (Becton Dickinson, San Jose, CA, USA). The proportion of cells in different phases of the cell cycle (top panel: G1 phase, mid panel: S phase, bottom panel: G2/M phase) is plotted against the recovery time. (×): heat‐shocked cells (●): primed cells. Values are means (±SE) of at least three independent experiments.
During the first 8 h of recovery the cell cycle distribution remained relatively constant. Therefore, cells in all cell cycle phases were completely arrested at their actual position in the cell cycle. However, 10 h after heat shock treatment a drastic change in the phase distribution took place. S phase cells slowly started to continue DNA‐synthesis while G1 and G2/M cells remained further arrested. After 16 h nearly all initial S phase cells had apparently completed replication and accumulated in G2/M. Sixteen hours is a relatively long duration for S phase, especially considering that unstressed C6 cells have a doubling time of 18–24 h (Sharma & Raj 1987; Martin & Regan 1991). This implies a prolongation of S phase after heat shock in part due to a complete block during the first 8 h. Even though the use of asynchronous cells is not suitable for an exact measurement of cell cycle delays estimation of S phase length in unstressed, synchronous C6 cultures confirms the delay in S phase progression seen here. Even if the initial 8 h without cell cycle redistribution is subtracted, the remaining 8 h could still be considered as a 4‐h delay because S phase in unstressed synchronous cultures can be accomplished in 4 h.
S phase cells almost completely accumulated in G2/M and led to a maximum of G2/M cells (50%) 14–16 h after heat shock. These changes in the distribution of cell cycle fractions suggest that G1 cells were arrested before the G1/S transition whereas cells in S or G2/M phase during heat treatment were arrested in G2/M. Cells seem to be simultaneously relieved from both blocks 18 h after heat shock, as observed by the increase of cells in S phase and the decrease of cells in G2/M phase. However, the G2/M fraction decreased to only 33% and then remained constant from 22 to 24 h of recovery indicating that not all cells were released from the G2/M block synchronously. It seems reasonable that the remaining fraction is made up by the initial S phase cells, which had accumulated in G2/M (32.4%), because the values are nearly identical.
Considering the synchronous release from the blocks and the checkpoint model of cell cycle regulation (Elledge 1996), the formation of the cell cycle blocks can be seen as a biphasic process. During the first 8 h (phase 1) all cells are arrested at their actual position in the cell cycle and do not progress at all. In the second phase of the cell cycle block (10–16 h) restricted cell cycle progression in all phases takes place. G1, S and G2/M cells continue progression through their actual cell cycle phase but thereafter arrest at the checkpoints in G1 or G2/M. Flow cytometry only reveals cell cycle progression without phase transition in S phase cells because progression through G1 and G2/M is not accompanied by changes in DNA content.
Flow cytometry also does not reveal where in G2 or M cells are arrested after hyperthermia. Microscopic examination of four, 6‐diamidino‐2‐phenyl‐indol‐dihydrochloride (DAPI)‐stained samples however, favours the view of a G2 arrest of cells, which had progressed through S or G2 phase during heat shock because there is no significant accumulation of cells with condensed chromatin after 16 h of recovery (Kühl, unpubl. results). Cells already in M phase during the heat treatment might either be arrested in mitosis or might represent the minimal fraction that dies in these experiments. In none of the samples did heat shock induce apoptotic cell death even though the apoptotic pathway is intact in this cell line. A DNA ladder as well as nuclear fragmentation can be induced e.g. by a 24‐h treatment with the kinase inhibitor staurosporine (200 nm). Furthermore, there was only a negligible loss of cells (< 2%) due to other modes of cell death (data not shown).
To evaluate the protective effects of a priming heat shock on randomly proliferating cultures, cells were shocked for 30 min at 44 °C and allowed to recover at 37 °C for 24 h before the second shock was applied. Figure 1 (primed, 0 h) shows cells immediately before the second heat shock. These cultures show a cell cycle distribution that is slightly different from randomly proliferating cultures not exposed to a priming heat shock treatment: the percentage of S phase cells is lower and that of G2/M cells higher. Corresponding to the distribution of unprimed cultures after 24 h of recovery this difference is due to heat‐induced phase prolongation after release from the blocks. Therefore, the status of the primed asynchronous cells might be subject to criticism in that they cannot be seen as randomly proliferating cells. However, the results obtained with heat‐shocked synchronous cultures up to 48 h after heat shock (Fig. 3) do not reveal a second accumulation or block from 24 to 48 h after heat shock. Considering this, primed asynchronous cells can be compared to unprimed cells even though they have a slightly different cell cycle distribution profile.
Figure 3.
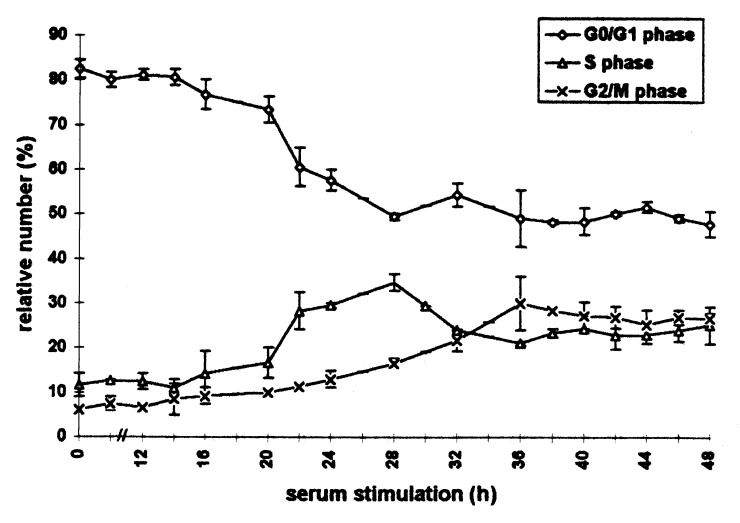
Cell cycle distribution of heat‐shocked C6 cells after serum stimulation analysed by flow cytometry for 48 h. Rat C6 glioma scells synchronized by serum deprivation were heat‐shocked (44 °C, 30 min) directly after serum stimulation and harvested at the indicated times. After flow cytometric measurement, quantitative analysis of DNA‐histograms was performed using the Lysis II and Cellfit research programs (Becton Dickinson, San Jose, California). The proportion of cells in different phases of the cell cycle is plotted against the time of serum stimulation. (○): G1 phase (△): S phase (×): G2/M phase. Values are means (±SE) of three independent experiments.
Heat shock induced the same pattern of arrests in primed as in unprimed cells. The onset of the phase redistribution as well as the release from the blocks, however, both occurred 6–8 h earlier in primed cells. These results clearly demonstrate that a priming heat shock renders cells thermotolerant as shown by the shortening of the heat‐induced cell cycle delays. The onset of the phase redistribution directly after the heat shock treatment implies that thermotolerance mainly protects C6 cells from the first phase of cell cycle arrest (see above) and allows cells to immediately progress towards the G1 or G2/M checkpoints.
The induction of thermotolerance did not exhibit cell cycle phase‐specific differences, as documented by the identical shifts of the two blocks. Furthermore, thermotolerant cells which were rescued from a longer G1 arrest did not accumulate in G2/M and vice versa. We therefore conclude that a priming heat shock in any of the cell cycle phases protects cells at least partly against hyperthermia‐induced arrests and enables cells to escape from the block earlier.
Heat shock effects on synchronized cells
Asynchronous cultures are not suitable for an exact determination of the heat shock or thermotolerance effects with respect to phase prolongation after release from cell cycle blocks. Therefore, we used C6 cells synchronized in G0 by serum starvation for further investigations.
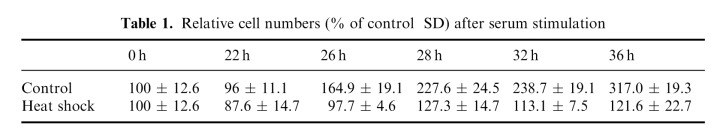
Flow cytometric measurement of C6 cells cultivated in medium with low serum content demonstrated a cell cycle arrest mainly in the G0/G1 phase (Fig. 2a (control) and 2b). Subsequent serum addition induced a relatively synchronous entry into the cell cycle. Within the first 8 h of cell cycle progression, the morphological differentiation observed during cultivation at low serum was reversed without detectable induction of DNA‐synthesis. The G1/S transition (right‐handed shift of the G1‐peak in DNA histograms) occurred almost synchronously after 12 h. The maximum of cells in S phase (about 50%) was observed after 14–16 h and was immediately followed by the first significant increase in the G2/M fraction (16 h). This indicates that S phase lasts about 4 h in C6 cells. The maximum of G2/M cells was detected after 20 h. This fraction then gradually declined to a minimum 28 h after serum addition. The initial synchrony decreased after this first cycle as shown by the smaller second increase of the S phase fraction, which began after 22 h. In order to determine the cell cycle length, we also counted the number of cells (Table 1). These counts confirmed the flow cytometric results and revealed that the main onset of cell division occurred about 24 h after serum stimulation. All cells finished the first division cycle within 28 h.
Figure 2.
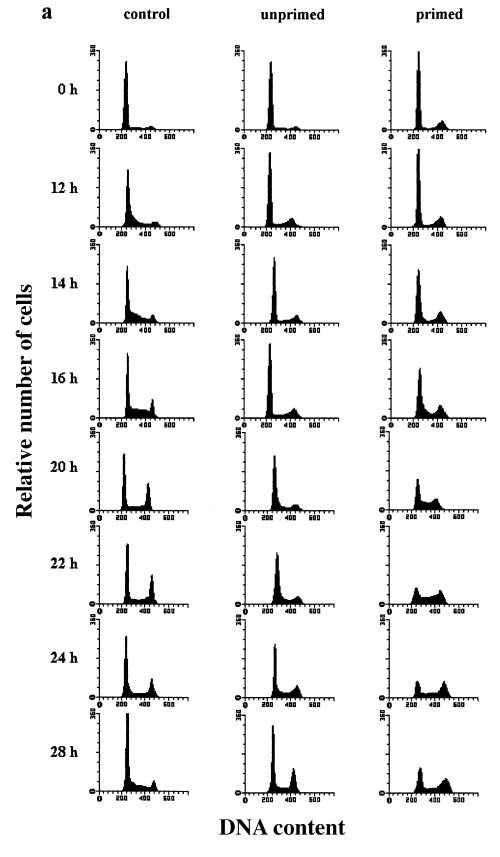
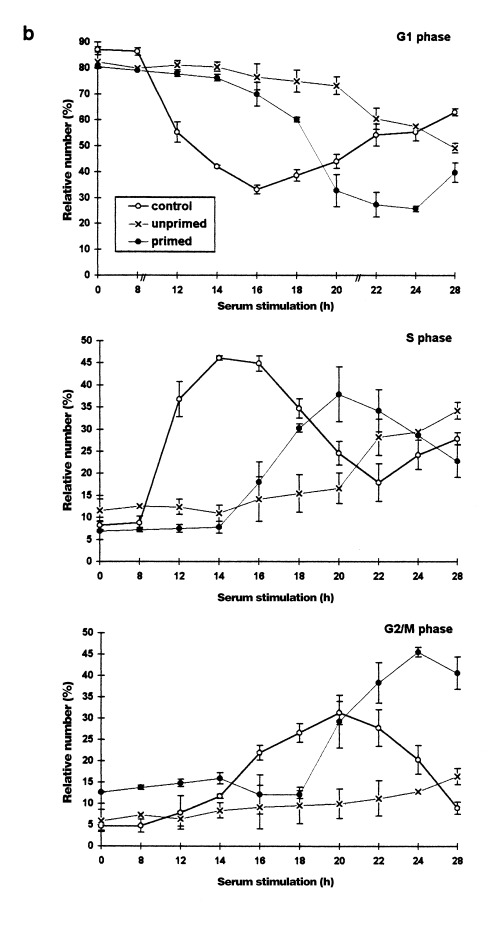
Heat shock effects on the cell cycle distribution of serum‐stimulated (synchronized) rat C6 cells analysed by flow cytometry. C6 cells synchronized by serum deprivation were stimulated to reenter the cell cycle by shifting the serum concentration to 20% (control cells). Unprimed cells were heat shocked (44 °C, 30 min) directly after serum stimulation. Primed cells were heat‐shocked (44 °C, 30 min) 24 h previously and then heat shocked again directely after serum stimulation. (a) Representative DNA‐histograms of cells harvested at the indicated times after serum stimulation. DNA‐content based on PI‐fluorescence (x axis), is plotted against relative number of cells (10 000 cells per histogram; yaxis). (b) Quantitative analysis of DNA‐histograms using the Lysis II and Cellfit research programs (Becton Dickinson). The proportion of cells in different phases of the cell cycle (top panel: G1 phase, mid panel: S phase, bottom panel: G2/M phase) is plotted against the time of serum stimulation. (○): control cells (×): heat‐shocked cells (●): primed cells. Values are means (±SE) of at least three independent experiments.
Table 1.
Relative cell numbers (% of control SD) after serum stimulation
To investigate the effects of heat shock on the kinetics of cell cycle entry and progression, cells were stressed for 30 min at 44 °C directly after growth stimulation. The results show that heat shock greatly prolonged the cell cycle and diminished the synchrony of the cells (Fig. 2a (unprimed) and 2b). In order to cover a complete cell cycle, heat‐shocked cells were analysed for up to 48 h after serum stimulation (Fig. 3). Compared to the control cells, the onset of DNA synthesis in heat‐shocked cells was shifted about 10 h – from 12 to about 22 h after serum addition. Furthermore, the S phase entry was less synchronous, as shown by the smaller maximum of S phase cells (38%) 28 h after growth stimulation. Subsequently, this fraction decreased and reached a more or less constant level from 32 up to 48 h after stimulation. Entry into G2/M phase occurred with a delay of 12 h. Comparison of the shifts in S phase and G2/M phase entry (10 and 12 h, respectively) indicates a prolongation of DNA‐synthesis of at least 2 h, which confirms the observation made with asynchronous cultures. Furthermore, the G2/M phase itself was considerably prolonged. The position of the maximum was shifted from 20 to 36 h after serum stimulation which indicates a prolongation of at least 4 h. Thereafter, the G1‐, S‐and G2/M fraction did not change significantly for up to 48 h. This shows a reduction of synchrony and also reveals that the G2/M phase prolongation in some cells exceeds 4 h.
In randomly proliferating cells, a priming shock was shown to induce thermotolerance, i.e. to shorten the cell cycle delays in response to a second shock. In the following series of experiments we wanted to investigate whether a priming shock delivered in the G0 phase also induces thermotolerance against cell cycle effects of a subsequent heat shock applied directly after serum stimulation. Therefore, synchronous cells were heat‐shocked in medium with low serum content for 30 min at 44 °C and were then allowed to recover at 37 °C. Twenty‐four hours after this initial heat shock, the cells were serum‐stimulated and heat‐shocked again under the same conditions.
The priming heat shock led to a slightly different state of arrest in the recovery phase. Compared to the arrested control cultures (Fig. 2a, top panels), the S phase fraction was lower and the G2/M fraction higher. This result shows that the few unarrested S phase cells accumulated in G2/M after the priming heat shock, consistent with the results in randomly proliferating cells. Figure 2a (primed) and 2b show that the heat shock effects observed in unprimed, synchronously proliferating cells were diminished or even reversed in primed cultures. In these cultures, cell cycle thermotolerance is documented by the shorter delays before S and G2/M phase entry. The beginning of DNA synthesis occurred 16 h after serum addition instead of 22 h in unprimed cells and was therefore delayed for only 4 instead of 10 h. In addition, the synchrony of the S phase resembled that of unstressed cells. The first significant increase in G2/M cells was observed 20 h after serum stimulation and was also delayed for only 4 h. Thus, no prolongation of S phase was detectable in primed cultures in contrast to unprimed heat‐shocked cells. Surprisingly, the restored synchrony of the G2/M entry in thermotolerant cells led to a particularly high maximum of cells in G2/M (> 45%) 24 h after serum stimulation. Even though primed cultures showed a higher proportion of G2/M cells before as well as during the first hours after serum addition, the increase of cells in this phase was probably not due to this fact. An explanation may be deduced from the observation that the G2/M fraction decreased from 14 to 18 h. This indicates that the cells arrested in G2/M before serum stimulation have already progressed into cell division and G1. Thus, the high G2/M maximum may be due to the fact that altogether more G1 cells were stimulated to progress through the cell cycle as implied by the larger and longer decline in the G1 fraction of primed cells.
Heat shock protein levels in randomly proliferating cells
One of the most well known heat shock responses is the induction of heat shock proteins (HSP). In mammalian cells, the major heat shock‐induced forms belong to the HSP70 family. Thus, these proteins are thought to play an important role during the recovery from stress (see De Maio 1999). In addition, in several studies high levels of HSP70 have been shown to be at least a major feature of thermotolerance (Venetianer et al. 1994; Thayer & Mirkes 1997; De Maio 1999). Therefore, we investigated the HSP70 expression in normal and thermotolerant C6 cells.
In rodent cells, the HSP70 family is mainly represented by the 70 and 68 kDa isoforms. The 70 kDa protein is homologous to HSP73 in HeLa cells. It is constitutively expressed and therefore, called heat shock cognate 70 (HSC70). HSP68 corresponds to HeLa HSP72, is detected only after stress in rodent cells and represents the most strongly heat‐inducible isoform (Fig. 4, Subjeck & Shyy 1986; Sainis et al. 1994; Venetianer et al. 1994). In order to compare the relative contents of HSC70 and HSP68, the latter is shown as a percentage of the HSC70 content of normal, unstressed cells in the graphs.
Figure 4.
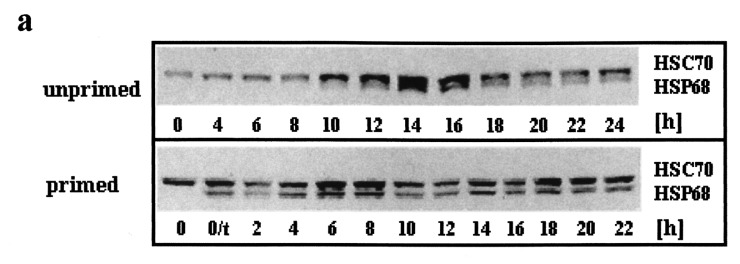
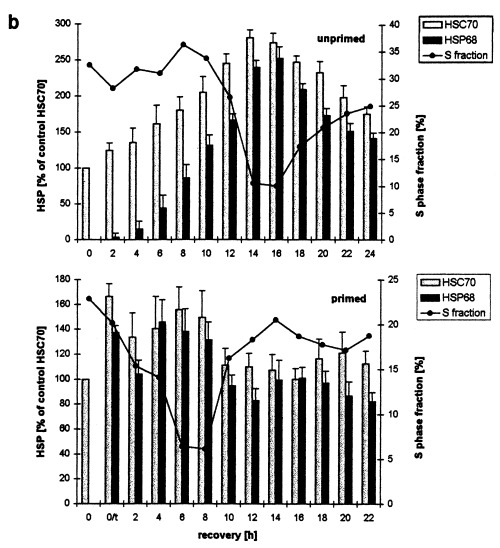
Western blot determination of HSC70 and HSP68 levels in asynchronously proliferating C6 cells after heat shock. Primed and unprimed cells (0 h) were heat shocked (30 min, 44 °C), harvested after the indicated recovery times at 37 °C and analysed by western blot. (a) Representative immunodetection using the SPA‐820 antibody that recognizes HSC70 as well as HSP68. (b) Western blots of unprimed (top panel) and primed C6 (lower panel) were quantitatively evaluated by video scanning measurement using the CREAM research software (Ver 4.1). Values of HSC70 (grey columns) and HSP68 (black columns) are relative amounts of pixel density given in percentage of the HSC70 value of unstressed, unprimed C6 cells (left ordinate) and are plotted against the recovery time. The fraction of cells in S phase is displayed as a line (right ordinate, see Figure 1 for other fractions). 0/t: primed C6 cells directly before the second heat shock. Values are means (±SE) of at least three independent experiments.
In unprimed, asynchronously proliferating C6 cells, the level of HSC70 increased continously for about 14–16 h after heat shock (Fig. 4a, unprimed and 4b, top panel, grey columns). After this period of time the relative level was about 275% when compared to the unstressed control (0 h). Thereafter, the HSC70 level decreased progressively after 24 h of recovery, the relative content however, was still about 180%. The induction of HSP68 occurred more slowly than that of HSC70. Only very low amounts were detectable during the first 4 h of recovery. During the next hours the amounts increased rapidly leading to a HSP68 maximum that was only slightly lower than that of HSC70 after 14–16 h. The decrease of HSP68 was slightly faster than that of HSC70: after 24 h of recovery HSP68 was reduced to 140%.
With respect to the flow cytometric determination of the kinetics of the S phase fraction (Fig. 4b, top panel, line) the changes in the HSP levels revealed temporal correlations. First, the induction of HSC70 and HSP68 started earlier than the onset of the cell cycle redistribution. Both HSPs were already induced during the first phase of the cell cycle arrest when C6 did not progress through the cell cycle at all. Maximal HSP levels 14–16 h after heat shock coincided with the highest degree of cell cycle redistribution, e.g. the minimum of the S phase fraction during the second phase of the cell cycle arrests. Finally, the subsequent reduction of both HSP levels was paralelled by the release of C6 cells from the cell cycle blocks.
To evaluate the possible protective effects of HSC70 and HSP68 on cell cycle progression after heat shock we also investigated their levels in primed C6 cells (Fig. 4a, primed and 4b, lower panel, grey columns). Directly before the second heat shock, primed cells displayed elevated levels of both HSPs due to the priming heat shock (0/t). When compared to the HSC70 content of unprimed control cells (0 h) the relative HSC70 and HSP68 levels had increased to about 170 and 140%, respectively. The second heat shock did not elevate the HSP levels considerably. Ten hours after heat shock HSC70 and HSP68 levels decreased to control levels and more or less remained at this level.
Even though the changes of HSP levels and the cell cycle redisribition (Fig. 4b lines) of primed and unprimed are clearly different, the reduction of HSP levels during the recovery in both cases parallels the release of C6 cells from the cell cycle block. Therefore, maximal HSP induction seem to be necessary in order to allow cells to escape from the cell cycle blocks.
Heat shock protein levels in synchronized cells
Figure 5 shows the kinetics of HSC70 and HSP68 levels in synchronous C6 cells after serum stimulation. We first investigated whether there is a cell cycle dependency in the HSC70 levels after serum stimulation without heat shock (Fig. 5a,b, top panels). During the first 14 h of serum‐stimulation, HSC70 levels remained approximately constant. A distinct increase of HSC70 was observed between 14 and 16 h. From 20 to 22 h the HSC70 level decreased drastically and reached a minimum of about 20–30%. Then, the HSC70 level increased again to about 140% after 28 h. Comparison of the changes in HSC70 levels with the cell cycle kinetics (Fig. 5b, top panel, line) revealed that the major HSC70 increase took place when cells were in mid and late S phase. The decline of HSC70 coincided with the G2/M maximum.
Figure 5.
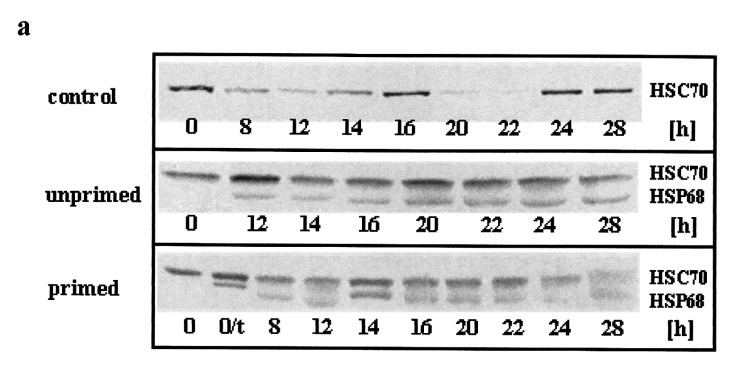
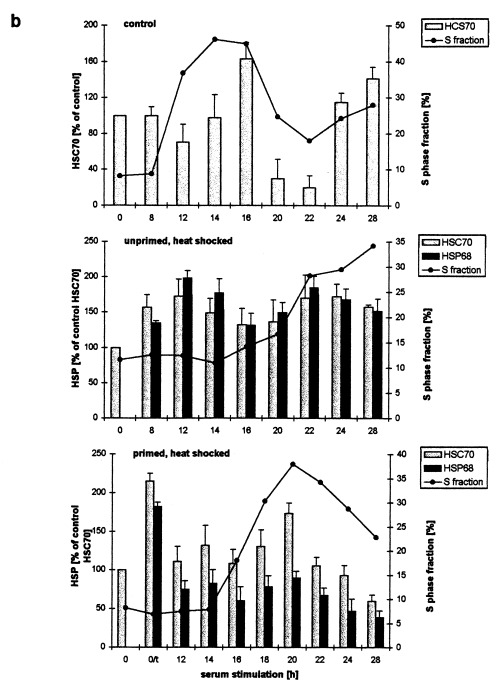
Western blot determination of HSC70 and HSP68 levels in synchronized C6 cells after serum stimulation and heat shock. C6 cells synchronized by serum starvation were serum stimulated (top panel) and heat shocked directly afterwards (middle and bottom panel). After the indicated times of serum stimulation cells were harvested and analysed by western blot. (a) Representative immunoblots using the SPA‐820 antibody that recognizes HSC70 and HSP68. (b) Western blots were quantitatively evaluated by video scanning measurement using the CREAM research software (Ver 4.1). Values of HSC70 (grey columns) and HSP68 (black columns) are relative amounts of pixel density given in percentage of the HSC70 value of unstressed, unprimed C6 cells and are plotted against the recovery time. The fraction of cells in S phase is displayed as a line (see Figure 2 for other fractions). 0/t: primed C6 cells directly before the second heat shock. Values are means (±SE) of at least three independent experiments.
After serum stimulation and subsequent heat shock treatment (30 min, 44 C) of unprimed C6 cells this pattern was completely changed (Fig. 5a,b, middle panels). HSC70 and, in addition, HSP68 increased after 8 h of serum stimulation (and recovery from the heat shock). Both HSPs reached their maximal level (175–200%) after 12 h and declined between 16 and 20 h to about 130–150%. After 22 h a second increase up to about 180% was observed, again followed by a slow decrease. The first HSP maximum occurred within the first phase of the cell cycle arrest when cells were arrested at their individual cell cycle position. The first significant increase of the S phase fraction followed after the decrease of HSP levels between 14 and 16 h (Fig. 5b, middle panel, line).
Shortly before the second heat shock treatment (30 min, 44 °C), synchronous, primed C6 cells (5a and b, bottom panels, 0/t) displayed high HSP levels due to the priming heat shock (HSP68 levels are given in percentage of HSC70 to allow a comparison). During the first 12 h of serum stimulation and recovery from the heat shock HSC70 and HSP68 levels both dropped drastically. HSC70 then remained rather constant up to 20 h when a significant increase took place. Thereafter, HSC70 decreased steadily to about 60% of the control level 28 h after serum stimulation and heat shock. HSP68 followed this pattern, the changes however, were much smaller and therefore, not significant. Only a small, insignificant decrease of HSPs was dectetable before the S phase entry. Like in control and unprimed cells however, a clear S phase specific increase was observed for HCS70 (Fig. 5b, bottom panel, line).
DISCUSSION
Heat shock‐dependent cell cycle blocks and delays in normal C6 cells
The flow cytometric DNA measurements show that heat shock applied to randomly proliferating C6 cells causes a biphasic cell cycle arrest of 18 h for all cell cycle fractions. During the first 10 h of recovery cells are not progressing through the cell cycle at all and are arrested at exactly the point where they were heat‐shocked (phase 1). However, after 10 h cells resume cell cycle progression and then become arrested at the cell cycle control points in G1 or G2/M (phase 2). G1 cells are arrested before the G1/S transition, whereas S and G2/M cells become arrested in G2/M. Due to methodical limitations we can only detect this progress for S phase cells which restart DNA synthesis and acumulate in G2/M from 12 to 16 h. However, the synchronous release of G1 and G2/M cells from their blocks after 18 h indicates that these cells progressed from 10 to 16 h and accumulated at the respective checkpoints.
The cell cycle blocks observed in G1 and G2/M in these experiments are in agreement with studies using a similar approach in other cell lines (Higashikubo et al. 1993; Sugano et al. 1995; Nitta et al. 1997). The parallel development of both blocks in a cell cycle phase‐dependent manner however, has only been described for CHO cells (Read et al. 1984; Hang & Fox 1995). This may be due to the focus on the G1/S transition in other studies. Deviations from this, possibly general, pattern of two cell cycle blocks, might be explained by cell line‐ or treatment‐specific differences in the arrest kinetics or by a transformation‐associated loss of a stringent G1 checkpoint as observed in CHO and HeLa cells, respectively (Read et al. 1984; Fox et al. 1985; Mackey et al. 1992; Van der Waal et al. 1997). The blocks in G1 and G2/M may also be descibed in terms of phase prolongation. Several studies have shown that heat shock prolongs all phases and transitions and that cell cycle phase‐specific differences often depend on the cell type and heat shock treatment (Rice et al. 1984; Coss 1986; Higashikubo et al. 1993).
In synchronized, serum‐stimulated C6 cells, heat shock delays progression into S and G2/M phase for 10 and 12 h, respectively. This indicates a G0 or G1 arrest and at least a two hour prolongation of S phase. The following G2/M phase is delayed for at least 4 h. On the basis of flow cytometric experiments it is not possible to distinguish whether the observed initial delay is due to a block in G0, before G0/G1 or in any of the parts of G1. Microscopic inspection of cells however, rather supports the assumption of a time shift of the early processes of cell cycle re‐entry (a G0, G0/G1 transition or an early G1 block): the morphological dedifferentiation which usually occurs in the first hours after serum stimulation is delayed after heat shock (data not shown). This is compatible with the notion that heat shock enhances differentiation in neuroblastoma cells, embryonic carcinoma cells and U937 leukaemia cells (Stoklosinski et al. 1992; Cellier et al. 1993; Maruyama et al. 1996).
Acquired cell cycle thermotolerance
The term ‘acquired thermotolerance’ was introduced to describe the ability of a priming stress to confer enhanced cellular resistence to a subsequent stress. With respect to the cell cycle, acquired thermotolerance was characterized only by an enhancement of cell survival (for review see Roti Roti et al. 1992). It was shown for example, that chronic heat shock treatment at 45 °C is lethal for asynchronous CHO and HeLa cells. However, when the cells had been exposed to a 35‐min heat shock at 45 °C followed by a 4–72 h recovery at 37 °C cell survival during the following chronic treatment was drastically enhanced (Read et al. 1984; Fox et al. 1985). Subsequent investigations have shown that thermotolerance can be induced by chronic and acute heat shock treatment and protects against heat‐killing due to subsequent heat exposures in both approaches (Read et al. 1984; Fox et al. 1985; Li et al. 1990; Mackey et al. 1992; Roti Roti et al. 1992).
No severe induction of cell death via loss of clonogenicity, necrosis or apoptosis was observed after hyperthermic treatment of C6 cells. To elucidate whether a priming heat shock may protect the ongoing cell cycle against heat‐induced delays, the cell cycle kinetics of primed cells (44 °C, 30 min followed by 24 h recovery) after a second heat shock (44 °C, 30 min) were determined. The heat shock‐induced cell cycle blocks were not abolished, but the first arrest phase (without changes in redistribution) was missing and therefore, resumption of cell cycle progression (cell cycle phase redistribution) as well as the release from the subsequent blocks occurred earlier in primed cells. In addition, no stress‐induced prolongation of S phase was detectable in primed synchronous cultures after delayed G0/G1 progression. Therefore, the priming heat shock conferred thermotolerance in terms of shortening the delays, thus representing a new manifestation of thermotolerance besides the enhancement of cell survival.
In agreement with several studies investigating thermotolerance protection from lethal heat shock (Read et al. 1984; Rice et al. 1984; Fox et al. 1985; Li et al. 1990), cell cycle thermotolerance of C6 cells developed in all cell cycle fractions. However, in contrast to some of the studies mentioned above major differences between the fractions such as lower thermotolerance of S phase cells were not observed.
The priming heat shock led to a transient redistribution of asynchronous cells mainly in G1 or G2/M after 16 h of recovery. At the time of the second shock (24 h) cells were again perturbed in all cell cycle phases, and formerly G1 or G2/M arrested cells were now in S or G2/M and G1, respectively. The second shock arrested cells in the other block but the duration of the arrests was shortened due to the thermotolerant state. Primed G1 cells were more resistent to an arrest in G2/M and vice versa, which clearly demonstrates that thermotolerance is independent of the phase in which the priming shock is applied. In addition, the assumption that thermotolerance is cell cycle phase‐independent was confirmed by the fact that protection of the cell cycle machinery can be induced in noncycling G0 cells.
For both synchronously and asynchronously proliferating cells only thermotolerant cells that had recovered for 24 h after the priming shock were used. In other studies (Read et al. 1984; Li et al. 1990; Mackey et al. 1992), thermotolerance against heat shock‐induced cell death developed very fast during the recovery from acute heat shock. For example, in CHO cells thermotolerance was detectable already 4–8 h after heat shock, became maximal after about 12 h and then decayed progressively up to 72 h (Fox et al. 1985). Therefore, slightly different thermotolerance kinetics may result also in C6 cells when using other recovery periods. However, we chose this interval because the cells are then perturbed in all cell cycle phases and at high HSP levels.
Heat shock protein levels and acquired thermotolerance
In rat C6 glioma cells as in other rodent cells (Subjeck & Shyy 1986), the moderately heat‐inducible HSC70 (heat shock cognate 70) is expressed constitutively whereas HSP68, the major stress‐inducible isoform, is synthesized only after stress. In synchronized serum‐stimulated C6 cells, HSC70 expression is enhanced during S phase (Zeise et al. 1998). This implies an S phase‐specific function of this isoform under physiological conditions.
Both HSC70 and HSP68 isoforms act as molecular chaperones, which faciliate folding of nascent and denatured proteins and support translocation of proteins into subcellular compartments (for review see Feige & Polla 1994). S phase‐specific expression of HSC70 and/or HSP70 isoforms as seen for synchronized C6 cells has been shown in several unstressed as well as heat‐shocked rodent and human cell lines (some of the latter constitutively expressing the human HSP68 homologue HSP70 or HSP72) (Sainis et al. 1994; Hang & Fox 1995; Taira et al. 1997, for further detail see Zeise et al. 1998). Moreover, in CV1 monkey kidney cells, induction of HSP72 mRNA was correlated with the G2 phase after stimulation of proliferation by infection with wt SV40 and adenovirus 5 (Sainis et al. 1994). However, a clear phase‐specific function of HSPs has not yet been identified.
After a 30‐min heat shock at 44 °C HSC70 is enhanced and HSP68 expression is induced in randomly proliferating as well as in synchronized C6 cells. In proliferating C6 cells, maximal relative HSP levels of about 275% are reached after 14–16 h of recovery from the heat shock. Thereafter, the decrease of both isoforms parallels the release of C6 cells from the cell cycle blocks. It is thus tempting to speculate that HSPs might be necessary to overcome the heat‐induced blocks.
The phenomenon of acquired thermotolerance has been attributed to the increased production, especially the 70 kDa family of heat shock proteins (Subjeck et al. 1982; De Maio 1999). In agreement with this theory, a multiplicity of agents which elicit stress protein synthesis also confer the thermotolerant state (Kampinga et al. 1995; Hedge et al. 1995). Furthermore, the kinetics of thermotolerance induction and decay correlates with the kinetics of stress protein synthesis and degradation (Subjeck et al. 1982; Mizzen & Welch 1988; Thayer & Mirkes 1997).
The redistribution of cell cycle phases in asynchronous C6 cells does not start until 8–10 h after heat shock when the HSP levels have increased drastically. Considering that heat shock is a stressor with a wide array of effects (see Welch 1992) this lag‐phase might arise as a consequence of inhibitory effects on basic metabolic processes such as transcription, protein synthesis and ATP generation. In HeLa cells for example, heat shock‐induced cell cycle phase redistributions occurs after a lag‐phase without obvious cell cycle progression; the onset of redistribution is found to be correlated with the removal of proteins that have accumulated in the nucleus after heat shock (Roti Roti et al. 1986). Thermotolerance on the other hand, is established after heat shock with respect to several investigated endpoints, e.g. energy depletion, transcription, splicing, translation, protein aggregation, nuclear morphology and cytoskeletal redistribution (Roti Roti, Uygur & Higashikubo, 1986; Mizzen & Welch 1988; Welch & Mizzen 1988). In several studies, a contribution of HSP70 isoforms in the establishment of these thermotolerance manifestations has been described (Subjeck et al. 1982; Welch & Mizzen 1988; Beck & De Maio 1994; Kampinga et al. 1995; Kim, Ouyang & Li, 1995; De Maio 1999). The elimination of the first arrest phase after heat shock seen in primed proliferating C6 cells may therefore, be based on the generally faster recovery of metabolic processes in thermotolerant cells due to HSP accumulation.
The timing of the changes of HSP levels in C6 cells however, also indicates a further role for these proteins for cell cycle progression after heat shock as well as for the development of cell cycle thermotolerance. The release of C6 cells from the cell cycle blocks (second arrest phase) follows the maximum of HSP levels in unprimed and primed C6 cells. Therefore, one may speculate that the HSP (chaperone) function is critical for overcoming the block. The resumption of cell cycle progression after the decline of both HSPs on the other hand, may also be explained by an inhibitory function of HSPs on one or more positive cell cycle regulator(s). However, no such targets of HSPs have as yet been identified. That specific targets of HSPs exist can be derived from the fact that HSP70 inhibits the stress‐activated protein kinase (JNK/SAPK) thereby conferring resistance to heat‐induced apoptosis. This function of HSP70 is not dependent on its chaperon function because ATPase‐and substrate binding domain mutations do not inhibit HSP70‐mediated kinase inhibition ((1997), (1998)).
Acknowledgements
We thank M. G. Vicker for helpful discussions and reading the manuscript.
REFERENCES
- Beck SC, De Maio A.(1994). Stabilisation of protein synthesis in thermotolerant cells during heat shock. Association of heat shock protein‐72 with ribosomal subunits of polysomes. J. Biol. Chem. 269,21803–21811. [PubMed] [Google Scholar]
- Benda P, Lightbody J, Sato G, Levine L, Sweet W.(1968). Differentiated rat glial cell strain in tissue culture. Science 161,370–371. [DOI] [PubMed] [Google Scholar]
- Cellier MFM, Taimi M, Chateau MT, Cannat A, Marti J.(1993). Thermal stress as an inducer of differentiation of U937 cells. Leukemia Res. 17,649–656. [DOI] [PubMed] [Google Scholar]
- Coss RA.(1986). Decay of thermal resistence following acute heating is independent of the G1‐ to S‐phase transition. Rad. Research 107,143–146. [PubMed] [Google Scholar]
- De Maio A.(1999). Heat shock proteins: facts, thoughts and dreams. SHOCK 11,1–12. [DOI] [PubMed] [Google Scholar]
- Elledge SJ.(1996). Cell cycle checkpoints: preventing an identity crisis. Science 274,1664–1671. [DOI] [PubMed] [Google Scholar]
- Feige U, BS Polla.(1994). Heat shock proteins: the hsp70 family. Experientia 50,979–986.7988673 [Google Scholar]
- Fox MH, Read RA, Bedford JS.(1985). The cell cycle dependence of thermotolerance. 3. HeLa cells heated at 45C. Rad. Res. 104,429–442. [PubMed] [Google Scholar]
- Gabai VL, Meriin AB, Mosser DD et al. (1997). Hsp70 prevents activation of stress kinases. J. Biol. Chem. 272,18033–18037. [DOI] [PubMed] [Google Scholar]
- Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY.(1998). Role of Hsp70 in regulation of stress‐kinase JNK: implications in apoptosis and aging. FEBS Lett 438,1–4. [DOI] [PubMed] [Google Scholar]
- Hang H, Fox MH.(1995). Expression of HSP70 induced in CHO cells by 45.0 C hyperthermia is cell cycle associated and DNA synthesis dependent. Cytometry 19,119–125. [DOI] [PubMed] [Google Scholar]
- Hedge RS, Zuo J, Voellmy R, Welch WJ.(1995). Short circuiting stress protein expression via tyrosine kinase inhibitor, herbimycin. Am. J. Cell. Physiol. 165,186–200. [DOI] [PubMed] [Google Scholar]
- Higashikubo R, White RA, Roti Roti JL (1993). Flow cytometric BrdUrd‐pulse chase study of heat‐induced cell cycle progression delays. Cell Prolif. 26,337–348. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Bristing JF, Stege GJ, Burgman PW, Konings AW.(1995). Thermal protein denaturation and protein aggegation in cells made thermotolearnt by various chemicals: role of heat shock proteins. Exp. Cell Res. 219,536–546. [DOI] [PubMed] [Google Scholar]
- Kim D, Ouyang H, Li GC.(1995). Heat shock protein HSP70 accelerates the recovery of heat‐shocked mammalian cells through its modulation of heat shock transcription factor HSF‐1. PNAS 92,2126–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Wong RS, Dewey WC.(1990). Thermal tolerance during S phase for cell killing and chromosomal abberations. Radiat. Res. 122,193–196. [PubMed] [Google Scholar]
- Mackey MA, Anolik SL, Roti Roti JL.(1992). Cellular mechanisms associated with the lack of chronic thermotolerance expression in HeLa S3 cells. Cancer Res. 52,1101–1106. [PubMed] [Google Scholar]
- Martin ML, Regan CM.(1991). Transient heat shock in mid‐G1‐phase of the C6 glioma cell cycle impairs entry into S‐phase. Toxicol. Lett. 59,197–202. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Umezawa A, Kusakari S, Kikuchi H, Nozaki M, Hata J‐I.(1996). Heat shock induced differentiation of human embyonal carcinoma cells into trophectoderm lineages. Exp. Cell Res. 224,123–127. [DOI] [PubMed] [Google Scholar]
- Mizzen LA, Welch WJ (1988). Characterisation of the thermotolerant cell. I. Effects on protein synthesis activity and the regulation of heat‐shock protein 70 expression. J. Cell Biol. 106,1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta M, Okamura H, Aizawa S, Yamaizumi M.(1997). Heat shock induces transient p53‐dependent cell cycle arrest at G1/S. Oncogene 15,561–568. [DOI] [PubMed] [Google Scholar]
- Read RA, Fox MH, Bedford JS.(1984). The cell cycle dependence of thermotolerance. CHO cells heatedat 45C. Rad. Res. 98,491–505. [PubMed] [Google Scholar]
- Rice GC, Gray JW, Dean PN, Dewey WC.(1984). Fluorescence‐activated cell sorting analysis of the induction and expression of acute thermal tolerance within the cell cycle. Cancer Res. 44,2368–2376. [PubMed] [Google Scholar]
- Roti Roti JL, Mackey MA, Higashikubo R.(1992). The effects of heat shock on cell proliferation. Cell Proliferation 25,89–99. [DOI] [PubMed] [Google Scholar]
- Roti Roti JL, Uygur N, Higashikubo R.(1986). Nuclear protein following heat shock: protein removal kinetics and cell cycle rearrangements. Rad. Res. 107,250–261. [PubMed] [Google Scholar]
- Sainis I, Angelidis C, Pagoulatos G, Lazaridis I.(1994). The hsc70 gene which is slightly induced by heat is the main virus inducible member of the hsp70 gene family. FEBS Lett 355,282–286. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Raj ABJ (1987). Transient increase in intracellular concentration of adenosine 3 5‐cyclic monophosphate results in morphological and biochemical differentiation of C6‐glioma cells in culture. J. Neurosci. Res. 17,135–141. [DOI] [PubMed] [Google Scholar]
- Stoklosinski A, Kruse H, Richter‐Landsberg C, Rensing L.(1992). Effects of heat shock on proliferation and differentiation of neuroblstoma (NIE 115) cells. Exp. Cell Res. 200,89–96. [DOI] [PubMed] [Google Scholar]
- Subjeck JR, Sciandra JJ, Johnson RJ.(1982). Heat shock proteins and thermotolerance: a comparisonof induction kinetics. Br. J. Radiol. 55,579–584. [DOI] [PubMed] [Google Scholar]
- Subjeck JR, Shyy T‐T (1986). Stress protein systems of mammalian cells. Am. J. Physiol.,C1–C17. [DOI] [PubMed]
- Sugano T, Nitta M, Ohmori H, Yamaizumi M.(1995). Nuclear accumulation of p53 in normal human fibroblasts is induced by various cellular stresses which evoke the heat shock response independently of the cell cycle. Jpn J. Cancer Res. 86,415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira T, Narita T, Iguchi‐Ariga SMM, Ariga H.(1997). A novel G1‐specific enhancer identified in the human heat shock protein 70 gene. Nuceic Acids Res. 25,1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer JM, Mirkes PE (1997). Induction of HSP72 and transient nuclear localisation of Hsp73 and Hsp72 correlate with the acquisition and loss of thermotolerance in postimplantation rat embryos. Dev. Dynamics 208,227–243. [DOI] [PubMed] [Google Scholar]
- Van der Waal R, Malyapa RS, Higashikubo R, Roti Roti JL.(1997). A comparison of the modes and kinetics of heat‐induced cell killing in HeLa and L5178Y cells. Radiat. Res. 148,455–462. [PubMed] [Google Scholar]
- Venetianer A, Pirity M, Hever‐Szabo A.(1994). The function of heat shock proteins in stress tolerance. Cell Biol. Int. 18,605–615. [DOI] [PubMed] [Google Scholar]
- Welch WJ.(1992). Mammalian stress response, cell physiology, structure and function of stress proteins, and implications for medicine and disease. Physiol. Rev. 72,1063–1081. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Mizzen LA.(1988). Characterisation of the thermotolerant cell. II. Effects on the intracellular distribution of heat‐shock protein 70, intermediate filaments, and small nuclear ribonucleoprotein complexes. J. Cell Biol. 106,1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RS, Kapp LN, Krishnaswamy G, Dewey WC.(1993). Critical steps for induction of chromosomal abberations in CHO cells heated in S phase. Radiat. Res. 133,52–59. [PubMed] [Google Scholar]
- Yonezawa M, Othsuka T, Matsui N et al. (1996). Hyperthermia induces apoptosis in malignant fibrous histiocytoma cells in vitro. Int. J. Cancer 66,347–351. [DOI] [PubMed] [Google Scholar]
- Zeise E, Kühl N, Kunz J, Rensing L.(1998). Nuclear translocation of the stress protein HSC70 during S phase in rat C6‐glioma cells. Cell Stress Chaperones 3,94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]