

Abstract.
We assessed the radiosensitivity of the grade III human glioma cell line U‐373MG by investigating the effects of radiation and the specific protein kinase C inhibitor, calphostin C on the cell cycle and cell proliferation. Irradiated glioma U‐373MG cells progressed through G1‐S and underwent an arrest in G2‐M phase. The radiosensitivity of U‐373MG cells to graded doses of either photons or electrons was determine by microculture tetrazolium assay. The data was fitted to the linear‐quadratic model. The proliferation curves demonstrated that U‐373MG cells appear to be highly radiation resistant since 8 Gy was required to achieve 50% cell mortality. Compared to radiation alone, exposure to calphostin C (250 n m) 1 h prior to radiation decreased the proliferation of U‐373MG by 76% and calphostin C provoked a weakly synergistic effect in concert with radiation. Depending on the time of application following radiation, calphostin C produced an additive or less than additive effect on cell proliferation. We postulate that the enhanced radiosensitivity observed when cells are exposed to calphostin C prior to radiation may be due to direct or indirect inhibition of protein kinase C isozymes required for cell cycle progression.
Introduction
Recently, considerable attention has been given to the possibility of utilizing protein kinase C (PKC) inhibitors such as calphostin C, UNC‐01, tamoxifen, hypericin and PKC antisense as chemotherapeutic adjuncts for the treatment of malignant gliomas (Ikemoto et al. 1995; Baltuch et al. 1993; Couldwell et al. 1993; 1994b, 1994a; Dean et al. 1994a; Dean & McCay 1994b; Pollack, Kawecki & Lazo 1996; Pollack & Kawecki 1997; Ahmad et al. 1994). PKC has been targeted in malignant gliomas since their rapid proliferation rates compared to nontransformed astrocytes have been functionally linked to inherently high levels of glioma PKC, resulting in excessive activation of PKC‐mediated pathways (Couldwell et al. 1991; Couldwell et al. 1994c; Baltuch & Yong 1996). Anti‐PKC therapy (Couldwell et al. 1994a) in combination with radiation may prove to be promising for controlling glioma proliferation since radiation therapy is used for the early treatment of localized gliomas. However, heterogeneity in intrinsic and acquired radiation resistance in gliomas is a major obstacle to the efficacy of radiation therapy (Yang et al. 1991) making adjunct therapies necessary.
Previous work with nonspecific PKC inhibitors (i.e. tamoxifen, isoquinoline sulphonamide H7 and staurosporine) revealed the possibility that PKC inhibitors may act as radiation sensitizers (1992b, 1992a; Zhang et al. 1992; Couldwell et al. 1993; Zhang et al. 1993a; Zhang et al. 1996). However, nonselective protein kinase inhibitors have an IC50 (concentration required for 50% inhibition) for PKC in the micromolar concentration range and possible inhibition of multiple kinase(s) could not be excluded (Mori et al. 1980). Consequently, these studies are being confirmed with specific PKC inhibitors, i.e. sangivamycin (Hallahan et al. 1992a), hypericin (Zhang et al. 1996), chelerythrine (Chumra et al. 1997) and UCN‐01 (Tsuchida & Urano 1997).
Inhibition of PKC may be beneficial since PKC activation is one of the initial events following ionizing radiation exposure and PKC is involved in radiation induced cell cycle arrest at checkpoints associated with genome integrity (Hallahan et al. 1991a; Hallahan et al. 1992b). The G2 phase arrest is important for cell survival since irradiated cells repair their DNA during this checkpoint and PKC inhibitors increase radiosensitivity by attenuating the radiation‐induced G2 arrest (Hallahan et al. 1992b; Zhang et al. 1993a; Bernhard et al. 1994a; Bernhard, McKenna & Muschel 1994b; Maity et al. 1997).
Current postulates for radiation‐induced PKC activation are that DNA damage may initiate PKC activation, or that radiation directly induces the oxidation of PKC (Hallahan et al. 1992b) or that radiolysis of cytosolic and bound water produces hydrogen peroxide, hydroxyl and superoxide radicals (Hutchinson 1985). These oxidants stimulate tyrosine kinase and lipid peroxidation (Nakajima & Yukawa 1996), i.e. hydrolysis of phospholipase C resulting in arachidonate, diacylglycerol and inositol triphosphate causing calcium mobilization and PKC activation (Nicotera et al. 1986). Radiation‐mediated signal transduction also provokes disturbances in the PKC‐phospholipase A2 pathway and affects the transcription of early response genes (i.e. TNF‐α, Egr‐1 and jun; 1991b, 1991a; Hallahan et al. 1992b) whose transcription can be blocked with PKC inhibitors. Ionizing radiation also promotes PKC‐α mRNA expression in Syrian hamster embryo cells in a dose and radiation type dependent manner (Woloschak et al. 1990).
Given the growing evidence for the potential therapeutic application of PKC inhibitors and the unknown radiosensitivity of human glioma U‐373MG cells, we investigated the effects of radiation and the PKC inhibitor, calphostin C on U‐373MG cell cycle and proliferation. Our studies focused on the U‐373MG cell line since we have previously characterized its total PKC activity (Acevedo‐Duncan & Zhang, 1994), ultrastructural movement of PKC‐α and PKC‐β (Acevedo‐Duncan et al. 1995), and effects of calphostin C on the U‐373MG cell cycle (Acevedo‐Duncan et al. 1997). Calphostin C, a specific perylene quinone, is derived from the fungus Cladosporium cladosporioides and is a selective and potent PKC irreversible inactivator with an IC50 of 0.05 µM (Kobayashi et al. 1989). The mechanism by which calphostin C inhibits PKC and its effects on various glioma cell lines has been described (Gopalakrishna, Chen & Gundimeda 1992; Ikemoto et al. 1995; Pollack & Kawecki 1997). Calphostin C inhibits both phorbol ester binding and phosphotransferase activity by binding to the regulatory domain of PKC without competing for phospholipids (Gopalakrishna et al. 1992). Calphostin C is activated by brief (min) exposure to light and may be of value in photodynamic therapy (Burns et al. 1991).
METHODS
Passage of human glioma cells
The U‐373MG cell line was obtained from the American Tissue Culture Collection (Rockville, MD, USA). Cells were seeded (1 × 106) and grown as monolayers in 75 cm2 flasks containing 90% minimal essential media (MEM), 10% fetal calf serum (FCS), 2 m m l‐glutamine and antibiotics (penicillin 10 U/ml and streptomycin 10 µg/ml) according to Ponten & MacIntyre (1968).
Cell cycle analysis of gliomas treated with electrons
Standard radiation procedures developed at the Moffitt Cancer Institute (Tampa, FL, USA) were used to irradiate U‐373MG cells. Flasks were exposed to 6 MeV electrons at a dose rate of 0.45 Gy/sec using a Siemens linear accelerator for the dose ranges and time periods indicated in the result section. Cell cycle analysis was performed as previously described (Acevedo‐Duncan et al. 1997). Briefly, cells were incubated in serum and removed at specific times, washed in phosphate buffered saline (PBS), trypsinized and fixed in cold ethanol. Single nuclei were prepared by incubating cells with 0.04% pepsin followed by 2N HCL and 0.1 m sodium borate. Nuclei from irradiated U‐373MG cells were stained with propidium iodide (PI) by modification of Yang et al. (1991). Nuclei were stained with PI to measure DNA content and the distributions of 40 000 nuclei were quantified using a FAC STARPlus, flow cytometer and CellFIT Cell Cycle Analysis program (Version 2.01.2; Becton Dickinson, San Jose, CA, USA).
Microculture tetrazolium (MTT) assay
The colorimetric assay containing 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT; Alley et al. 1988) was selected to measure cell proliferation following irradiation. The MTT assay does not quantify cell survival and is not a substitute for the standard clonogenic assay. The rationale for selecting the MTT assay as opposed to the standard clonogenic assay which measures clonogen survival was based on a glioma radiosensitivity study (Ramsey, Ward & Bleehen 1992). The study compared the clonogenic assay with the more rapid MTT assay and concluded that results from the MTT assay can be correlated with the clonogenic assay (Ramsey et al. 1992). Additionally, there has been increasing use of MTT assay for radiosensitivity testing (Carmichael et al. 1987; Price & McMillian 1990; Ramsey et al. 1992; Zhang et al. 1992; Zhang et al. 1993a; Zhang et al. 1996) and the MTT assay has been shown to correlate with viable glioma cell numbers (Zhang et al. 1996). The procedures used to measure the effects of radiation and/or calphostin C on cell proliferation have been described (Acevedo‐Duncan et al. 1997). Briefly, cells were plated on 24‐well plates at a density of 104 cells/well and irradiated and/or incubated with various concentrations of calphostin C under fluorescent light. Following, initial exposure to calphostin C, additional calphostin C was not applied or removed during the 7 day incubation period. On day 7, MTT (0.014 mg/ml) was added and after 4 h incubation at 37 °C, media was aspirated and dimethlysulfoxide (DMSO) was added to solubilize the MTT‐formazan product. Following mixing, absorbance was read at a wavelength of 570 nm using a plate reader (MR 700 Dynex; Dynatech Laboratories Inc., Chatilly, VA, USA).
Output of linear accelerator
The Moffitt linear accelerator is used for patient radiation therapy and its output is checked daily using an ionization chamber. The dose rate of photons and electrons was verified within 2% and 3%, respectively, which is very acceptable.
Statistics
Statistical determination and fits to the linear quadratic equation (L‐Q equation) was by Student’s t‐test using Minitab program (Mininc. State College, PA, USA).
RESULTS
Effects of radiation on U‐373MG cell cycle progression
The consequences of cell cycle modulators on the cell cycle depend on the position of the cell cycle phase in which they are applied (Sinclair 1968; Allalunis‐Turner, Barron & Day 1997). To establish the cell cycle effects of irradiating U‐373MG cells in G1 we attempted a reversible G0/G1 arrest by serum starvation. U‐373MG were seeded at 5 × 105 cells/flask and incubated at 37 °C in media plus 10% serum for 71 h. Subsequently, cells were washed with and incubated in serum free media for 24 h. Cells were washed again, incubated in media containing 10% serum and harvested at the indicated times for flow cytometric analysis. As shown in Fig. 1a, serum deprivation for 24 h produced a cell population consisting of G0/G1, 75%; S, 12%; and G2M, 13%. Since serum starvation did not produce a synchronous G0/G1 cell population, we examined the cell cycle effects of radiation on a high G0/G1 semisynchronous cell population. These results are consistent with published data indicating that it is difficult to completely arrest transformed cells (Cacace et al. 1993). Electrons were arbitrarily chosen to generate the radiation data presented in 1, 3 because the photon and electron proliferation curves in Fig. 2 were similar. Figure 1 illustrates the cell cycle distributions of (85% confluent) U‐373MG asynchronous control cells and cells treated with electrons (6.76 Gy). As shown in Fig. 1a, radiation did not produce a G1 arrest. The absence of a radiation induce G1 arrest is expected since the p53 tumor suppressor is mutated in U‐373MG and there is no functional p53 pathway (Russell et al. 1995) which contributes in part to the intrinsic radiation resistance found in U‐373MG cells. However, compared to controls, radiation (electrons, 6.76 Gy) provoked a G2M arrest as seen by the prolonged and augmented G2M phase (Fig. 1b).
Figure 1.
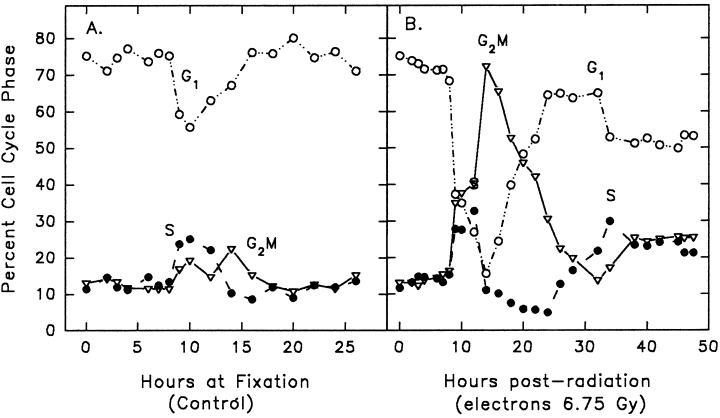
Cell cycle distribution of U‐373MG as a function of time. Cells were serum starved for 24 h and serum stimulated to enter the cycle. Subsequently, cells were sham irradiated (control, a) or treated with electrons (6.75 Gy, b). Symbols represent G1 (○), S (●), and G2M (▿). Data is representative of two independent experiments. Number of events collected was 40 000 per time point and treatment group.
Figure 3.

Survival of U‐373MG treated with calphostin C and radiation. ● Represents the survival of U‐373MG as a function of calphostin C concentration. ● Represents the survival of U‐373MG irradiated with electrons (6.76 Gy) in the presence of varying concentrations of calphostin C. Irradiation was with 6 MeV using a dose rate of 0.45 Gy/sec for a duration of exposure of 15 s. Additive model is depicted by the dotted line and □. Data below the additive model is weakly synergistic. Data are from two independent experiments with 12 control replicates and 3 experimental replicates per experiment. Exposure times were (a) 1 h prior to radiation (b) 9 h (c) 14 h (d) 20 h, and (e) 37 h post radiation, respectively.
Figure 2.
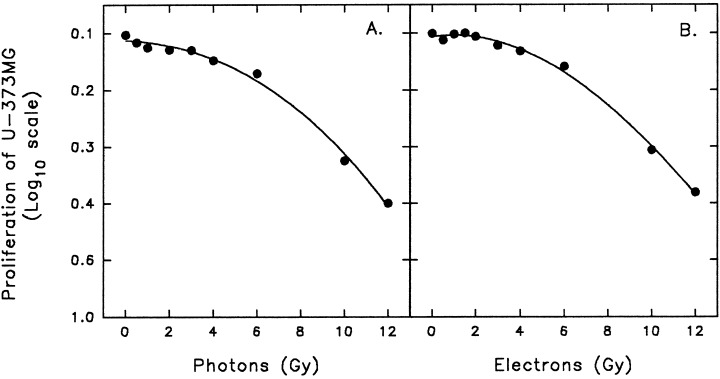
Radiation dose–response curve of U‐373MG glioma cells. (a) photons; (b) electrons. Control cells were sham irradiated. The data is a representative experiment and are expressed as the means ± SEM of triplicate determinations. SEM error bars are present but not visible due to the reproducibility of data.
Effects of radiation on U‐373MG proliferation
The effects of radiation on U‐373MG cell proliferation was evaluated by microculture tetrazolium (MTT) assay which quantifies cell number by tetrazolium dye reduction (Alley et al. 1988). Glioma U‐373MG were seeded into 24‐well plates at a density of 104 cells/well and cells were irradiated 24 h post cell plating. To compare cell proliferation following irradiation with either photons or electrons, cells were irradiated with either 6 mV or 6 MeV (Siemens linear accelerator; Siemens Medical Systems Inc., Iselin, NJ, USA) using either a dose rate for photons (0.035 Gy/sec) or for electrons (0.45 Gy/sec) and a single‐dose exposure ranging from 0 to 10 Gy. Cells were then incubated at 37 °C and on day seven, MTT was added and following a 4‐h incubation, cells were solubilized and absorbance measured. Photons and electrons produced the same radiobiological effects on U‐373MG cell proliferation (Fig. 2). The proliferation curves demonstrate that the U‐373MG are radiation resistant since 50% cell kill was achieved with 8 Gy. The data shown in Fig. 1. fitted a shouldered survival curve and the Dq (quasi‐threshold dose), Do (mean lethal dose; slope = 1/Do), n (extrapolation number; ability of cells to accumulate sublethal XRT damage), and D̄ (mean intrinsic radiation sensitivity), was calculated (Weichselbaum, Hallahan & Chen 1992). For the photon survival curve, we calculated Dq = 4.4 Gy, D0 = 5.8 Gy, D̄ = 8 Gy, n = 2.14, α = 0.03 Gy−1 and β = 0.007 Gy−2. For the electron survival curve Dq = 4.5 Gy, D0 = 5.7 Gy, D̄ = 4.5 Gy, n = 2.18, α = 0.03 Gy−1 and β = 0.007 Gy−2. The survival curves were constructed using a log 10 scale for proliferation of the U‐373MG cells and the dose of electrons and photons is in Gy. The extrapolation number (n) was calculated to be 2.14 and 2.18 for the electron survival curve. These n‐values reflect a relatively wide shoulder of 4.4 Gy (electron; Dq) and 4.5 (photons; Dq). However, the D0 is much larger than normal (usually 1–2 Gy).
Effects of radiation and calphostin C on U‐373MG proliferation
Since the effectiveness of PKC inhibitors may depend on the time of application of the PKC inhibitor following radiation, we examined the effect on radiosensitivity of various calphostin C concentrations at 1 h prior to radiation and at 9, 14, 20 and 37 h post radiation (Fig. 3). Cells were serum deprived for 24 h and prepared as in Fig. 1. The initial cell population at time zero consisted of G0/G1, 70% ± 0.828 SEM; S, 14% ± 0.523 SEM; and G2M, 13% ± 0.832 SEM (n = 10 replicates). Calphostin C was activated by exposing calphostin C treated cells to flourescent light for 5 min. Once cells were treated with calphostin C the media was not changed throughout the entire 7 day incubation period.
Figures 3a and 3d show that calphostin C (250 n m) at 1 h prior to radiation and in the 20 h controls decreased proliferation to similar levels (0.34 ± 0.050 SEM and 0.29 ± 0.062, respectively). Another pattern in the effectiveness of calphostin C in reducing proliferation is seen at 9, 14 and 37 h of calphostin C (250 n m) treatment. In these cells proliferation decreased to 0.51 ± 0.07 SEM, 0.52 ± 0.06 SEM and 0.52 ± 0.06 SEM, respectively. As stated in the discussion section, future flow cytometric studies will address the exact cell cycle phase at specific times of calphostin C application.
The radiation data demonstrated that compared to irradiated controls, pretreatment with calphostin C (100 n m) for 1 h decreased cell proliferation by 26% (P = 0.048; Fig. 3a). When the proliferation of irradiated controls are compared to the proliferation of irradiated and calphostin C (250 n m) treated cells, calphostin C (250 n m) enhanced cell radiosensitivity by 76% (P = 0.0001; Fig. 3a). These results indicate that calphostin C at either 100 n m or 250 n m is weakly synergistic with radiation. An additive model is depicted on the graphs and shows the predicted values for an additive radiosensitivity response. Any data below the additive model is weakly synergistic. The results show that following radiation and calphostin C pretreatment (100 n m or 250 n m), only 0.49 (± 0.06 SEM) and 0.158 (± 0.043 SEM) of the cells survived, respectively. If calphostin C’s effect was additive with radiation, the predicted survival at 100 n m calphostin C would be 0.62 [0.92 (100 n m calphostin C alone) X 0.67 (radiation alone)]. At 250 n m calphostin C, the predicted survival would be 0.22 [0.33 (100 n m calphostin C alone) X 0.67 (radiation alone)]. In both cases, calphostin C (100 n m and 250 n m) increased the actual cell radiosensitivity by 26% and 39%, respectively.
In each of the other time points (9, 14, 20 and 37 h post radiation), examination of the overall relationship between calphostin C exposure and radiosensitivity were consistently less than the enhanced radiation sensitivity obtained when cells were pretreated with calphostin C 1 h prior to radiation (Figure 3b–e). The changes in cell proliferation following calphostin C incubation at 9 h (control) or 9 h post radiation are shown in Fig. 3b. Calphostin C (250 n m) at either 9 h (control) or 9 h post radiation was not as effective in killing cells compared to calphostin C pretreatment, 1 h prior to radiation.
DISCUSSION
In this study we investigated the radiosensitivity of U‐373MG cells by measuring the effects of radiation and the specific PKC inhibitor, calphostin C on cell proliferation. We found that irradiated U‐373MG cells progressed through G1‐S and underwent a G2M arrest (Fig. 1). As in many human cancers, the absence of a radiation‐induced G1 arrest in U‐373MG cells is due in part to a mutated p53 tumor suppressor protein (Russell et al. 1995) whose normal function is to permit apoptosis in cells that have become tumorigenic. The cyclin‐dependent kinase inhibitor, p21 and the DNA damage‐gene GADD45 also play a role in the G1 arrest (El‐Deiry et al. 1994). The G2M arrest following exposure to genotoxic agents is a checkpoint for cellular repair of DNA damage and may involve suppression of cyclin B mRNA and/or protein and tyrosine phoshorylation of p34cdc2 (Maity, McKenna & Muschel 1994).
Our calphostin C proliferation data illustrate that there is a relationship between the time of calphostin C (100 n m and 250 n m) exposure and its antiproliferative effect (Fig. 3). Cells appear to be more sensitive to calphostin C if they receive a single application of calphostin C (100 n m or 250 n m) 1 h prior to radiation or if they are exposed to calphostin C (250 n m) at 20 h post initiation of the experiment. As stated in the Results section, at time zero, cells had a high G0/G1 cell population (G0/G1, 70% ± 0.828 SEM; S, 14% ± 0.523 SEM; and G2M, 13% ± 0.832 SEM; n = 10 replicates). Therefore, 20 h post commencement of the experiment, cells probably had a high G0/G1 cell population and similar results were observed. Future flow cytometric studies from our laboratory will address which exposure time(s) and cell cycle phase(s) are most sensitive to calphostin C.
We previously reported that calphostin C inhibited U‐373MG PKC activity, and that high concentrations of calphostin C (400 n m or 500 n m) in the absence or presence of serum blocked progression through either S phase or G2M, respectively (Acevedo‐Duncan et al. 1997). In the aforementioned study, a single exposure to calphostin C at 100 n m did not arrest cells in S phase. Additionally, we have shown herein that calphostin C at 100 n m did not completely inhibit U‐373MG proliferation (Fig. 3). Our results appear to differ from those of Ikemoto et al. (1995) who reported that calphostin C (100 n m) for 16–24 h is capable of producing apoptotic DNA fragmentation and cell death. However, our results cannot be compared to those of Ikemoto et al. (1995) due to differences in the experimental methodology. They quantified cell viability using trypan blue exclusion while we used the MTT assay to measured cell proliferation seven days post calphostin C incubation. Alternatively, Pollack & Kawecki (1997) reported that repeated daily exposures to calphostin C (60–125 n m) for 4 days completely blocked glioma proliferation. In our previous study, we also reported that TPA treatment (0.1–1.0 µM; for 27 h with and without serum) elevated the number of cells blocked in S phase and decreased U‐373MG cell proliferation (Acevedo‐Duncan et al. 1997). The difference on the effects of calphostin C at 100 n m and TPA on S phase progression are due to their mechanism of action. Prolonged TPA treatment initially activates PKC isozymes and then depletes PKC (Couldwell et al. 1991; Pollack et al. 1991), while calphostin C (100 n m) inhibits the regulatory domain of PKC by irreversible oxidative inactivation (Gopalakrishna et al. 1992).
Treatment with the PKC inhibitor calphostin C (250 n m) prior to radiation, additively enhanced the radiosensitivity of U‐373MG cells by decreasing cell proliferation by 76% (Fig. 3). We speculate that the enhanced radiosensitivity presented herein for cells treated with calphostin C (250 n m) prior to radiation may be due to the temporal inhibition of PKC isozymes required directly or indirectly for cell proliferation or DNA repair following irradiation. Support for the latter postulate comes from data reported by 1993b, 1993a. They showed that inhibition of rat C6 glioma PKC activity by prolonged treatment with TPA or the nonspecific PKC inhibitor staurosporine prior to irradiation, enhanced the radiation‐induced DNA damage and attenuated the repair of DNA injury. However, the other studies conflict with this hypothesis. Hallahan et al. (1992a) found that treatment of JSQ‐3 squamous carcinoma cells with X‐irradiation and the PKC inhibitor sangivamycin resulted in no difference in DNA single‐ and double‐strand breaks as measured by DNA alkaline and neutral elutions. Future work with U‐373MG cells will determine if in this glioma cell line, PKC isozymes function directly or indirectly to repair DNA post irradiation.
Acknowledgements
This project was supported in part by the Research Service of the Veterans Administration, Edmond J. and Jean Spence Foundation Inc., The Batchelor Foundation, Charles A. Lauffer Trust, and Catalina Marketing Corporation. We also acknowledge the contribution of the Flow Cytometry Core Facility at the H. Lee Moffitt Cancer Center & Research Institute.
References
- Acevedo‐Duncan M, Collins J, Zhang R, Haller E, Chalfant C, Cooper DR (1995) In‐situ effects of interferon on human glioma protein kinase C‐α and ‐β ultrastructural localization. Cell Growth Differentiation 6, 1353. [PubMed] [Google Scholar]
- Acevedo‐Duncan M & Zhang R (1994) Heterogeneity of protein kinase C activity in human U‐373 and G‐26 glioma cells. Biochem. Biophys. Res. Commun. 205, 127. [DOI] [PubMed] [Google Scholar]
- Acevedo‐Duncan M, Zhang R, Cooper DR, Greenberg HM (1997) Effects of interferon and PKC modulators on human glioma protein kinase C, cell proliferation and cell cycle. Neurochem. Res. 22, 775. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Mineta T, Martuza RL, Glazer RI (1994) Antisense expression of protein kinase C a and tumorigencity of human glioma cells. Neurosurgery 35, 904. [DOI] [PubMed] [Google Scholar]
- Allalunis‐Turner J, Barron GM, Day RS III (1997) Intact G2 phase checkpoint in cells of a human cell line lacking DNA‐dependent protein kinase activity. Radiat. Res. 147, 284. [PubMed] [Google Scholar]
- Alley MC, Scudiero DA, Monks A et al. (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture assay. Cancer Res. 48, 589. [PubMed] [Google Scholar]
- Baltuch GH, Couldwell WT, Villemure J‐G, Yong VW (1993) Protein kinase C inhibitors suppress cell growth in established and low‐passage glioma cell lines. A comparison between staurosporine and tamoxifen. Neurosurgery 33, 495. [DOI] [PubMed] [Google Scholar]
- Baltuch GH & Yong VW (1996) Signal transduction for proliferation of glioma cells in vitro occurs predominantly through a protein kinase C‐mediated pathway. Brain Res. 710, 143. [DOI] [PubMed] [Google Scholar]
- Bernhard EJ, Maity A, Muschel RJ, McKenna WG (1994a) Increased expression of cyclin B1 mRNA coincides with diminished G2‐phase arrest in irradiated HeLa cells treated with staurosporine or caffeine. Radiat. Res. 140, 393. [PubMed] [Google Scholar]
- Bernhard EJ, Mckenna WG, Muschel RJ (1994b) Cyclin expression and G2‐phase delay after irradiation. Radiat. Res. 138, s64. [PubMed]
- Burns RF, Miller FD, Merriman RM et al. (1991) Inhibition of protein kinase C by calphostin C is light‐dependent. Biochem. Biophys. Res. Commun. 176, 288. [DOI] [PubMed] [Google Scholar]
- Cacace A, Guadagno SN, Krauss RS, Han EK, Kolch W, Weinstein IB (1993) The epsilon isoform of protein kinase C is an oncogene when overexpressed in R6‐fibroblasts. Oncogene 8, 2095. [PubMed] [Google Scholar]
- Carmichael J, Degraff WG, Gazdar AF, Minna JD, Mitchell JB (1987) Evaluation of a tetrazolium‐based semiautomated colorimetric assay: assessment of radiosensitivity. Cancer Res. 47, 943. [PubMed] [Google Scholar]
- Chumra SJ, Mauceri HJ, Advani S et al. (1997) Decreasing the apoptotic threshold of tumor cells through protein kinase C inhibition and sphingomyelinase activation increases tumor killing by ionizing radiation. Cancer Res. 57, 4340. [PubMed] [Google Scholar]
- Couldwell W, Antel JP, Apuzzo MLJ, Yong VW (1994b) Inhibition of growth of established human glioma cell lines by modulators of the protein kinase‐C system. J. Neurosurg. 73, 594. [DOI] [PubMed] [Google Scholar]
- Couldwell WT, Gopalakrishna R, Hinton DR et al. (1994c) Hypericin: a potential antiglioma therapy. Neurosurgery 35, 705. [DOI] [PubMed] [Google Scholar]
- Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE (1994a) Protein kinase C inhibitors induce apoptosis in human glioma cell lines. FEBS Lett 345, 43. [DOI] [PubMed] [Google Scholar]
- Couldwell WT, Uhm JH, Antel JP, Yong VW (1991) Enhanced protein kinase C activity correlates with the growth rate of malignant gliomas in vitro. Neurosurgery 29, 880. [DOI] [PubMed] [Google Scholar]
- Couldwell WT, Weiss MH, Degiorgio CM et al. (1993) Clinical and radiographic response in a minority of patients with recurrent malignant gliomas treated with high‐dose Tamoxifen. Neurosurgery 32, 485. [DOI] [PubMed] [Google Scholar]
- Dean NM & McKay R (1994b) Inhibition of protein kinase C‐α expression in mice after systemic administration of phosphorothiate antisense oligodeoxynucleotides. Proc. Natl. Acad. Sci. 91, 11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean NM, McKay RM, Condon TP, Bennett CF (1994a) Inhibition of protein kinase C‐α expression in human A549 cells by antisense oligonucleotides inhibit induction of intercellular adhesion 1 (ICAM‐1) mRNA by phorbol esters. J. Biol. Chem. 269, 16416. [PubMed] [Google Scholar]
- El‐Deiry WS, Harper JW, O'Conner PM et al. (1994) WAF1/CIP1 is induced in p53‐mediated G1 arrest and apoptosis. Cancer Res. 54, 1169. [PubMed] [Google Scholar]
- Gopalakrishna R, Chen ZH, Gundimeda U (1992) Irreversible oxidative inactivation of protein kinase C by photosensitive inhibitor calphostin C. FEBS Lett. 186, 246. [DOI] [PubMed] [Google Scholar]
- Hallahan DE, Sukhatme VP, Sherman ML, Huberman E, Kufe DW, Eichselbaum RR (1991b) Tumor necrosis factor gene expression is mediated by protein kinase C following activation by ionizing radiation. Cancer Res. 51, 4565. [PubMed] [Google Scholar]
- Hallahan DE, Sukhatme VP, Sherman ML, Virudachalam S, Kufe D, Weichselbaum RR (1991a) Protein kinase C mediates X‐rays inducibility of nuclear signal transducers Egr‐1 and jun. Proc. Natl. Acad. Sci. USA 88, 2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallahan DE, Virudachalam S, Grdina D, Weichselbaum RR (1992b) The isoquinoline sulfonamide H7 attenuates radiation‐mediated protein kinase C activation and delays the onset of x‐ray induced G2 arrest. J. Radiat. Oncol. Biol. Phys. 24, 687. [DOI] [PubMed] [Google Scholar]
- Hallahan D, Virudachalam S, Schwartz JL, Panje N, Mustafi R, Weichselbaum RR (1992a) Inhibition of protein kinases sensitizes human tumor cells to Ionizing radiation. Radiat. Res. 129, 345. [PubMed] [Google Scholar]
- Hutchinson F (1985) Chemical changes induced in DNA by ionizing radiation. Prog. Nucl Acid Res. Mol. Biol. 32, 115. [DOI] [PubMed] [Google Scholar]
- Ikemoto H, Eiichi T, Matsumoto T, Nakano A, Furuyama J‐I (1995) Apoptosis of human glioma cells in response of calphostin C, a specific protein kinase C inhibitor. J. Neurosurg. 83, 1006. [DOI] [PubMed] [Google Scholar]
- Kobayashi E, Nakano H, Morimoto M, Tamaoki T (1989) Calphostin C (UNC‐1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 159, 548. [DOI] [PubMed] [Google Scholar]
- Maity A, Kao GD, Muschel RJ, Mckenna WG (1997) Potential molecular targets for manipulating the radiation response. J. Radiat. Oncol. Biol. Phys. 37, 639. [DOI] [PubMed] [Google Scholar]
- Maity A, McKenna WG, Muschel RJ (1994) The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother. Oncol. 31, 1. [DOI] [PubMed] [Google Scholar]
- Mori T, Takai Y, Minamikuchi R, Yu B, Nishizuka Y (1980) Inhibitory action of chlorpromazine, dibucaine, and other phospholipid‐interacting drugs on calcium‐activated, phospholipid‐dependent protein kinase. J. Biol. Chem. 255, 8378. [PubMed] [Google Scholar]
- Nakajima T & Yukawa O (1996) Radiation‐induced translocation of protein kinase C through membrane lipid peroxidation in primary cultured rat hepatocytes. Int. J. Radiat. Oncol. Biol. Phys. 70, 473. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Hartzell P, Baldi C, Svensson S‐A, Bellomo G, Orrenius S (1986) Cystamine induces toxicity in hepatocytes through the elevation of cytosolic Ca2+ and the stimulation of a nonlysosomal proteolytic system. J. Biol. Chem. 261, 14628. [PubMed] [Google Scholar]
- Pollack IF & Kawecki S (1997) The effect of calphostin C, a potent photodependent protein kinase C inhibitor, on the proliferation of glioma cells in vitro. J. Neuro-Oncol. 31, 255. [DOI] [PubMed] [Google Scholar]
- Pollack IF, Kawecki S, Lazo JS (1996) Blocking of glioma proliferation in‐vitro and in‐vivo and potentiating the effects of BCNU and cisplatin: UCN‐0, a selective protein kinase C inhibitor. J. Neurosurg. 84, 1024. [DOI] [PubMed] [Google Scholar]
- Pollack IF, Randell MS, Kristofik MP, Kelly RH, Selker RG, Vertosick FT (1991) Response to low‐passage human malignant gliomas in vitro to stimulation and selective inhibition of growth‐factor mediated pathways. J. Neurosurg. 75, 284. [DOI] [PubMed] [Google Scholar]
- Ponten J & MacIntyre EH (1968) Long term culture of normal and neoplastic human glia. Act. Pathol. Microbiol. Scand. 74, 465. [DOI] [PubMed] [Google Scholar]
- Price P & McMillian TJ (1990) Use of the tetrazolium assay in measuring the response of human tumor cells to ionizing radiation. Cancer Res. 50, 1392. [PubMed] [Google Scholar]
- Ramsey J, Ward R, Bleehen NM (1992) Radiosensitivity testing of human malignant gliomas. Int. J. Radiat. Oncol. Biol. Phys. 24, 675. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Ye Y‐W, Waber PG, Shuford M, Schold SC Jr., Nisen PD (1995) p53 mutation, O6‐alkylguanine DNA alkytransferase activity, and sensitivity to procarbazine in human brain tumors. Cancer 75, 1339. [DOI] [PubMed] [Google Scholar]
- Sinclair WK (1968) Cyclic X‐ray in mammalian cells in vitro . Radiation Res. 33, 620. [PubMed] [Google Scholar]
- Tsuchida E & Urano M (1997) The effect of ucn‐01 (7‐hydroxystaurosporine), a potent inhibitor of protein kinase C, on fractionated radiotherapy or daily chemotherapy of a murine fibrosarcoma. Int. J. Radiat Oncol. Biol. Phys. 39, 1153. [DOI] [PubMed] [Google Scholar]
- Weichselbaum RR, Hallahan DE, Chen GTY (1993) Physical and Biological Basis to Radiation Oncology In: Holland J, Frye E, Bast RC, Kufe D, Morton DL, Weichselbaum RR, eds. Cancer Medicine. Malvern: Lea and Febiger, pp539–566. [Google Scholar]
- Woloschak GE, Chang‐Liu C‐M, Shearin‐Jones P (1990) Regulation of protein kinase C by ionizing radiation. Cancer Res. 50, 3963. [PubMed] [Google Scholar]
- Yang X, Darling JL, McMillian TJ, Peacock JH, Steel GG (1991) Heterogeneity of radiosensitivity in a human glioma cell line. Int. J. Radiat. Oncol. Biol. Phys. 22, 103. [DOI] [PubMed] [Google Scholar]
- Zhang W, Hara A, Sakai N, Andoh T, Yamada H, Nozawa Y (1993b) Radiosensitization and inhibition of deoxyribonucleic acid repair in rat glioma cells by long‐term treatment with 12‐O‐tetradecanoylphorbol 13‐acetate. Neurosurgery 32, 432. [DOI] [PubMed] [Google Scholar]
- Zhang W, Hinton DR, Surnock AA, Couldwell WT (1996) Malignant glioma sensitivity to radiotherapy, high‐dose Tamoxifen, and Hypericin: Corroborating clinical response in vitro: Case report. Neurosurgery 38, 587. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yamada H, Sakai N, Niikawa S, Nozawa Y (1992) Enhancement of radiosensitivity by tamoxifen in C6 glioma cells. Neurosurgery 31, 725. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yamada H, Sakai N, Nozawa Y (1993a) Sensitization of C6 glioma cells to radiation by staurosporine, a potent protein kinase C inhibitor. J. Neuro-Onco. 15, 1. [DOI] [PubMed] [Google Scholar]