

Abstract
Abstract. Objectives: Cisplatin (cisPt) is used as a chemotherapeutic agent for the treatment of a variety of human tumours; more recently, it has been demonstrated that tumour cell exposure to cisPt ultimately results in apoptosis, but the mechanism by which nuclear cisPt/DNA generates the cytoplasmic cascade of events involved has not been clarified. We have investigated the effects of cisPt on proliferation in the neuronal cell line B50, with particular attention being given to understand whether mitochondria are a target of cisPt and their involvement in the apoptotic process. Materials and methods: Rat neuronal B50 cells were used to investigate the mechanisms of cisPt‐induced cytotoxicity; this line has been used as a model system for neurotoxicity in vivo. Results: Changes in proliferation, induction of apoptosis, activation of caspase‐3 and DNA fragmentation were observed in the cells, as well as morphological and biochemical alterations of mithocondria. Activation of caspase‐9 confirmed that mitochondria are a target of cisPt. Conclusion: CisPt exerts cytotoxic effects in the neuronal B50 cell line via a caspase‐dependent pathway with mitochondria being central relay stations.
INTRODUCTION
Cisplatin (cis‐dichlorodiammine platinum (II), cisPt) is a DNA‐damaging agent that is widely used in chemotherapy (for review, see Cohen & Lippard 2001). The target for cisPt is DNA, with which it binds efficiently to form a variety of monoadducts and cross‐links, either between adjacent bases on the same strand of DNA or on opposing strands (Zwelling et al. 1979; Fichtinger‐Schepman et al. 1985). These DNA lesions contribute to the cytotoxicity of cisPt, because they block DNA replication and promote cell death. In addition, several cellular proteins bind preferentially to cisPt‐damaged DNA, which could stimulate transduction pathways and ultimately signal apoptosis.
Recent findings suggest that ERK (extracellular signal‐regulated kinase) activation plays a dynamic role in mediating cisPt‐induced apoptosis in human glioma cells, and functions upstream to mitochondrial dysfunction (Jeong et al. 2002; Choi et al. 2004); caspase activation to initiate the apoptotic signal is also dependent on ERK (Gerschenson et al. 2001). On the other hand, activation of ERK could be associated with enhanced cell survival of cisPt‐treated cells (Hayakawa et al. 1999; Cui et al. 2000; Persons et al. 2000).
The chemotherapeutic effect of cisPt relies primarily on its ability to induce apoptosis in tumour cells (Ormerod et al. 1994a,b). Moreover, it has largely been demonstrated that apoptotic cell death is linked to cisPt neurotoxicity of glial cells (Fehlauer et al. 2000; Sawada et al. 2000) and neurons (Liu et al. 1998; Fischer et al. 2001). Induction of apoptosis has been recently shown in proliferating neural cells of the external granule cell layer of developing cerebellum in cisPt‐treated rats (Pisu et al. 2005). These findings can be included in the more general debate concerning neurotoxic effects of cisPt in the central nervous system (Schiffer et al. 1996) and the peripheral nervous system (Cavaletti et al. 1992; McDonald et al. 2005). However, the mechanism(s) whereby cisPt kills the cells is not fully understood. Apoptosis triggered by cisPt is generally considered to be generated by blocking replication and transcription, and an involvement of reactive oxygen intermediates (Sorenson & Eastman 1988, Sorenson et al. 1990).
Recent studies suggest that mitochondria play an important role in regulating calcium homeostasis and subsequently the apoptotic process (Chang & Reynolds 2006; Foster et al. 2006).
During apoptosis, Bax, a pro‐apoptotic member of the Bcl‐2 protein family, undergoes translocation to mitochondria (Bedner et al. 2000), leading to its accumulation in the mitochondrial intermembrane space; this appears to be a critical event in determining the release of cytochrome c into the cytosol (Yang et al. 1997) that triggers the irreversible steps of apoptosis, namely, the activation of caspases and initiation of degradation of many proteins. Inactive caspase‐9 zymogene is known to be localized in mitochondrial intermembrane spaces where it is involved in monitoring mitochondrial damage associated with cytochrome c release and subsequent activation of procaspase‐3 (Ritter et al. 2000). Moreover, changes in mitochondrial membrane permeabilization can lead to activation of caspase‐9 and subsequent activation of caspase‐3 (Li et al. 1997; Srinivasula et al. 1998). Active caspase‐3 cleaves several substrates, and results in fragmentation of chromosomal DNA into multiples of 180 base pairs and the morphological changes characteristic of apoptosis (Wyllie et al. 1980). Our team has already described membrane potential (ΔΨm) changes that precede typical morphological alterations of apoptosis, in HeLa cells (Bottone et al. 2007). In contrast, anti‐apoptotic Bcl‐2 family members, such as Bcl‐2 itself, play a pivotal protective role by preserving mitochondrial structure and function, preventing onset of mitochondrial permeability transition, and inhibiting release of cytochrome c into the cytosol (Park et al. 2002). A role for cytoskeletal actin in the apoptotic processes has been suggested. Actin could contribute to initiation of apoptosis by enabling cytosolic pro‐apoptotic proteins to be carried to mitochondria by the cytoskeleton‐driven trafficking system (Shirabe et al. 1997; Thomas et al. 2007).
The aim of this study was to investigate the mechanisms involved in cisPt‐induced cytotoxicity, in rat neuroblastoma B50 cell line that has been used previously as a model system for neurotoxicity in vivo. We studied the effects of cisPt on cell proliferation and its mechanism of induction of apoptosis, paying particular attention to whether mitochondria are affected by cisPt and their involvement in the apoptotic process. To achieve this, we correlated expression of Bcl‐2, translocation of Bax that induces changes in mitochondrial permeability, changes in cytoskeleton organization, activation of caspase‐9, and its entry into the execution phase of apoptosis via expression of caspase‐3.
MATERIALS AND METHODS
Cells and treatments
Rat B50 neuroblastoma cells (ATCC, Rockville, MD, USA) were cultured in 75 cm2 flasks in Dulbecco's minimal essential medium supplemented with 10% foetal bovine serum, 1% glutamine, 100 U penicillin and streptomycin (Celbio, Milan, Italy) in a 5% CO2 humidified atmosphere. Twenty‐four hours before experiments, cells were seeded on glass coverslips for fluorescence microscopy, or grown in 75 cm2 plastic flasks for flow cytometric analysis.
To induce apoptosis, cells were incubated in 40 µm cisPt (Teva Pharma, Milan, Italy) for 20 h at 37 °C. This concentration was chosen considering our in vivo experimental design (i.e. a single injection of 3.4 mm in 10‐day‐old rats); this dose corresponds to the dose most commonly used in the chemotherapy (Bodenner et al. 1986).
Cell cycle analysis and bromodeoxyuridine incorporation
Before the end of treatment, cells were pulse‐labelled with 40 µm of 5‐bromo‐2'‐deoxyuridine (BrdU) (Sigma Aldrich, Milan, Italy) for 30 min at 37 °C. They were then detached by mild trypsinization (to obtain single‐cell suspensions to be processed for flow cytometry), fixed in 70% ethanol, and incubated for 20 min at room temperature in 2 N HCl to partially denature DNA. After neutralization with 0.1 m sodium tetraborate (pH 8.2) for 3 min, samples were washed in phosphate‐buffered saline (PBS), and were incubated for 1 h with monoclonal antibody against BrdU (Beckton Dickinson, San Jose, CA, USA) diluted 1 : 20 in PBS. After two washings with PBS, samples were incubated for 1 h with FITC‐conjugated antimouse secondary antibody (Dako, Glostrup, Denmark), diluted 1 : 20 in PBS. Cells were washed three times with PBS and were stained with 5 µg/mL propidium iodide (PI) containing 100 U/mL of RNase A (Sigma Aldrich). Dual parameter measurements of green‐versus‐red fluorescence signals were taken by a flow cytometry using a Partec PAS III (Münster, Germany) equipped with argon laser excitation (power 200 mW) at 488 nm, 510–540 nm interference filter for the detection of FITC green fluorescence, and a 610‐nm long‐pass filter for PI red fluorescence detection. Cells were considered as positive when their green fluorescence values exceeded background threshold. At least 20 000 cells/sample were measured; five independent experiments were carried out and the average of scores was used. Values are expressed as the mean ± SEM and differences were compared using Student's t‐test.
Bromodeoxyuridine immunolabelling experiments were also performed on cells grown on coverslips.
Identification of apoptotic cells
Cells were detached by mild trypsinization as before, incubated with FITC‐conjugated annexin V (3 µL/106 cells) (Bender MedSystem, Prodotti Gianni, Milan, Italy) and were counterstained with 2 µg/mL PI. After 10 min incubation, dual‐parameter flow cytometric analysis was performed with the flow cytometer Partec PAS III, equipped with argon laser excitation (power 200 mW) at 488 nm, 510–540 nm interference filter for the detection of FITC green fluorescence, and a 610‐nm long‐pass filter for PI red fluorescence detection. Five independent experiments were carried out and the average of the scores was used. Values are expressed as the mean ± SEM and differences were compared using Student's t‐test.
Immunocytochemical staining for activated caspase‐3 and caspase‐9
Cells on coverslips were fixed with acetone for 10 min, rehydrated with PBS and were incubated with primary polyclonal antibodies recognizing the active form of caspase‐3 (diluted 1 : 50 in PBS) or the active form of caspase‐9 (diluted 1 : 50 in PBS), in a humidified chamber for 1 h at room temperature. Cells were washed with PBS and incubated with Alexa 488‐conjugated antirabbit antibody (Molecular Probes, Invitrogen, Milan, Italy); slides were then incubated with 1 µg/mL PI for 5 min at room temperature, washed with PBS and then mounted in Mowiol (Calbiochem, Inalco, Milan, Italy), for confocal microscopy analysis.
TUNEL reaction
Cells on coverslips were fixed with 4% formaldehyde for 30 min at room temperature and then with 70% ethanol for 24 h at –20 °C. Samples were incubated for 1 h at 37 °C with 50 µL TUNEL mixture, according to the manufacturer's instructions (Boehringer, Mannheim, Germany). After washings with PBS, cells were counterstained with Evan's Blue (0.1% in PBS, Sigma Aldrich) and were mounted upside down on a non‐fluorescent glass slide in a drop of Mowiol (Calbiochem).
Double immunocytochemical detection of mitochondria and cytoskeletal components
Cells on coverslips were fixed with 4% formaldehyde for 30 min at room temperature and with 70% ethanol for 24 h at –20 °C; then they were processed for a double immunoreaction for mitochondria and microtubules. Mitochondria were labelled with an autoimmune serum (1 : 200 in PBS) recognizing the 70 kDa E2 subunit of the pyruvate dehydrogenase complex (a kind gift of the IRCCS San Matteo, Pavia, Italy), and were revealed with Alexa 488‐conjugated antihuman antibody (Molecular Probes, Invitrogen). Specimens were then labelled with Alexa 594‐conjugated phalloidin (1 : 40 in PBS) (Molecular Probes, Invitrogen). All incubations were performed for 60 min at room temperature. Sections were counterstained for DNA with 0.1 µg/mL Hoechst 33258, washed with PBS and then mounted in Mowiol (Calbiochem), for confocal microscopy analysis.
Immunocytochemical staining for Bcl‐2 and Bax
Cells on coverslips were fixed with 4% formaldehyde for 30 min at room temperature and with 70% ethanol for 24 h at –20 °C. Samples were incubated with polyclonal anti‐Bcl‐2 antibody (dilution 1 : 50 in PBS) (Cell Signaling and Technology, Celbio, Milan, Italy), and revealed with Alexa 488‐conjugated antirabbit antibody (Molecular Probes, Invitrogen) or with polyclonal anti‐Bax antibody (dilution 1 : 50 in PBS) (Cell Signaling and Technology) revealed with Alexa 488‐conjugated antirabbit antibody (Molecular Probes, Invitrogen). Cells were then labelled with Alexa 594‐conjugated phalloidin (1 : 40 in PBS) (Molecular Probes, Invitrogen). All incubations were performed for 60 min at room temperature. Sections were counterstained for DNA with 0.1 µg/mL Hoechst 33258, washed with PBS and mounted in Mowiol (Calbiochem), for fluorescence microscopy analysis or confocal microscopy analysis.
Double immunocytochemical detection of Bax and mitochondria
Cells on coverslips were fixed with 4% formaldehyde for 30 min at room temperature and with 70% ethanol for 24 h at –20 °C. Samples were incubated with polyclonal anti‐Bax antibody (dilution 1 : 50 in PBS) (Cell Signaling and Technology) and then with Alexa 594‐conjugated antirabbit antibody (Molecular Probes, Invitrogen). Mitochondria were labelled with autoimmune serum (dilution 1 : 200 in PBS) recognizing the 70 kDa E2 subunit of the pyruvate dehydrogenase complex, revealed with Alexa 488‐conjugated antihuman antibody (Molecular Probes, Invitrogen). All the incubations were performed for 60 min at room temperature. Sections were counterstained for DNA with 0.1 µg/mL Hoechst 33258, washed with PBS and finally mounted in Mowiol (Calbiochem), for confocal microscopy analysis.
Measurement of mitochondrial membrane potential with JC‐1
Changes in mitochondrial membrane potential were monitored using JC‐1 (5,5V,6,6V‐tetrachloro‐1,1V,3,3V‐tetraethylbenzimidazolcarbocyanine iodide) (Molecular Probes, Invitrogen). JC‐1 emits either green or red fluorescence, depending on the mitochondrial membrane potential; the green signal indicates depolarized mitochondria and the red signal indicates polarized mitochondria (Reers et al. 1991). Thus, the shift from red to green fluorescence is considered a reliable indication of a drop in mitochondrial membrane potential. Cells grown in flasks were harvested by mild trypsination with 0.25% trypsin in PBS containing 0.05% ethylenediaminetetraacetic acid and were incubated in culture medium with 2 µm JC‐1 for 20 min at 37 °C in the dark. After two washes with PBS at 37 °C, cells in suspension were analysed on flow cytometry using a Partec PAS III equipped with argon ion laser with 20 mW output power at 488‐nm excitation and with 530/30 nm and 585/42 nm band‐pass emission filters. Data were analysed using FlowMax software from the same company.
Confocal fluorescence microscopy
For confocal laser scanning microscopy, Leica TCS‐SP system (Leica, Heidelberg, Germany) mounted on a Leica DMIRBE‐inverted microscope was used. For fluorescence excitation, an Ar/UV laser at 364 nm was used for Hoechst 33258, an Ar/Vis laser at 488 nm was used for FITC and an He/Ne laser at 543 nm was used for Alexa 594. Spaced (0.5 µm) optical sections were recorded using a 63× oil immersion objective. Images were collected in the 1024 × 1024 pixel format, stored on a magnetic mass memory and processed by Leica confocal software.
Fluorescence microscopy
An Olympus BX51 microscope equipped with a 100‐W mercury lamp was used under the following conditions: 330–385 nm excitation filter (excf), 400‐nm dichroic mirror (dm), and 420‐nm barrier filter (bf), for Hoechst 33258; 450–480 nm excf, 500‐nm dm, and 515 nm bf, for JC‐1. Images were recorded with an Olympus Camedia C‐5050 digital camera and stored on a PC by the Olympus software, for processing and printing.
RESULTS
Cell cycle perturbation
The initial findings confirmed that cisPt is an efficient cytostatic reagent involving the cell cycle. Viability of B50 cells after cisPt treatment was determined by the trypan blue dye exclusion test: in controls, the percentage of trypan blue‐positive cells was 2 ± 0.9%, in cisPt‐incubated cells it was 36.2 ± 1.7%. Immunocytochemical experiments after BrdU incorporation showed a decrease in the number of cells in the S phase after cisPt treatment (Fig. 1a,b). Flow cytometric measurements showed that the percentage of cells in the S phase decreased from 23.8 positive cells in control samples to 8.3 positive cells after cisPt treatment (Fig. 1c–e); cytofluorimetric analysis of DNA after PI confirmed these changes in the cell cycle (Fig. 1f,g). Moreover, after incubation with cisPt, a peak sub‐G1 component was present, this represents a population of cells with reduced DNA staining, indicating probable presence of DNA fragmentation followed by apoptosis (Fig. 1g, arrow) (Table 1).
Figure 1.
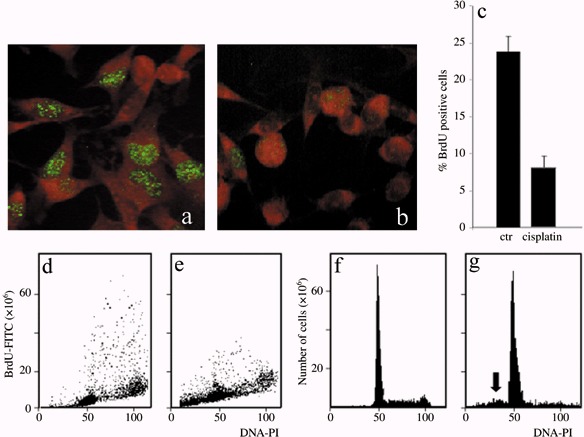
Immunocytochemical reaction for BrdU in B50 cells: positive (green fluorescence) control sample (a) and after treatment with cisPt (b). Cytofluorimetric analysis of BrdU in B50 control cells (d) and treated with cisPt (e). The histogram represents the average of five independent experiments (c). Cytofluorimetric histograms of DNA content after propidium iodide (PI) staining in control B50 cells (f). The sub‐G1 peak (arrow) demonstrates that apoptotic cells are present after cisPt treatment (g).
Table 1.
Percentage of cells in control and treated samples in each phase of the cell cycle
| Phases of cell cycle | Cell of CTR (%) | Cell after cisPt (%) |
|---|---|---|
| Sub‐G1 | 19.64 ± 2.3 | |
| G1 | 65.0 ± 0.7 | 45.18 ± 0.4 |
| S | 20.2 ± 1.3 | 09.00 ± 1.2 |
| G2 | 14.5 ± 1.2 | 25.20 ± 0.9 |
CTR, control.
Apoptosis
Presence of apoptosis after cisPt treatment was investigated to demonstrate that cell cycle perturbation and apoptosis are inextricably linked in this system, too. Appearance of phosphatidylserine residues (normally hidden within the plasma membrane) on the surface of the cell is a parameter that can be used to detect and measure apoptosis as cell membrane integrity is lost as the apoptotic process progresses. Using DNA‐specific viability dyes, such as PI, it was possible to distinguish early apoptotic cells (annexin V‐positive cells), late apoptotic cells (annexin V‐ and PI‐positive cells) and dead cells (PI‐positive cells). After treatment with cisPt, there was an increase in apoptotic B50 cells as revealed by annexin V positivity (Fig. 2a–c) of the early apoptotic fraction (7.08%) and late apoptotic fraction (15.9%) of cells. The finding showed the relationships of entry of cells into the execution phase of apoptosis, analysed by the immunoreaction for activated caspase‐3. In cisPt‐treated B50 cells, there were numerous cells with cytoplasmic labelling for activated caspase‐3 (Fig. 2d,e); these also showed nuclei with typical morphological apoptotic appearance as shown by the TUNEL reaction (Fig. 3a,b).
Figure 2.
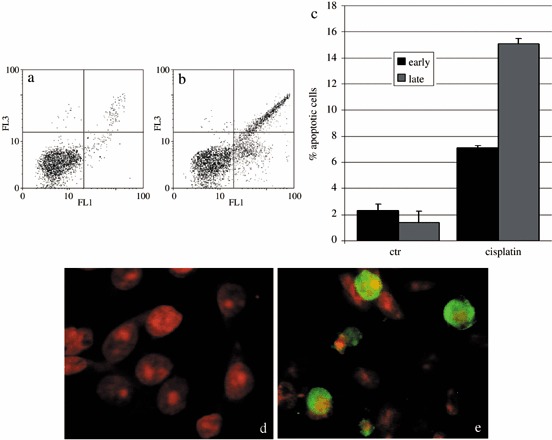
Dual parameter cytogram of FITC‐labelled annexin V (in abscissa) versus propidium iodide (PI) staining (ordinate). Double‐negative, non‐apoptotic cells fall in the lower left quadrant, while early and late apoptotic cells fall in the lower and upper right quadrant, respectively. There was an increase of early and late apoptotic cells in cisPt‐treated cells (b). The histogram represents the average of five independent experiments (c). Immunocytochemical reaction for activated caspase‐3 in control (d) and cisPt‐treated cells (e, green fluorescence). DNA was counterstained with PI.
Figure 3.
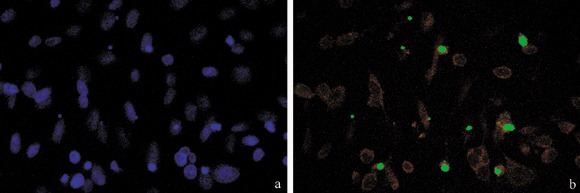
Treatment with cisPt induced caspase activation, and DNA fragmentation demonstrated by TUNEL assay. TUNEL‐positive cells (b, green fluorescence) were counterstained with Evan's blue (red fluorescence); in (a) DNA was counterstained with Hoechst 33258.
Morphology of mitochondria and cytoskeleton
In comparison to untreated controls (Fig. 4a), morphology and intracellular distribution of mitochondria in treated B50 cells were altered and their presence was denser around nuclei (Fig. 4b). This finding fitted with reorganization of the actinic cytoskeleton that, differently from controls (Fig. 4a), showed evident alterations in treated B50 cells (Fig. 4b).
Figure 4.

Confocal microscopy of dual immunolabelling of mitochondria (green fluorescence) and actinic cytoskeleton (red fluorescence) in B50 cells. Compared to untreated controls (a), in cisPt‐treated cells (b), mitochondria clustered around the nucleus and formed dense masses in the cytoplasm. Control cells (a) revealed a filamentous actin skeleton; in (b) cisPt induced disruption of filamentous actin structures and assembly of depolymerized actin in peripheral regions of cytoplasm close to the cell membrane. DNA was counterstained with Hoechst 33258. Confocal microscopy: immunocytochemical detection of Bcl‐2 (green fluorescence) in control cells (c) and after cisPt treatment (d). After treatment with cisPt, Bcl‐2 protein levels increased significantly in the nucleus. The cytoskeleton was labelled with Alexa 594‐conjugated phalloidin (red fluorescence).
Activation of intrinsic apoptotic mitochondrial pathway
In comparison to control samples (Fig. 4c), B50 cells after treatment, had increased positivity of Bcl‐2, observed mainly in the nucleus (Fig. 4d). In cells in which the apoptotic programme was advanced, as shown by the morphological appearance of apoptotic nuclei, there was increased immunopositivity for Bax (Fig. 5b) around condensed chromatin, while in control cells reactivity was diffusely distributed (Fig. 5a). Analysis by immunofluorescence confocal microscopy demonstrated that apoptotic Bax translocation from the cytosol was confined to mitochondria (Fig. 5c,d), as the two fluorescence labels colocalized (Fig. 5e). The three images (Fig. 5e′,e″,e′″) represent fusion of red/green fluorescence measured in three sequential sections of the same cell. Co‐localization of Bax and mitochondria in treated cells was observed in cells with typical apoptotic nuclei. Functional variations shown by changes of mitochondrial potential after cisPt treatment corresponded to morphological changes. B50 control cells (Fig. 6a) showed an equilibrium from red to green fluorescence. After cisPt treatment (Fig. 6b), the two‐colour cytofluorimetric analysis showed that the fluorescence switched from aggregated yellow–orange (JC‐1 aggregates) to fluorescent green (JC‐1 monomer), indicating significant mitochondrial membrane depolarization (Li et al. 1999). A significant increase in caspase‐9 cytoplasmic immunopositivity was noticed in cells after cisPt exposure (Fig. 7a,b), which was due to activation of the mitochondrial apoptotic pathway. We observed that not only cells with condensed chromatin were positive, but also those showing no clear morphological signs of apoptosis.
Figure 5.
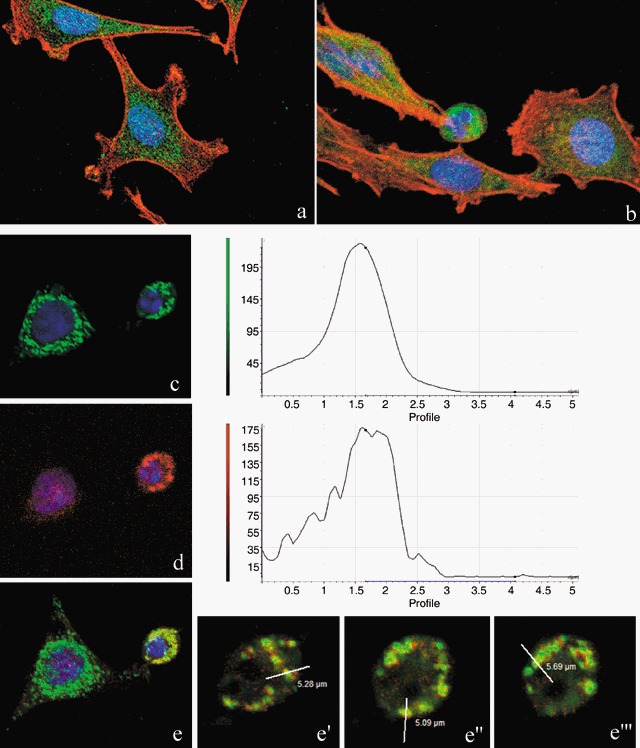
Confocal microscopy. Immunocytochemical detection of Bax (green fluorescence) in control cells (a) and after cisPt treatment (b). Cytoskeleton was labelled with Alexa 594‐conjugated phalloidin (red fluorescence) and DNA was counterstained with Hoechst 33258. Double immunoreaction for mitochondria (green fluorescence, c) and Bax (red fluorescence, d) in cisPt‐treated B50 cells, DNA counterstained with Hoechst 33258. The three different sections (e′, e″, e′″) represent the fluorescence distribution determined for sections of the cell, as indicated in the red/green fusion image (e). After treatment, Bax translocates from its predominantly cytoplasmic location to mitochondria and induces activation of apoptosis.
Figure 6.
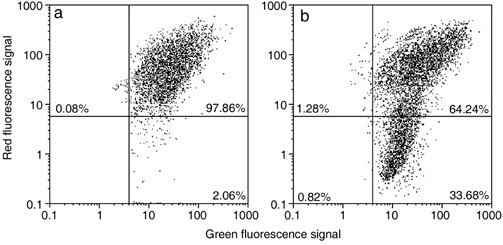
Cytometric analysis of green‐versus‐red fluorescence of JC‐1 showing effects of cisPt treatment (b) on mitochondrial potential: compared to control (a), there is an increase of cell fraction with high green fluorescence and a relatively low red fluorescence.
Figure 7.
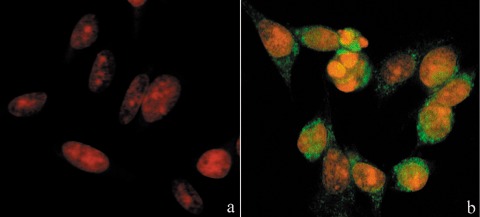
Immunocytochemical detection of activated caspase‐9 (green fluorescence) in cisPt‐treated cells (b). In (a) untreated cells, DNA counterstained with propidium iodide (PI).
DISCUSSION
It is generally accepted that the primary cytotoxic mechanism of action of cisPt is DNA damage with subsequent induction of apoptosis (Sekiguchi et al. 1996; Krajci et al. 2006), although the precise mechanisms inducing this were not fully understood. Apoptosis is an incontestable result, not only of the chemotherapeutic effects of cisPt in tumour cells, but also of neurotoxic action of the drug in normal proliferating cells, such as those of the external granule cell layer during cerebellar development (Pisu et al. 2005). An acute, direct action on differentiating Purkinje cells has also been reported, regarding in particular, metabolism enzymes (Bernocchi et al. 1990), cytoskeleton components and calcium‐binding proteins (Scherini & Bernocchi 1994). Presence of other degenerative alterations of Purkinje cells (Scherini & Bernocchi 1994) and developmental morphogenic molecules (Pisu et al. 2003, 2004; Avella et al. 2006) after cisPt treatment does not exclude involvement of the signal transduction pathways in cell apoptosis/degeneration.
We employed the B50 neuronal cell line from rat central nervous system that retains many morphological and chemical characteristics of neural tissues. Use of these cells, which can grow in tissue culture in relatively large quantities as a uniform cell population, is ideal for understanding expression of different cytoplasmic proteins involved in the apoptotic cascade.
The main effect of cisPt is to cause arrest of the cell cycle with a specific checkpoint at the boundary of G2 and M phases; these events have been suggested to be involved in apoptosis induction in a variety of proliferating cells (Eastman 1991). Here, we report that cisPt exerts cytotoxic effects in neuronal B50 cells via a caspase‐dependent pathway with mitochondria being central relaying stations. It has been described (Bottone et al. 2007) that morphological modification of mitochondria represent a significant parameter linked to functional alterations and apoptosis. On the other hand, mitochondria are vital organelles for cell survival, with the central nervous system particularly depending on them for regulating calcium homeostasis. In turn, calcium serves as a regulator of several enzymatic activities and cell processes, including apoptosis (for review, see Chang & Reynolds 2006).
We have observed that the mitochondria‐dependent pathway is regulated by the Bcl‐2 family of proteins. Increase of Bcl‐2 expression in B50 cells after cisPt may be related to the possibility that Bcl‐2 prevents cell death by blocking release of cytochrome c (Yang et al. 1997). It has been described that Bcl‐2, which inhibits apoptosis, promotes survival of various cells (Tsujimoto 1989; Garcia et al. 1992) and its function is based on maintenance of membrane potential and depression of the permeability transition. However, it has also been demonstrated that Bcl‐2 can act in a pro‐apoptotic fashion, in relation to its subcellular localization (Portier & Taglialatela 2006); nuclear compartment‐associated Bcl‐2 seems to promote, rather than protect, cells from apoptosis. Moreover, after cisPt treatment, we noticed a loss in mitochondrial membrane potential that has been described to precede the release of cytochrome c (Sanchez‐Alcazar et al. 2000). To understand this event, we monitored redistribution of Bax from the cytosol to the mitochondria, since it has been described that translocation of Bax to mitochondria can cause loss of their membrane potential (ΔΨm) and a release of proteins, such as cytochrome c (Eskes et al. 1998; Jurgensmeier et al. 1998), into the intermembrane space (Nechushtan et al. 2001). We found translocation of Bax to mitochondria of cells showing nuclei with clear apoptotic morphology. Thus, Bax may neutralize anti‐apoptotic members of the Bcl‐2 family (Yin et al. 1994; Han et al. 1996; Zha et al. 1996), bind and regulate mitochondrial proteins, such as voltage‐dependent anion channel (Narita et al. 1998) and adenine nucleotide transporter (Marzo et al. 1998), or act as a pore‐forming protein by homo‐oligomerization to release apoptosis‐activating factors (Antonsson et al. 1997, 2001; Schlesinger et al. 1997; Eskes et al. 1998; Mikhailov et al. 2001). In different models, Bax crosses mitochondria membranes at the termination of apoptotic activation; addition of Bax to mitochondria has been shown to be sufficient to trigger cytochrome c release and subsequent caspase activation (Bossy‐Wetzel et al. 1998; Jurgensmeier et al. 1998). Caspase‐9 appears to be a functionally important initiator of the apoptotic cascade (Costantini et al. 2002). We observed a high percentage of caspase‐9‐positive cells after cisPt treatment and noticed that most of them did not show clear apoptotic morphology, demonstrating that activation of caspase‐9 precedes condensation of chromatin (Faleiro & Lazebnik 2000). As a consequence of the activation of caspase‐9, we found some caspase‐3‐positive cells with a clearly apoptotic nucleus, demonstrating that the apoptotic programme had reached the final stage. Caspase‐3 cleaves inhibitor of caspase‐activated DNase and allows caspase‐activated DNase to translocate to the nucleus to degrade DNA, producing the characteristic apoptotic phenotype of cell shrinkage, membrane blebbing, chromatin condensation and oligonucleosomal DNA fragmentation until cell death is reached (Janicke et al. 1998; Budihardjo et al. 1999; Earnshaw et al. 1999).
We have demonstrated that cisPt insult leads to activation of a mitochondrial‐dependent caspase cascade that is responsible for the irreversible process towards apoptotic cell death, as shown by TUNEL positivity, a specific marker signalling that the apoptotic programme has been decided (Soldani et al. 2001). Apoptotic induction also triggered reorganization of the F‐actin (filamentous actin) network with an increase in its association with mitochondria, observed before mitochondrial fission and nuclear condensation.
Finally, with regard to diverse mitochondrial functions and their integration into various cellular signalling pathways, it is not surprising that alterations in mitochondrial physiology are currently being considered as pivotal events in several neurodegenerative diseases (for review, see Foster et al. 2006). Many pharmacological tools have been used to study mitochondrial function and dysfunction. In this context, drug discovery and development are essential approaches that may lead to the emergence of new possibilities for therapeutic intervention in the central nervous system pathology.
ACKNOWLEDGEMENTS
This work was supported by the University of Pavia (Fondo di Ateneo per la Ricerca, FAR 2005).
Thanks are due to Dr. Claudia Alpini (IRCCS Policlinico S. Matteo, Pavia) for the kind gift of human autoimmune serum recognizing mitochondria and to Dr. Patrizia Vaghi for confocal micrographs taken at the Centro Grandi Strumenti of the University of Pavia.
REFERENCES
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC (1997) Inhibition of Bax channel‐forming activity by Bcl‐2. Science 277, 370–372. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Sanchez B, Martinou JC (2001) Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276, 11615–11623. [DOI] [PubMed] [Google Scholar]
- Avella D, Pisu MB, Roda E, Gravati M, Bernocchi G (2006) Reorganization of the rat cerebellar cortex during postnatal development following cisplatin treatment. Exp. Neurol. 201, 131–143. [DOI] [PubMed] [Google Scholar]
- Bedner E, Li X, Kunicki J, Darzynkiewicz Z (2000) Translocation of Bax to mitochondria during apoptosis measured by laser scanning cytometry. Cytometry 41, 83–88. [PubMed] [Google Scholar]
- Bernocchi G, Scherini E, Nano R (1990) Developmental patterns in the rat cerebellum after cis‐dichlorodiammineplatinum treatment. Neuroscience 39, 179–188. [DOI] [PubMed] [Google Scholar]
- Bodenner DL, Dedon PC, Keng PC, Katz JC, Borch RF (1986) Selective protection against cis‐diamminedichloroplatinum (II)‐induced toxicity in kidney, gut, and bone marrow by diethyldithiocarbamate. Cancer Res. 46, 2751–2755. [PubMed] [Google Scholar]
- Bossy‐Wetzel E, Newmeyer DD, Green DR (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD‐specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottone MG, Soldani C, Fraschini A, Alpini C, Croce AC, Bottiroli G, Pellicciari C (2007) Enzyme‐assisted photosensitization with rose Bengal acetate induces structural and functional alteration of mitochondria in HeLa cells. Histochem. Cell Biol. 127, 263–271. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Tredici G, Marmiroli P, Petruccioli MG, Barajon I, Fabbrica D (1992) Morphometric study of the sensory neuron and peripheral nerve changes induced by chronic cisplatin (DDP) administration in rats. Acta Neuropathol. 84, 364–371. [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ (2006) Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Neurobiol. 80, 241–268. [DOI] [PubMed] [Google Scholar]
- Choi BK, Choi CH, Oh HL, Kim YK (2004) Role of ERK activation in cisplatin‐induced apoptosis in A172 human glioma cells. Neurotoxicology 25, 915–924. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Lippard SJ (2001) Cisplatin: from DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 67, 93–130. [DOI] [PubMed] [Google Scholar]
- Costantini P, Bruey JM, Castedo M, Metivier D, Loeffler M, Susin SA, Ravagnan L, Zamzami N, Garrido C, Kroemer G (2002) Pre‐processed caspase‐9 contained in mitochondria participates in apoptosis. Cell Death Differ. 9, 82–88. [DOI] [PubMed] [Google Scholar]
- Cui W, Yazlovitskaya EM, Mayo MS, Pelling JC, Persons DL (2000) Cisplatin‐induced response of c‐jun N‐terminal kinase 1 and extracellular signal – regulated protein kinases 1 and 2 in a series of cisplatin‐resistant ovarian carcinoma cell lines. Mol. Carcinog. 29, 219–228. [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424. [DOI] [PubMed] [Google Scholar]
- Eastman A (1991) Analysis and quantitation of the DNA damage produced in cells by the cisplatin analog cis‐[3H]dichloro (ethylenediamine) platinum (II). Anal Biochem. 197, 311–315. [DOI] [PubMed] [Google Scholar]
- Eskes R, Antonsson B, Osen‐Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou JC (1998) Bax‐induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 143, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faleiro L, Lazebnik Y (2000) Caspases disrupt the nuclear–cytoplasmic barrier. J. Cell Biol. 151, 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlauer F, Barten‐Van Rijbroek AD, Stalpers LJ, Leenstra S, Lindeman J, Tjahja I, Troost D, Wolbers JG, Van Der Valk P, Sminia P (2000) Additive cytotoxic effect of cisplatin and X‐irradiation on human glioma cell cultures derived from biopsy‐tissue. J. Cancer Res. Clin. Oncol. 126, 711–716. [DOI] [PubMed] [Google Scholar]
- Fichtinger‐Schepman AM, Van Der Veer JL, Den Hartog JH, Lohman PH, Reedijk J (1985) Adducts of the antitumor drug cis‐diamminedichloroplatinum (II) with DNA: formation, identification and quantitation. Biochemistry 24, 707–713. [DOI] [PubMed] [Google Scholar]
- Fischer SJ, McDonald ES, Gross L, Windebank AJ (2001) Alterations in cell cycle regulation underlie cisplatin induced apoptosis of dorsal root ganglion neurons in vivo . Neurobiol. Dis. 8, 1027–1035. [DOI] [PubMed] [Google Scholar]
- Foster KA, Galeffi F, Gerich FJ, Turner DA, Muller M (2006) Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog. Neurobiol. 79, 136–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Martinou I, Tsujimoto Y, Martinou JC (1992) Prevention of programmed cell death of sympathetic neurons by the bcl‐2 proto‐oncogene. Science 258, 302–304. [DOI] [PubMed] [Google Scholar]
- Gerschenson M, Paik CY, Gaukler EL, Diwan BA, Poirier MC (2001) Cisplatin exposure induces mitochondrial toxicity in pregnant rats and their fetuses. Reprod. Toxicol. 15, 525–531. [DOI] [PubMed] [Google Scholar]
- Han Z, Chatterjee D, Early J, Pantazis P, Hendrickson EA, Wyche JH (1996) Isolation and characterization of an apoptosis‐resistant variant of human leukemia HL‐60 cells that has switched expression from Bcl‐2 to Bcl‐xL. Cancer Res. 56, 1621–1628. [PubMed] [Google Scholar]
- Hayakawa J, Ohmichi M, Kurachi H, Ikegami H, Kimura A, Matsuoka T, Jikihara H, Mercola D, Murata Y (1999) Inhibition of extracellular signal‐regulated protein kinase or c‐Jun N‐terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J. Biol. Chem. 274, 31648–31654. [DOI] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG (1998) Caspase‐3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357–9360. [DOI] [PubMed] [Google Scholar]
- Jeong HG, Cho HJ, Chang IY, Yoon SP, Jeon YJ, Chung MH, You HJ (2002) Rac1 prevents cisplatin‐induced apoptosis through down‐regulation of p38 activation in NIH3T3 cells. FEBS Lett. 518, 129–134. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajci D, Mares V, Lisa V, Bottone MG, Pellicciari C (2006) Intranuclear microtubules are hallmarks of an unusual form of cell death in cisplatin‐treated C6 glioma cell. Histochem. Cell Biol. 125, 183–191. [DOI] [PubMed] [Google Scholar]
- Li E, Bedner X, Gorczyca W, Melamed MR, Darzynkiewicz Z (1999) Analysis of apoptosis by laser scanning cytometry. Cytometry 35, 181–195. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- Liu W, Staecker H, Stupak H, Malgrange B, Lefebvre P, Van De Water TR (1998) Caspase inhibitors prevent cisplatin‐induced apoptosis of auditory sensory cells. Neuroreport 9, 2609–2614. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeir JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031. [DOI] [PubMed] [Google Scholar]
- McDonald ES, Randon KR, Knight A, Windebank AJ (2005) Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol. Dis. 18, 305–313. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, Saikumar P (2001) Bcl‐2 prevents Bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem. 276, 18361–18374. [DOI] [PubMed] [Google Scholar]
- Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y (1998) Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 95, 14681–14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ (2001) Bax and Bak coalesce into novel mitochondria‐associated clusters during apoptosis. J. Cell Biol. 153, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod MG, O’Neill CF, Robertson D, Harrap KR (1994a) Cisplatin induces apoptosis in a human ovarian carcinoma cell line without concomitant internucleosomal degradation of DNA. Exp. Cell Res. 211, 231–237. [DOI] [PubMed] [Google Scholar]
- Ormerod MG, Orr RM, Peacock JH (1994b) The role of apoptosis in cell killing by cisplatin: a flow cytometric study. Br. J. Cancer 69, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, De Leon M, Devarajan P (2002) Cisplatin induces apoptosis in LLC‐PK1 cells via activation of mitochondrial pathways. J. Am. Soc. Nephrol. 13, 858–865. [DOI] [PubMed] [Google Scholar]
- Persons DL, Yazlovitskaya EM, Pelling JC (2000) Effect of extracellular signal‐regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 275, 35778–35785. [DOI] [PubMed] [Google Scholar]
- Pisu MB, Guioli S, Conforti E, Bernocchi G (2003) Signal molecules and receptors in the differential development of cerebellum lobules. Acute effects of cisplatin on nitric oxide and glutamate systems in Purkinje cell population. Brain Res. Dev. Brain Res. 145, 229–240. [DOI] [PubMed] [Google Scholar]
- Pisu MB, Roda E, Avella D, Bernocchi G (2004) Developmental plasticity of rat cerebellar cortex after cisplatin injury: inhibitory synapses and differentiating Purkinje neurons. Neuroscience 129, 655–664. [DOI] [PubMed] [Google Scholar]
- Pisu MB, Roda E, Guioli S, Avella D, Bottone MG, Bernocchi G (2005) Proliferation and migration of granule cells in the developing rat cerebellum: cisplatin effects. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 287, 1226–1235. [DOI] [PubMed] [Google Scholar]
- Portier BP, Taglialatela G (2006) Bcl‐2 localized at the nuclear compartment induces apoptosis after transient overexpression. J. Biol. Chem. 281, 40493–40502. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB (1991) J‐aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30, 4480–4486. [DOI] [PubMed] [Google Scholar]
- Ritter PM, Marti A, Blanc C, Baltzer A, Krajewski S, Reed JC, Jaggi R (2000) Nuclear localization of procaspase‐9 and processing by a caspase‐3‐like activity in mammary epithelial cells. Eur. J. Cell Biol. 79, 358–364. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Alcazar JA, Ault JG, Khodjakov A, Schneider E (2000) Increased mitochondrial cytochrome c levels and mitochondrial hyperpolarization precede camptothecin‐induced apoptosis in Jurkat cells. Cell Death Differ. 7, 1090–1100. [DOI] [PubMed] [Google Scholar]
- Sawada M, Nakashima S, Banno Y, Yamakawa H, Takenada K, Shinoda J, Nishimura Y, Sakai N, Nozawa Y (2000) Influence of Bax or Bcl‐2 overexpression on the ceramide‐dependent apoptotic pathway in glioma cells. Oncogene 20, 3508–3520. [DOI] [PubMed] [Google Scholar]
- Scherini E, Bernocchi G (1994) cisDDP treatment and development of the rat cerebellum. Prog. Neurobiol. 42, 161–196. [DOI] [PubMed] [Google Scholar]
- Schiffer J, Walach N, Lushkov G, Nyska A, Gur R, Pollak L (1996) Effect of the combination of cisplatinum and radiation in the rabbit's brain. Neurol. Res. 18, 454–456. [DOI] [PubMed] [Google Scholar]
- Schlesinger MJ, Ryan C, Chi MM, Carter JG, Pusateri ME, Lowry OH (1997) Metabolite changes associated with heat shocked avian fibroblast mitochondria. Cell Stress Chaperones 2, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi I, Suzuki M, Tamada T, Shinomiya N, Tsuru S, Murata M (1996) Effects of cisplatin on cell cycle kinetics, morphological change, and cleavage pattern of DNA in two human ovarian carcinoma cell lines. Oncology 53, 19–26. [DOI] [PubMed] [Google Scholar]
- Shirabe S, Fang WH, Schwartz JP (1997) Altered intemediate filament expression in human neuroblastoma cells transformed by a growth‐promoting agent derived from schizophrenis CSF. Cell. Mol. Neurobiol. 17, 1–11. [DOI] [PubMed] [Google Scholar]
- Soldani C, Bottone MG, Pellicciari C, Scovassi AI (2001) Two‐colour fluorescence detection of Poly (ADP‐Ribose) Polymerase‐1 (PARP‐1) cleavage and DNA strand breaks in etoposide‐induced apoptotic cells. Eur. J. Histochem. 45, 389–392. [DOI] [PubMed] [Google Scholar]
- Sorenson CM, Eastman A (1988) Mechanism of cis‐diamminedichloroplatinum (II)‐induced cytotoxicity: role of G2 arrest and DNA double‐strand breaks. Cancer Res. 48, 4484–4488. [PubMed] [Google Scholar]
- Sorenson CM, Barry MA, Eastman A (1990) Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J. Natl. Cancer Inst. 82, 749–755. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes‐Alnemri T, Alnemri ES (1998) Autoactivation of procaspase‐9 by Apaf‐1‐mediated oligomerization. Mol. Cell 1, 949–957. [DOI] [PubMed] [Google Scholar]
- Thomas SG, Hwang S, Li S, Straiger CJ, Franklin‐Tong VE (2007) Actin depolymerization is sufficient to induce programmed cell death in self‐incompatible pollen. JCB 174, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y (1989) Stress‐resistance conferred by high level of Bcl‐2 alpha protein in human B lymphoblastoid cell. Oncogene 4, 1331–1336. [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF, Currie AR (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251–306. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl‐2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl‐2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369, 321–323. [DOI] [PubMed] [Google Scholar]
- Zha H, Aime‐Sempe C, Sato T, Reed JC (1996) Proapoptotic protein Bax heterodimerizes with Bcl‐2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 271, 7440–7444. [DOI] [PubMed] [Google Scholar]
- Zwelling LA, Anderson T, Kohn KW (1979) DNA‐protein and DNA interstrand crosslinking by cis‐ and trans‐Pt(II)(NH3)2Cl2 in L1210 cells. Cancer Res. 39, 365–369. [PubMed] [Google Scholar]