

Abstract
Objectives
A number of previous studies has provided evidence that the well‐known anti‐bacterial quinolones may have potential as anti‐cancer drugs. The aim of this study was to evaluate potential anti‐tumour activity and selectivity of a set of 6‐aminoquinolones showing some chemical similarity to naphthyridone derivative CX‐5461, recently described as innovative anti‐cancer agent.
Materials and methods
In‐house quinolones 1‐8 and ad hoc synthesized derivatives 9‐13 were tested on Michigan Cancer Foundation‐7 (MCF‐7) breast cancer cells and mesenchymal progenitor (MePR2B) cell lines, analysing their effects on the cell cycle and cell death using FACS methodology. Activation of p53 was evaluated by western blotting.
Results
Benzyl esters 4, 5 and their amide counterparts 12, 13 drastically modulated MCF‐7 cell cycles inducing DNA fragmentation and cell death, thus proving to be potential anti‐tumour compounds. When assayed in non‐tumour MePR2B cells, compounds 4 and 5 were cytotoxic while 12 and 13 had a certain degree of selectivity, with compound 12 emerging as the most promising. Western blot analysis revealed that severe p53‐K382ac activation was promoted by benzylester 5. In contrast, amide 12 exerted only a moderate effect which was, however, comparable to that of suberoylanilide hydoxamic acid (SAHA).
Conclusions
Taken together, these results further reinforce evidence that quinolones have potential as anti‐cancer agents. Future work will be focused on understanding compound 12 mechanisms of action, and to obtain more potent and selective compounds.
Introduction
According to the World Health Organization (WHO), cancer represents a leading cause of morbidity and mortality in the world (1). This burden has prompted WHO to launch the Global Action Plan for Prevention and Control of Non‐communicable Diseases 2013–2030, that aims to reduce by 25% premature mortality due to cancer, and other non‐communicable diseases (2,3). In 2012, 8.2 million people died of cancer and approximately 12 million new cases were diagnosed. In the next two decades, increase in cancer incidence will be even more serious (70%) 1, 2, 3. Some major and well‐documented predispositions to the development of cancer are: high body mass index, low fruit and vegetable intake, lack of physical activity and tobacco and alcohol misuse; viral infections such as by HCV, HBV and HPV strongly contribute to spread of their related cancers. It is well known that some cancers can be partly prevented by modifying and avoiding risk factors, and early diagnosis strongly improves survival of patients. However, people with advanced disease need effective therapy and, especially for chemotherapy, the drugs arsenal has to be continuously renewed due to development of resistance. Cancer treatment involves physical, chemical and biological approaches 4, 5. Among the biological approaches, introduction of biodrugs has revolutionized therapeutic protocols; sadly, such drugs also have weak aspects such as high production costs, high side effects, low bioactivity, high dosage and immunogenic response 6, 7. This means that more potent, well‐tolerated and innovative drugs are still necessary to combat cancer. Thus, classes of compounds with previous impact in related and unrelated cancer fields are investigated for anti‐proliferative and apoptotic effects 8, 9, 10.
The well‐known antibiotic quinolones have been extensively developed as anti‐bacterial agents targeting DNA gyrase, prokaryotic counterpart of topoisomerse II 11, and besides their canonical anti‐bacterial activity, several studies have provided evidence that quinolones may also have potential as anti‐cancer drugs 12, 13. Indeed, suitable functionalization of the quinolone scaffold has allowed shifting of inhibitory activity from bacterial uses towards mammalian topoisomerase II (14,15), with vosaroxin (also called voreloxin) as the most promising anti‐cancer agent 16. Subsequently, anti‐cancer quinolone‐based compounds able to inhibit the STAT3 pathway 17, 18 or human protein kinase CK2 (18), have been reported. In the last few years, there has been renewed interest in anti‐bacterial fluoroquinolones as promising anti‐cancer agents. In particular, a variety of studies has been focused on novel anti‐tumour mechanisms of action such as modulation of TAK1/TAB 2 interaction 19 and RNA interference pathways (20), just to mention a few. Moreover, Cylene Pharmaceuticals has reported the discovery of naphthyridone analogue, CX‐5461 (Fig. 1), as an innovative anti‐cancer agent, inhibitor of RNA Pol I‐mediated transcription with in vivo activity in tumour growth efficacy models 21.
Figure 1.
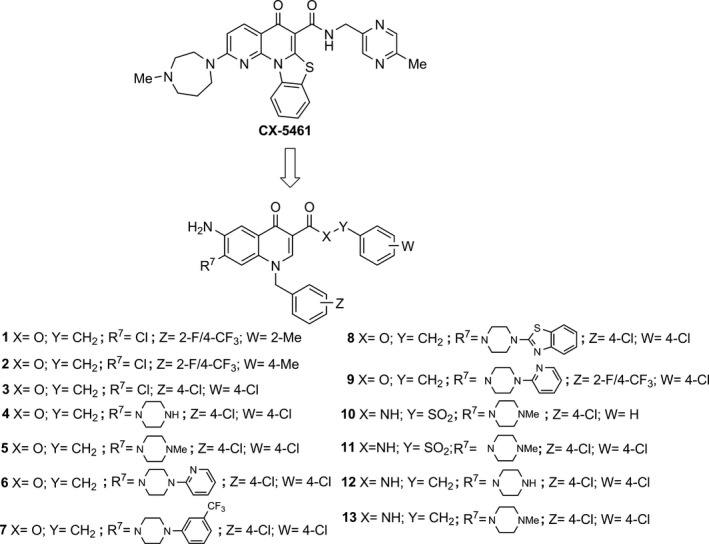
Chemical structures of CX‐5461 and 6‐aminoquinolones 1‐13. In‐house 6‐aminoquinolones (1‐8) showing chemical similarity to CX‐5461, and ad hoc designed and newly synthesized derivatives 9‐13.
For many years, our team has been focusing on the 6‐aminoquinolone chemotype as a privileged scaffold to obtain novel chemotherapeutic agents such as anti‐HIV, 22, 23, 24, 25, 26 anti‐HCMV, 27, 28 anti‐HCV 29 and anti‐bacterial 30, 31, 32 compounds.
With this historical backdrop, we have evaluated tumour cell population growth inhibitory capability for 6‐aminoquinolones 1‐8 29, previously reported as anti‐HCV agents, as well as for newly synthesized 6‐aminoquinolone derivatives 9‐11, given their chemical similarity with anti‐cancer agent CX‐5461 21 (Fig. 1). Anti‐cancer activity of 6‐aminoquinolones 1‐11 was evaluated in the breast cancer cell line Michigan Cancer Foundation‐7 (MCF‐7). Next, a further two quinolone derivatives 12 and 13 were designed and synthesized guided by interesting biological results obtained for their ester counterparts 4 and 5. Cytoxicity to non‐tumour mesenchymal progenitor 2B stem cells (MePR2B) was evaluated for the compounds, showing highest toxicity in the cancer settings. Results of this preliminary pharmacological survey for the whole set of 6‐aminoquinolones, and synthesis of derivatives 9–13 are reported here.
Materials and methods
Synthesis of target compounds 1–13
Compounds 1–8 were prepared according to a procedure previously reported by our group 29, while newly synthesized compounds 9–13 were prepared as described below.
General chemistry
All starting materials were commercially available, unless otherwise indicated. Reagents and solvents were purchased from common commercial suppliers and were used as such. Organic solutions were dried over anhydrous Na2SO4 and evaporated to dryness using a rotary evaporator at low pressure. All reactions were routinely checked using thin‐layer chromatography (TLC) on silica gel 60F254 (Merck), and visualized using UV or iodine. Flash column chromatography separations were carried out on Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Electrotermal model 9100) and are uncorrected. Yields were of purified products and were not optimized. Reactions conducted under microwave irradiation were carried out employing a microwave reactor Biotage Initiator™ 2.0 version 2.3 build 6250. H NMR spectra were recorded at 200 or 400 MHz (BrukerAvance DRX‐200 or 400, respectively). Chemical shifts are given in ppm (δ) relative to TMS and spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR and spectral data are consistent with the assigned structures. Compounds 9–13 were ≥95% pure, determined by LC/MS using Agilent 1290 Infinity System apparatus equipped with diode array detector (DAD) from 190 to 640 nm. Purity was revealed at 280.4 ± 2 nm using an Agilent Elipse Plus RRHD C18 (2.1 mm × 100 mm, 1.8 μm particle size column) reverse phase with gradient 10–100% acetonitrile with 0.1% formic acid (channel B) in water with 0.1% formic acid (channel A) at 0.45 ml/min. Injection volume was 0.5 μl and column temperature of 40 °C. Peak retention times are given in minutes. Detection mass was based on electrospray ionization (ESI) in positive polarity using Agilent 1290 Infinity System equipped with a MS detector Agilent 6550UHD Accurate Mass Q‐TOF.
4‐Chlorobenzyl 6‐amino‐1‐[2‐fluoro‐4‐(trifluoromethyl)benzyl]‐4‐oxo‐7‐[4‐(2‐pyridinyl)‐1‐piperazinyl]‐1,4‐dihydro‐3‐quinolinecarboxylate (9). A stirred mixture of quinolone carboxylic acid 22 (0.5 g, 0.94 mmol), 4‐chloro‐benzylchloride (0.22 g, 0.18 ml, 1.4 mmol) and K2CO3 (0.25 g, 1.8 mmol) in N,N‐dimethylformamide (DMF) (60 ml) was heated at 60°C for 30 h. The solvent was concentrated under vacuum to half volume and the mixture was poured into ice water and neutralized with 2N HCl; then NaCl saturated solution (3 ml) was added until a precipitate was observed. Solids so obtained were filtered, washed in cyclohexane and dried. The crude product was thus purified by flash column chromatography eluting with CHCl3/MeOH (93:7) to give compound 9 (0.313 g, 50% yield) as a whitish‐pink solid: mp 277–279 °C. 1H‐NMR (400 MHz, DMSO‐d6) δ: 2.90–2.95 and 3.60–3.67 (each m, 4H, piperazine CH2), 5.23 (bs, 2H, NH2), 5.27 (s, 2H, OCH2), 5.75 (s, 2H, NCH2), 6.62–6.65 (m, 1H, pyridine‐H), 6.85 (d, J = 8.4 Hz, 1H, pyridine‐H), 6.90 (s, 1H, H‐8), 7.40–7.45 (m, 3H, aromatic‐H), 7.45–7.55 (m, 5H, H‐5 and aromatic‐H), 7.75 (d, J = 11.3 Hz, 1H, N‐benzyl‐H‐3), 8.20 (d, J = 4.0 Hz, 1H, pyridine‐H), 8.77 (s, 1H, H‐2). HRMS (ESI) m/z [M+Na]+ calcd for C34H28ClF4N5O3: 688.1715, found: 688.1720; LC‐MS: 3.769 min.
6‐Amino‐1‐(4‐chlorobenzyl)‐7‐(4‐methyl‐1‐piperazinyl)‐4‐oxo‐N‐(phenylsulfonyl)‐1,4‐dihydro‐3‐quinolinecarboxamide (10). A stirred solution of nitro derivative 24 (0.50 g, 0.8 mmol) in DMF (90 ml) was hydrogenated over a catalytic amount of Raney Ni at room temperature and atmospheric pressure for 3 h. The mixture was then filtered over Celite and filtrate was evaporated to dryness. After purification by flash column chromatography eluting with CHCl3/MeOH (80:20), compound 10 (48% yield) was obtained as a pale brown solid: mp 229–231 °C. 1H‐NMR (400 MHz, DMSO‐d6) δ: 2.15 (s, 3H, CH3), 2.40–2.50 and 2.70–2.90 (each m, 4H, piperazine‐CH2), 5.40 (s, 2H, NH2), 5.60 (s, 2H, NCH2), 7.00 (s, 1H, H‐8), 7.20–7.30 (m, 2H, benzyl‐H‐2 and H‐6), 7.35–7.40 (m, 2H, benzyl‐H‐3 and H‐5), 7.50 (s, 1H, H‐5), 7.55–7.60 (m, 3H, H‐3′, H‐4′ and H‐5′), 8.00–8.10 (m, 2H, H‐2′ and H‐6′), 8.90 (s, 1H, H‐2), 14.25 (bs, 1H, NHSO2). HRMS (ESI) m/z [M+H]+ calcd for C28H28ClN5O4S: 565.1551, found: 565.1557; LC‐MS: tR 2.815 min.
6‐Amino‐1‐(4‐chlorobenzyl)‐N‐[(4‐chlorophenyl)sulfonyl]‐7‐(4‐methyl‐1‐piperazinyl)‐4‐oxo‐1,4‐dihydro‐3‐quinolinecarboxamide (11). Starting from 25, compound 11 was prepared employing the same procedure as reported for derivative 10 (reaction time 3.5 h). After purification by flash column chromatography eluting with CHCl3/MeOH, compound 11 (50% yield) was obtained as a yellow solid: mp 258–260 °C. 1H‐NMR (200 MHz, DMSO‐d6) δ: 2.15 (s, 3H, NCH3), 2.30–2.45 and 2.75–2.85 (each m, 4H, piperazine‐CH2), 5.27 (bs, 2H, NH2), 5.60 (s, 2H, NCH2), 6.95 (s, 1H, H‐8), 7.20–7.25 (m, 2H, benzyl‐H‐3 and H‐5), 7.30–7.40 (m, 2H, benzyl‐H‐2 and H‐6), 7.50 (s, 1H, H‐5), 7.60–7.70 (m, 2H, H‐3′ and H‐5′), 7.90–8.00 (m, 2H, H‐2′ and H‐6′), 8.80 (s, 1H, H‐2), 13.50 (bs 1H, NHSO2). HRMS (ESI) m/z [M+H]+ calcd for C28H27Cl2N5O2: 600.1240, found: 600.1244; LC‐MS: tR 3.137 min.
6‐Amino‐N,1‐bis(4‐chlorobenzyl)‐4‐oxo‐7‐(1‐piperazinyl)‐1,4‐dihydro‐3‐quinolinecarboxamide (12). In a microwave oven tube and neat conditions, ester intermediate 19 (0.2 g, 0.45 mmol) and 4‐chlorobenzylamine (1.40 g, 1.2 ml, 9.9 mmol) were mixed and the tube plugged. The mixture was irradiated at 120 °C for 40 min., employing the following experimental parameters: pressure 5 bar, cooling off, FHT on, pre‐stirring 30 s, normal absorption, potency: 150 W. The solution was then poured into ice water and basified with NaOH 10% (pH = 12) giving a precipitate which was filtered, treated with cyclohexane and dried. The crude product was then crystallized by EtOH to provide compound 12 (0.80 g, 33% yield) a pale brown solid: mp 254–255 °C. 1H‐NMR (400 MHz, DMSO‐d6) δ: 2.60–2.75 and 2.75–2.80 (each m, 4H, piperazine CH2), 4.55 (d, J = 5.9 Hz, 2H, NHCH2), 5.30 (bs, 2H, NH2), 5.60 (s, 2H, NCH2), 6.90 (s, 1H, H‐8), 7.25–7.30 (m, 2H, aromatic‐H), 7.35–7.40 (m, 6H, aromatic‐H), 7.50 (s, 1H, H‐5), 8.80 (s, 1H, H‐2), 10.57 (t, J = 5.9 Hz, 1H, NH). HRMS (ESI) m/z [M+H]+ calcd for C28H27Cl2N5O2: 536.1621, found: 536.1625; LC‐MS: tR 2.915 min.
6‐Amino‐N,1‐bis(4‐chlorobenzyl)‐7‐(4‐methyl‐1‐piperazinyl)‐4‐oxo‐1,4‐dihydro‐3‐quinolinecarboxamide (13). Starting from 20, compound 13 was prepared employing the same procedure as reported for derivative 19. After recrystallization by EtOH, amino derivative 13 (35% yield) was obtained as a yellow solid: mp 291–293 °C. 1H‐NMR (200 MHz, DMSO‐d6) δ: 2.20 (s, 3H, NCH3), 2.30–2.40 and 2.90–3.00 (each m, 4H, piperazine CH2), 4.55 (d, J = 5.6 Hz, 2H, NHCH2), 5.20 (bs, 2H, NH2), 5.54 (s, 2H, NCH2), 7.00 (s, 1H, H‐8), 7.25–7.30 (m, 2H, aromatic‐H), 7.30–7.40 (m, 6H, aromatic‐H), 7.50 (s, 1H, H‐5), 8.85 (s, 1H, H‐2), 10.60 (t, J = 5.8 Hz, 1H, NH). HRMS (ESI) m/z [M+H]+ calcd for C29H29Cl2N5O2: 550.1777, found: 550.1782; LC‐MS: tR 2.940 min.
Experimental procedures for preparation of intermediates 16–25 and corresponding analytical data are in the Supporting Information section.
Biological assays
Cell line
Human breast cancer MCF‐7 cells (ATCC) were propagated in Dulbecco's modified Eagle's medium (DMEM) with 10% foetal bovine serum (FBS) (Sigma, St. Louis, MO, USA), 2 mm L‐glutamine (Euroclone, Pero MI, Italy) and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin) 33. Cells were stimulated with respective drugs for 24, 48 and 24 h + 24 h (drugs were added again post 24 h stimulation).
Mesenchymal progenitor (MePR) cell line from amniotic fluid‐derived human mesenchymal stem cells (hMSCs) were cultured as reported previously 34.
FACS, cell cycle analysis and caspase assays
Compounds 1–13 were tested at final concentration and incubation times, as reported in the manuscript figures 2–7. Propidium iodide (PI; Sigma Aldrich, St. Louis, MO, USA) was used for cell cycle staining. Briefly, breast cancer oestrogen‐responsive cell line MCF‐7 and MePR2B were grown in DMEM medium for 48 h. They were then pelleted before being re‐suspending in phosphate‐buffered saline (PBS) containing PI (50 mg/mL; Invitrogen, Waltham, MA, USA), RNAse A (0.1 mg/ml; Sigma Aldrich) and Triton X‐100 (0.05%; Sigma Aldrich) followed by cytofluorometric analysis using a FACSCalibur (Becton Dickinson, Erembodegem, Belgium) 34.
Positive control suberoylanilide hydoxamic acid (SAHA) 35, 36 (Merck) was dissolved in DMSO (Sigma Aldrich) and used at 5 μm final concentration, at indicated incubation times as reported in the figures 2–7.
Caspase activity was monitored in MCF‐7 cells stimulated for 16 h with compounds 4,5,12 and 13 at 10 μm. Caspase B‐BRIDGE Kits supplied with cell‐permeable fluorescent substrates was used according to the manufacturer's instructions. Fluorescent substrates used were FAM‐DEVD‐FMK for caspase‐3/7; FAM‐LETD‐FMK for caspase 8; and SR‐LEHD‐FMK for caspase 9. Cells were analysed using Cell Quest software applied to a FACScalibur (BD). Experiments were performed in biological duplicates and values expressed in mean ± SD from two independent experiments.
Protein extraction
Cell were washed, pelleted and resuspended in lysis buffer (50 mm Tris‐HCl pH 7.4, 150 mm NaCl, 1% NP40, 10 mm NaF, 1 mm PMSF and protease inhibitor cocktail). The lysis reaction was performed for 20 min at 4 °C. Samples were centrifuged at 17 950 g for 30 min at 4 °C and protein concentration was determined by Bradford assay (Bio‐Rad, Hercules, CA, USA).
Western blotting
Of total protein extract, 50 μg was loaded on 10% SDS‐PAGE. Nitrocellulose filters were stained with Ponceau red (Sigma Aldrich) as the additional control for equal loading. ERK1 antibody was from Santa‐Cruz and p53‐K382ac antibody was from Millipore. Semi‐quantitative analysis was performed using ImageJ software 37, 38.
Results
Synthesis of 6‐aminoquinolones 9‐13
Synthetic procedures for tested compounds 1‐8 have been reported in a previous publication, 29 whereas synthesis of the new 6‐aminoquinolones 9‐13 was accomplished as outlined in Scheme 1. In particular, reaction of 7‐chloro‐6‐nitro‐quinolone ethyl ester 14 29 with piperazine or 4‐methyl‐1‐piperazine in DMF gave, in good yield, compounds 16 and 17 respectively. Employing the same procedure, quinolone 15 was reacted with 1‐(2‐pyridyl)piperazine to provide derivative 18. Starting from nitro intermediates 16‐18, catalytic hydrogenation of the nitro group afforded C‐6 aminoquinolones 19‐21 in moderate to high yields. Nucleophilic substitution of amino ethyl esters 19 and 20 with neat 4‐chlorobenzylamine and using microwave irradiation at 120 °C, advantageously afforded target amides 12 and 13 respectively. On the other hand, amino ethyl ester 21 was hydrolysed into corresponding 3‐carboxylic acid 22 using refluxing 5% aqueous NaOH, and successively esterified in DMF employing 4‐chlorobenzyl chloride and K2CO3 as base, to provide target derivative 9 at a moderate yield. Starting from ester 17, compound 23 was obtained after basic hydrolysis, then coupled to benzenesulfonamide or 4‐chlorobenzenesulfonamide using 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide (EDAC) and 4‐dimethylaminopyridine (DMAP) in DMF, to afford in moderate yields, acylsulfonamides 24 and 25 respectively. Finally, catalytic reduction of nitro group of intermediates 24 and 25 led to target derivatives 10 and 11 in moderate yields.
Scheme 1.
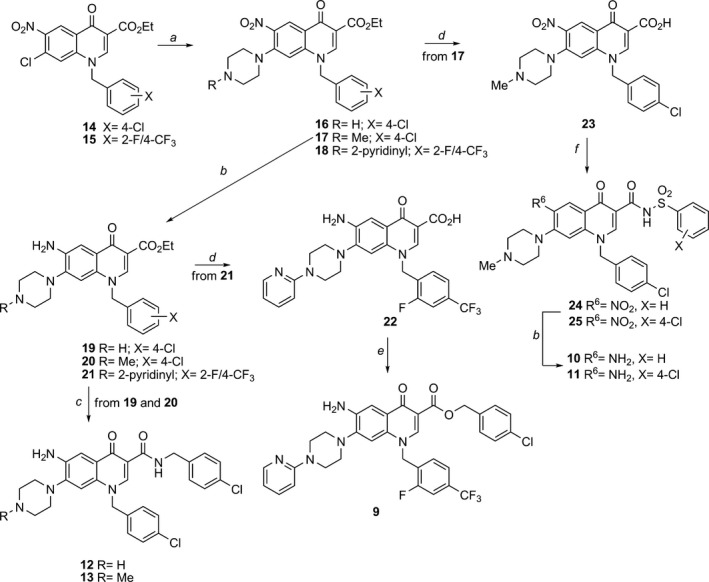
Synthesis of target compounds 9‐13. Reagents and conditions: (a) appropriate piperazine, DMF, 80 °C; (b) H2, Raney Ni, DMF, rt; (c) 4‐chloro‐benzylamine, MW, neat, 120 °C; (d) 5% NaOH, EtOH, reflux; (e) 4‐chloro‐benzylchloride, K2 CO 3, DMF, 60 °C; (f) appropriate aryl‐sulfonylamide, EDAC, DMAP, dry DMF, 70 °C.
Cell cycle analysis and Pre‐G1 fraction in MCF‐7 cells after treatment with compounds 1‐13
In the first assay, compounds 1‐11 were tested at 10 μm, and cell cycle and pre‐G1 fraction were analysed after 24, 48 and 24 + 24 h exposure. SAHA at 5 μm was included in all assays as positive control (35,36). Among the tested compounds 1‐11, only derivatives 4 and 5 significantly altered the cell cycle, especially at 24 and 24 + 24 h, causing high pre‐G1 phase cell induction (Fig. 2). Strong effects observed made statistical analysis difficult, acting in a negative way (asterisks in Fig. 2). In particular, 6‐aminoquinolones 4 and 5 after 24 + 24 h increased percentages up to 80–90% in cells with DNA fragmentation in pre‐G1 (Fig. 2b).
Figure 2.
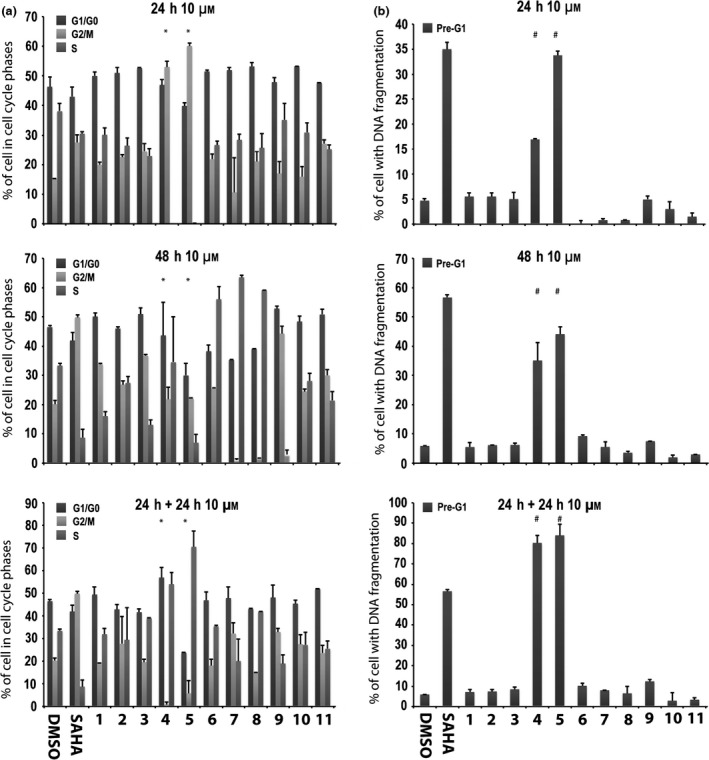
Preliminary screening in MCF‐7 cells. Cell cycle analysis (a) and pre‐G1 phase (b) in MCF‐7 cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with compounds 1–11 at 10 μm for 24, 48 and 24 + 24 h. All data expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance P‐value: 0.001 with the exception of * and # marked as 0.05.
In a successive study, only the two active compounds 4 and 5 were tested at 5 and 10 μm at 12, 24 and 48 h. In this experiment, they induced high percentages of cells in G1/G0 phase at both 5 and 10 μm (Fig. 3a). DNA fragmentation was observed at 10 μm but derivatives 4 and 5 did not show any significant effect at the lowest concentration (i.e. 5 μm) (Fig. 3b). Only compound 4 at 5 μm and after 48 h caused nearly 20% of DNA fragmentation in pre‐G1 (Fig. 3b).
Figure 3.
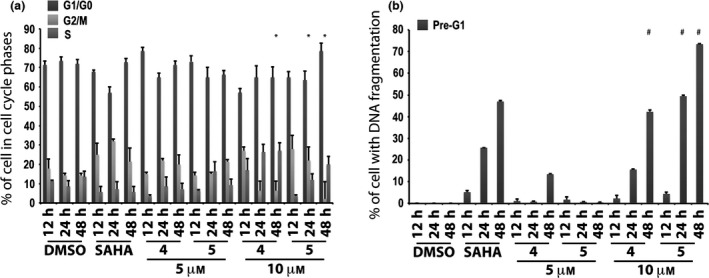
Biological evaluation in MCF‐7 cells for compounds 4 and 5. Cell cycle analysis (a) and pre‐G1 phase (b) in MCF‐7 cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with compounds 4 and 5 at 5 and 10 μm for 12, 24 and 48 h. All data expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance P‐value: 0.001 except * and # marked 0.05.
Derivatives 12 and 13 were assayed at 5 and 10 μm using MCF‐7 cells, and cell cycle and pre‐G1 fraction were analysed after 24, 48 and 24 + 24 h exposure.
Tested compounds strongly altered cell cycles with marked G1/G0 phase cell accumulation throughout treatment time and with both concentrations employed (Fig. 4a). 6‐Aminoquinolones 12 and 13 caused strong cell death in pre‐G1 fractions at 10 μm inducing 70–90% DNA fragmentation at 48 and 24 + 24 h (Fig. 4b). When reducing concentration to 5 μm, only derivative 12 resulted in still being active (60–70% of DNA fragmentation at 48 and 24 + 24 h) (Fig. 4b).
Figure 4.
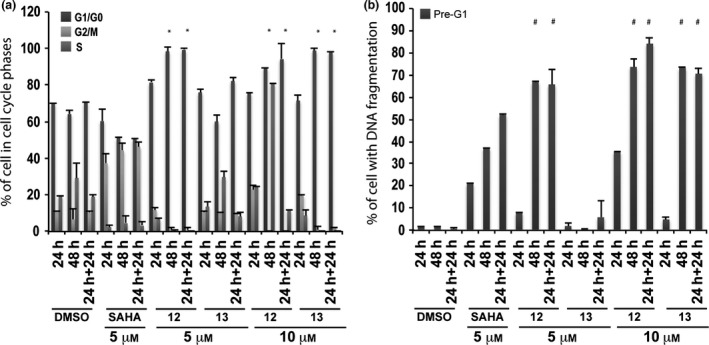
Biological evaluation in MCF‐7 cells for compounds 12 and 13. Cell cycle analysis (a) and pre‐G1 phase (b) in MCF‐7 cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with compounds 12 and 13 at 5 and 10 μm for 24, 48 and 24 + 24 h. All data are expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance is associated to the P‐value: 0.001 with the exception of * and # marked with a 0.05.
Cell cycle analysis and pre‐G1 fraction in non‐tumour MePR2B cells after treatment with compounds 4, 5, 12 and 13
To analyse cell cycles and pre‐G1 fractions in the non‐tumour model, active 6‐aminoquinolones 4 and 5 were tested in non‐tumour MePR2B cells at 10 and 15 μm at 24, 48 and 24 + 24 h, including SAHA at 5 μm as positive control (Fig. 5). Significant toxicity was observed for both quinolones with stronger effect for compound 5, highlighting non‐specific effects (Fig. 5b).
Figure 5.
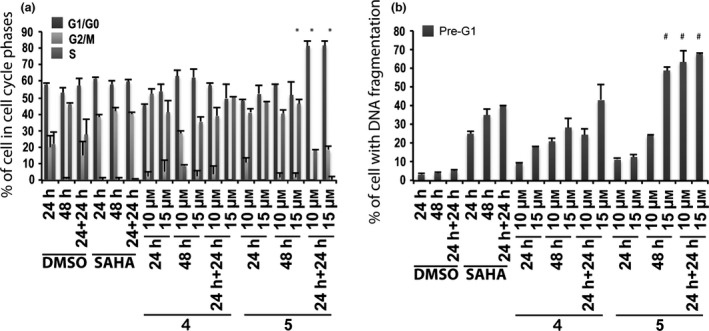
Biological evaluation in MePR2B cells for compounds 4 and 5. Cell cycle analysis (a) and pre‐G1 phase (b) in MePR2B cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with compounds 4 and 5 at 10 and 15 μm for the indicated time. All data are expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance P‐value: 0.001 except * and # marked as 0.05.
Derivatives 12 and 13 were evaluated in non‐tumour MePR2B cells at 10 μm, and cell cycles and pre‐G1 fraction were analysed after 24, 48 and 24 + 24 h of compounds exposure (Fig 6). These compounds displayed weak cytotoxic effects with about 20% of cells in pre‐G1 phase showing DNA fragmentation at 24 and 24 + 24 h (Fig. 6b).
Figure 6.
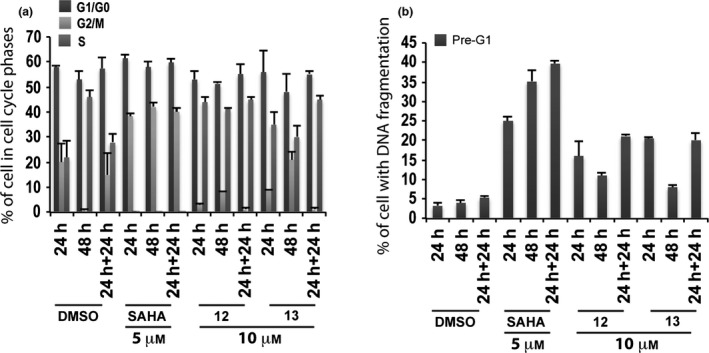
Biological evaluation in MePR2B cells for compounds 12 and 13. Cell cycle analysis (a) and pre‐G1 fraction (b) in MePR2B cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with compounds 12 and 13 at 10 μm for 24, 48 and 24 + 24 h. All data are expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance P‐value: 0.001.
Dose–response curve of pre‐G1 fraction in MCF‐7 cells treated with compounds 4, 5, 12 and 13
MCF‐7 cells were treated with growing concentrations (0.5–15 μm) of compounds 4, 5, 12 and 13 and dose–effect curves were generated over 24 h exposure (Fig. 7). 50% Cell death was observed at 15 μm concentration for derivatives 4 and 5 (Fig. 7a). In contrast, quinolone 12 produced 50% cell death induction at concentrations between 10–15 μm, meanwhile derivative 13 did not act on cell death at 24 h (Fig. 7b).
Figure 7.
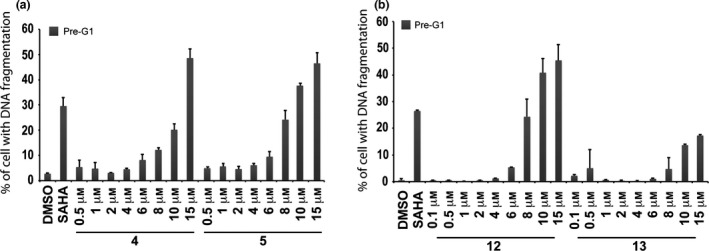
Dose‐response curves for compounds 4, 5, 12 and 13. Dose–response curve of pre‐G1 fraction in MCF‐7 cells untreated (DMSO, negative control), treated with SAHA (positive control) at 5 μm and with growing concentration of compounds 4, 5 (a) and 12, 13 (b) for 24 h.
p53‐K382ac activation after treatment with compounds 4, 5, 12 and 13
p53‐K382ac activation, compared to p53 total protein level, in MCF‐7 cells treated at 10 μm for 24 h with compounds 4, 5, 12 and 13 was investigated by western blot assay. Due to high cell debris derived from compound‐induced cell death, especially from compounds 4, 12 and 13, protein extraction was highly affected (Fig. 8a). To better estimate the entity of p53‐K382ac activation, a semi‐quantitative analysis was applied using ImageJ 37, 38 software that revealed p53‐K382ac activation in all samples, at least comparable with SAHA (compounds 12‐13), with greater effect for benzylester 4 and a 30‐fold induction for compound 5 (Fig. 8b).
Figure 8.
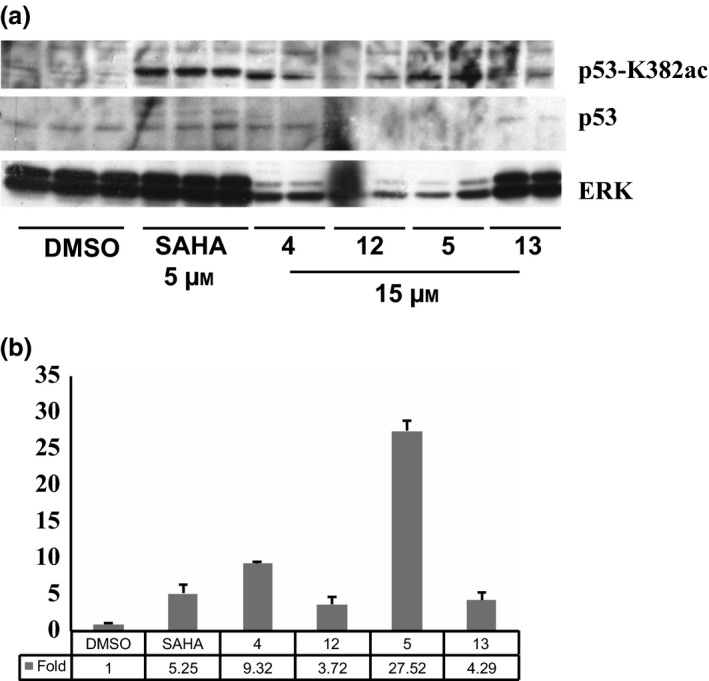
p‐53‐K382ac Activation. Western blot analysis (a) of p53‐K382ac activation after treatment with indicated compounds compared to total p53 and ERK normalization; (b) ImageJ analysis of relative p‐53K382ac abundance in western blots.
Given the increase of p53‐K382ac post drug stimuli, we investigated potential dose‐dependent effect mediated by compounds 4 and 12.
By western blot analysis, p53‐K382ac was found to be higher for both the drugs used at 15 and 10 μm for 24 h (Fig. 9). This increase was not present at lower concentrations.
Figure 9.
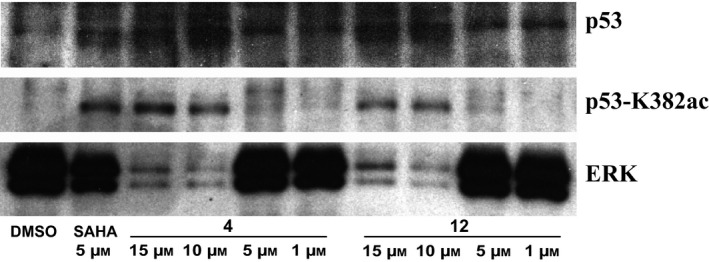
Western blot analysis of p53‐K382ac activation after treatment with indicated compounds compared to total p53 and ERK levels at dose‐dependent manner.
To assess cell death mediated by compounds 4, 5, 12 and 13, we evaluated their potential action on activity of initiator caspases 8 and 9, and of effector caspases 3/7, using flow cytometry. Results of analyses post 16 h stimulation at 10 μm show activation of both initiator caspases 8 and 9 and activation of effector caspases 3/7, suggesting involvement of diverse apoptotic pathways (Fig. 10).
Figure 10.
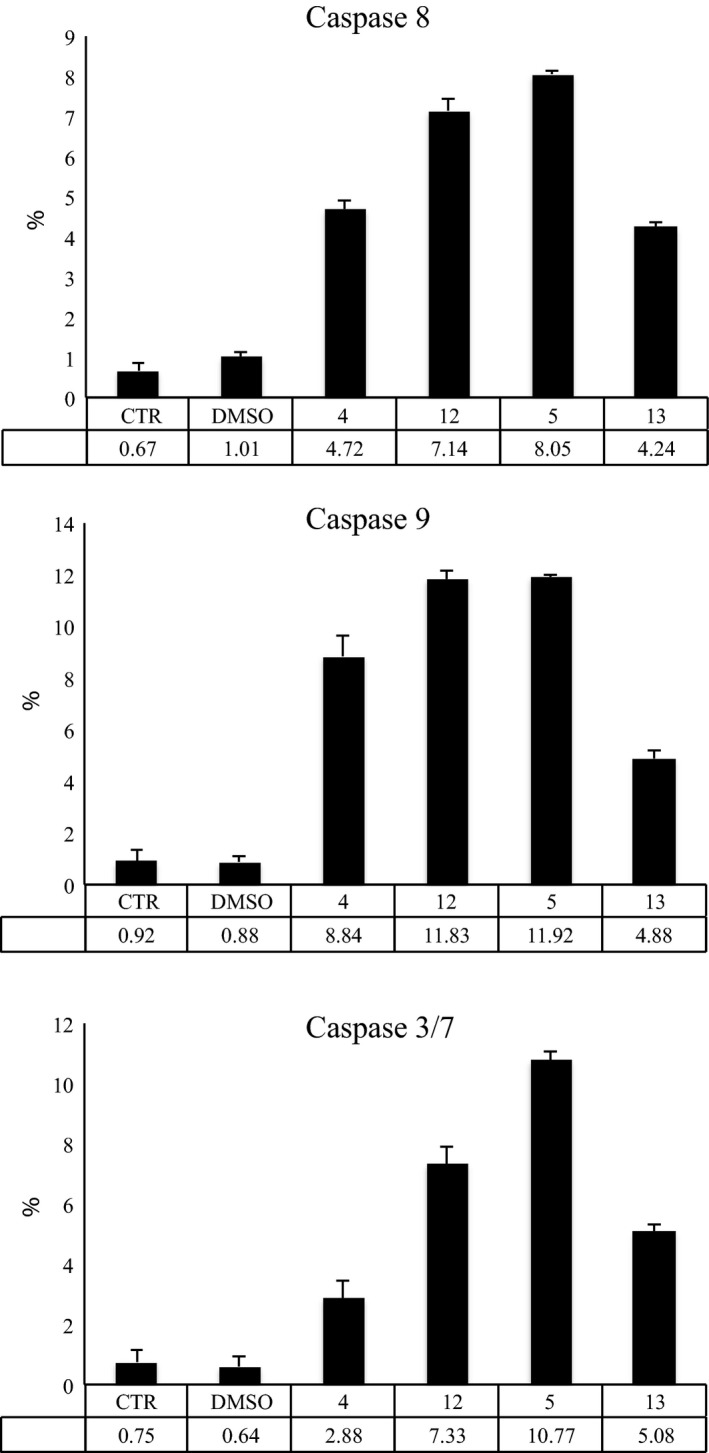
Caspase activation. Caspase 8, 9 and 3/7 assays, reported as % of activation on caspases 8, 9 and 3/7 after treatment with compounds 4, 5, 12 and 13 for 16 h.
Furthermore, given activation seen for p53 and activation of several caspases, we also investigated p21 levels after treatment with compounds 4 and 12 for 24 h, in a dose‐dependent manner. p21 increased after treatment with compound 4 at lower concentrations (5 and 1 μm), suggesting that p21 was involved in apoptotic drug mechanisms (Fig. 11).
Figure 11.
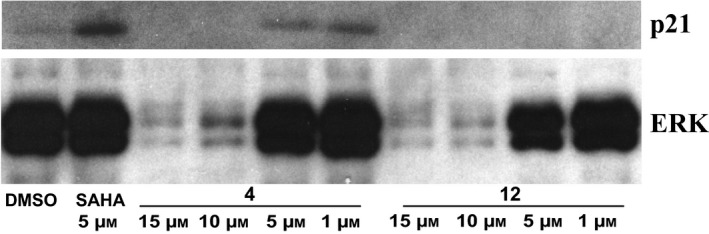
p21 Activation. Western blot analysis of p21 protein level after treatment with the indicated compounds. SAHA was used as positive control for p21 increase and ERKs for normalization.
Discussion
Cancer is a multi‐mechanistic disease that has many features, and requires wide‐ranges of approach for its treatment, control and prevention. Numerous studies have provided evidence that quinolones may have potential as anti‐cancer drugs 12, 13. The 6‐aminoquinolone chemotype is a privileged scaffold to obtain novel anti‐bacterial and anti‐viral agents 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. Besides this activity, and taking into account the chemical similarity of 6‐aminoquinolones 1‐8 29 with CX‐5461 21, we initially decided to evaluate their tumour cell population growth inhibitory capability along with newly synthesized 6‐aminoquinolone derivatives 9‐11 (Fig. 1).
Thus, 6‐aminoquinolones 1‐11 were tested to assess their action on cell cycle progression and cell death in the MCF‐7 cell line at 10 μm concentration (Fig. 2). Alteration in cell cycle (Fig. 2a) and pre‐G1 phase (Fig. 2b) was observed with compounds 4 and 5, which also displayed time‐dependent action. At 24 + 24 h, derivatives 4 and 5 were found to increase percentages of cells (up to 80–90%) with DNA fragmentation in pre‐G1 (Fig. 2b). Considering that 4 and 5 showed cell death effects depending on time of stimulation, whether or not a lower compound concentration could still ensure biological activity, was examined. Thus, we tested these two quinolone derivatives in the MCF‐7 cell line at 5 μm, and compared results to previously used concentration (10 μm) and positive control SAHA. In this second experiment, percentages of cells in each cell cycle phase were quite different from those observed in the first assay (compare Fig. 3a with Fig. 2a at 24 h and 48 h). Analysis of the pre‐G1 phase showed that compounds 4 and 5 induced high DNA fragmentation at 10 μm, as already observed, but unfortunately resulted in being ineffective at 5 μm (Fig. 3b). Only a slight increase in DNA fragmentation in pre‐G1 was observed for compound 4 at 5 μm and after 48 h (Fig. 3b).
All the remaining compounds exerted a somewhat similar effect on cell cycle progression when compared to derivatives 4 and 5 (Fig. 2a) but showed very different behaviour in the pre‐G1 phase (Fig. 2b). In particular, derivatives with a chlorine atom at C‐7 position (1‐3) as well as those with an arylpiperazine (6‐9) or a C‐3 acylsulfonamide moiety (10 and 11) resulted in being significantly less toxic in the time‐dependent experiments and were unable to affect the pre‐G1 cell phase (Fig. 2b). Data collected highlighted that, although all tested quinolones 1‐11 showed chemical features similar to CX‐5461, only its strictest analogues 4 and 5 proved to be active. In particular, contemporary presence of a benzyl ester function at C‐3 position of the quinolone scaffold and a basic heterocyclic side chain at C‐7 position (the piperazine in compound 4 and N‐4‐methylpiperazine in compound 5 respectively) were cytotoxic to MCF‐7 cells.
In a subsequent step, it became interesting to understand whether cell death observed could be attributed to intrinsic compound cytotoxicity, or to specific anti‐cancer effects. To address this question, derivatives 4 and 5 were tested on the MePR2B cell line used as non‐tumour model 34. Compared to controls, significant toxicity was observed for both the quinolones with a stronger effect for compound 5, highlighting a non‐specific effect (Fig. 5b). To assess the dose‐dependent effect on pre‐G1 induced MCF‐7 cells, a curve was generated for compounds 4 and 5 over 24 h exposure, and 50% cell death was observed at 15 μm concentration for both compounds (Fig. 7a). At the same concentration and time, compounds 4 and 5 produced higher percentages (about 50% for both compounds) of death in MCF‐7 cells rather than in non‐tumour MePR2B cells (compare Fig. 5b with Fig. 7a).
Guided by the promising results obtained with quinolones 4 and 5 on cell death in MCF‐7 cells and with the aim to reduce toxicity to non‐tumour cells, two additional derivatives 12 and 13 were designed, synthesized and tested in biological assays. The design strategy was oriented towards replacement of the liable benzylester moiety with the more stable benzylamide counterpart, resembling the linkage present in CX‐5461 (Fig. 1).
Biological results for the two newly synthesized derivatives are discussed below. First, we verified cell cycle progression effects and potential modulation of the pre‐G1 phase in the MCF‐7 cell line (Fig. 4). Compounds 12 and 13 induced strong cell death at 10 μm with 70–90% DNA fragmentation at 48 and 24 + 24 h (Fig. 4b). Only derivative 12 resulted still active (60–70% of DNA fragmentation at 48 and 24 + 24 h) when the concentration was reduced to 5 μm (Fig. 4b). It is worth noting that at 48 and 24 + 24 h, pre‐G1 phase induced by quinolone 12 was comparable to SAHA, when using the same compound concentration (Fig. 4b). When tested on non‐tumour MePR2B cells at 10 μm concentration, the new derivatives displayed only weakly cytotoxic effects with about 20% of cells in pre‐G1 phase showing DNA fragmentation at 24 and 24 + 24 h (Fig. 6b). This effect is remarkable considering the strong DNA fragmentation induced by the quinolones in the MCF‐7 cells at the same 10 μm compound concentration (compare Fig. 4b with Fig. 6b). Thus, results obtained in the two different cell lines highlighted that quinolones 12 and 13 had a certain anti‐cancer selective action. Of note, the biological data acquired in MePR2B non‐tumour cells also suggested that the replacement of the benzylester moiety (4 and 5) with the benzylamide counterpart (12 and 13) led to reduced cytotoxic effects (compare Fig. 5b with Fig. 6b).
To determine 12 and 13 induced cell death IC50s, the dose‐dependent effect at 24 h in MCF‐7 was evaluated (Fig. 7b). Quinolone 12 caused 50% cell death induction at concentration between 10–15 μm, meanwhile derivative 13 did not cause cell death at 24 h confirming the previous results. In this scenario, p53‐K382ac activation in response to compounds 12 and 13 was investigated via Western blot including the ester counterparts 4 and 5 for comparative purpose (Fig. 8a). Indeed, p53‐K382ac has been shown to enhance p53 transcriptional activity associated with cell cycle arrest and apoptosis 39. p53‐K382ac activation was observed in all samples in dose‐dependent manner demonstrating a greater effect for benzylesters with a 30‐fold induction for compound 5 and a still remarkable induction for the compounds 4, 12, and 13 (Fig. 8b). The plethora of effects observed post drug stimuli brought us to investigate whether the cell death was an apoptotic effect. The fact that we detected activation in caspase activity, suggested the idea that compounds 4, 5, 12 and 13 induce apoptosis. It is possible that activation of p53 and p21 induction contributed to the induction of cell death.
In conclusion, in the present study, we have explored the possibility that a series of known and newly synthesized 6‐aminoquinolones 1‐13 might act inhibiting tumour cell growth and affect cell cycle progression. The C‐3 benzylamide quinolone 12 showed the best biological profile displaying a certain selectivity in inhibiting tumour cell proliferation and inducing cell death in cancer cells rather than normal cells. However, compound 12 had the ability to increase p53‐K382ac activation, which was comparable to SAHA. Interestingly, benzylester 5 caused a strong effect on p53 acetylation and caspase 3/7, 8 and 9 activation. Overall, this preliminary biological survey prompts us to clarify the mechanism of action and especially to optimize this 6‐aminoquinolone class with the final aim to obtain more potent and selective compounds.
Supporting information
Synthetic procedures for the preparation of intermediates 16‐25.
Acknowledgements
This work was supported by EU, Blueprint project no. 282510 (LA); the Italian Flag Project EPIGEN (LA); PRIN‐2012 (LA).
References
- 1. Cancer. Available at http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 23 March 2015).
- 2. Global Action Plan for the Prevention and Control of NCDs 2013‐2020. Available at http://www.who.int/nmh/events/ncd_action_plan/en/ (accessed 23 March 2015).
- 3. Ullrich A, Miller A (2014) Global response to the burden of cancer: The WHO approach. Am. Soc. Clin. Oncol. Educ. Book e311–5 EdBook_AM.2014.34.e311. [DOI] [PubMed] [Google Scholar]
- 4. Agostinelli E, Vianello F, Magliulo G, Thomas T, Thomas TJ (2015) Nanoparticle strategies for cancer therapeutics: nucleic acids, polyamines, bovine serum amine oxidase and iron oxide nanoparticles. Int. J. Oncol. 46, 5–16. [DOI] [PubMed] [Google Scholar]
- 5. Rosen EM, Day R, Singh VK (2015) New approaches to radiation protection. Front Oncol. 4, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou L, Xu N, Sun Y, Liu X (2014) Targeted biopharmaceuticals for cancer treatment. Cancer Lett. 352, 145–151. [DOI] [PubMed] [Google Scholar]
- 7. Kono K (2014) Current status of cancer immunotherapy. J. Stem Cells Regen. Med. 10, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gupta SC, Sung B, Prasad S, Webb LJ, Aggarwal BB (2013) Cancer drug discovery by repurposing: teaching new tricks to old drugs. Trends Pharmacol. Sci. 34, 508–517. [DOI] [PubMed] [Google Scholar]
- 9. Cragg GM, Grothaus PG, Newman DJ (2014) New horizons for old drugs and drug leads. J. Nat. Prod. 77, 703–723. [DOI] [PubMed] [Google Scholar]
- 10. Vlahopoulos S, Critselis E, Voutsas IF, Perez SA, Moschovi M, Baxevanis CN et al (2014) New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr. Drug Targets 15, 843–851. [DOI] [PubMed] [Google Scholar]
- 11. Mitscher LA (2005) Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 105, 559–592. [DOI] [PubMed] [Google Scholar]
- 12. Paul M, Gafter‐Gvili A, Fraser A, Leibovici L (2007) The anti‐cancer effects of quinolone antibiotics? Eur. J. Clin. Microbiol. Infect. Dis. 26, 825–831. [DOI] [PubMed] [Google Scholar]
- 13. Sharma PC, Chaudhary M, Sharma A, Piplani M, Rajak H, Prakash O (2013) Insight view on possible role of fluoroquinolones in cancer therapy. Curr. Top. Med. Chem. 13, 2076–2096. [DOI] [PubMed] [Google Scholar]
- 14. Ahmed A, Daneshtalab M (2012) Nonclassical biological activities of quinolone derivatives. J. Pharm. Pharm. Sci. 15, 52–72. [DOI] [PubMed] [Google Scholar]
- 15. Mugnaini C, Pasquini S, Corelli F (2009) The 4‐quinolone‐3‐carboxylic acid motif as a multivalent scaffold in medicinal chemistry. Curr. Med. Chem. 16, 1746–1767. [DOI] [PubMed] [Google Scholar]
- 16. El‐Amm J, Tabbara IA (2014) Vosaroxin for acute myeloid leukemia. Clinc Invest 4, 147–152. [Google Scholar]
- 17. Xu J, Cole DC, Chang CPB, Ayyad R, Asselin M, Hao W et al (2008) Inhibition of the signal transducer and activator of transcription‐3 (STAT3) signalling pathway by 4‐oxo‐1‐phenyl‐1,4‐dihydroquinoline‐3‐carboxylic acid esters. J. Med. Chem. 51, 4115–4121. [DOI] [PubMed] [Google Scholar]
- 18. Golub AG, Yakovenko OY, Bdzhola VG, Sapelkin VM, Zien P, Yarmoluk SM (2006) Evaluation of 3‐carboxy‐4(1H)‐ quinolones as inhibitors of human protein kinase CK2. J. Med. Chem. 49, 6443–6450. [DOI] [PubMed] [Google Scholar]
- 19. Kan JY, Hsu YL, Chen YH, Chen TC, Wang JY, Kuo PL (2013) Gemifloxacin, a fluoroquinolone antimicrobial drug, inhibits migration and invasion of human colon cancer cells. Biomed. Res. Int. 2013, 1597868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melo S, Villanueva A, Moutinho C, Davalos V, Spizzo R, Ivan C et al (2011) Small molecule enoxacin is a cancer‐specific growth inhibitor that acts by enhancing TAR RNA‐binding protein 2‐mediated microRNA processing. Proc. Natl Acad. Sci. USA 108, 4394–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haddach M, Schwaebe MK, Michaux J, Nagasawa J, O'Brien SE, Whitten JP et al (2012) Discovery of CX‐5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Med. Chem. Letters 3, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cecchetti V, Parolin C, Moro S, Pecere T, Filipponi E, Calistri A et al (2006) 6‐Aminoquinolones as new potential anti‐HIV agents. J. Med. Chem. 43, 3799–3802. [DOI] [PubMed] [Google Scholar]
- 23. Tabarrini O, Stevens M, Cecchetti V, Sabatini S, Dell'Uomo M, Manfroni G et al (2004) Structure modifications of 6‐aminoquinolones with potent anti‐HIV activity. J. Med. Chem. 47, 5567–5578. [DOI] [PubMed] [Google Scholar]
- 24. Tabarrini O, Massari S, Daelemans D, Stevens M, Manfroni G, Sabatini S et al (2008) Structure−activity relationship study on anti‐HIV 6‐desfluoroquinolones. J. Med. Chem. 11, 5454–5458. [DOI] [PubMed] [Google Scholar]
- 25. Tabarrini O, Massari S, Cecchetti V (2010) 6‐Desfluoroquinolones as HIV‐1 tat‐mediated transcription inhibitors. Future Med. Chem. 2, 1161–1180. [DOI] [PubMed] [Google Scholar]
- 26. Massari S, Daelemans D, Manfroni G, Sabatini S, Tabarrini O, Pannecouque C et al (2009) Studies on anti‐HIV quinolones: new insights on the C‐6 position. Bioorg. Med. Chem. 17, 667–674. [DOI] [PubMed] [Google Scholar]
- 27. Loregian A, Mercorelli B, Muratore G, Sinigalia E, Pagni S, Massari S et al (2010) The 6‐aminoquinolone WC5 inhibits human cytomegalovirus replication at an early stage by interfering with the transactivating activity of viral immediate‐early 2 protein. Antimicrob. Agents Chemother. 54, 1930–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Massari S, Mercorelli B, Sancineto L, Sabatini S, Cecchetti V, Gribaudo G et al (2013) Design, synthesis, and evaluation of WC5 analogues as inhibitors of human cytomegalovirus immediate‐early 2 protein, a promising target for anti‐HCMV treatment. Chem. Med. Chem. 8, 1403–1414. [DOI] [PubMed] [Google Scholar]
- 29. Manfroni G, Cannalire R, Barreca ML, Kaushik‐Basu N, Leyssen P, Winquist J et al (2014) The versatile nature of the 6‐aminoquinolone scaffold: identification of submicromolar hepatitis C virus NS5B inhibitors. J. Med. Chem. 57, 1952–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cecchetti V, Clementi S, Cruciani G, Fravolini A, Pagella PG, Savino A et al (1995) 6‐aminoquinolones: a new class of quinolone antibacterials? J. Med. Chem. 38, 973–982. [DOI] [PubMed] [Google Scholar]
- 31. Wise R, Pagella PG, Cecchetti V, Fravolini A, Tabarrini O (1995) In vitro activity of MF 5137, a new potent 6‐aminoquinolone. Drugs 49, 272–273. [DOI] [PubMed] [Google Scholar]
- 32. Cecchetti V, Fravolini A, Lorenzini MC, Tabarrini O, Terni P, Tao X (1996) Studies on 6‐aminoquinolones: synthesis and antibacterial evaluation of 6‐amino‐8‐methylquinolones. J. Med. Chem. 39, 436–445. [DOI] [PubMed] [Google Scholar]
- 33. Denis JG, Franci G, Altucci L, Aurrecoechea JM, de Lera ÁR, Álvarez R (2015) Synthesis of 7‐alkylidene‐7,12‐dihydroindolo[3,2‐d]benzazepine‐6‐(5H)‐ones (7‐alkylidene‐paullones) by N‐cyclization‐oxidative Heck cascade and characterization as sirtuin modulators. Org. Biomol. Chem. 13, 2800–2810. [DOI] [PubMed] [Google Scholar]
- 34. Miceli M, Franci G, Dell'Aversana C, Ricciardiello F, Petraglia F, Carissimo A et al (2013) MePR: a novel human mesenchymal progenitor model with characteristics of pluripotency. Stem Cells Dev. 22, 2368–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA et al (2002) The histone deacetylase inhibitor SAHA arrests cancer cell growth, up‐regulates thioredoxin‐binding protein‐2, and down‐regulates thioredoxin. Proc. Natl Acad. Sci. USA 99, 11700–11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franci G, Miceli M, Altucci L (2010) Targeting epigenetic networks with polypharmacology: a new avenue to tackle cancer. Epigenomics 6, 731–742. [DOI] [PubMed] [Google Scholar]
- 37. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collins TJ (2007) ImageJ for microscopy. Biotechniques 43, 25–30. [DOI] [PubMed] [Google Scholar]
- 39. Reed SM, Quelle DE (2014) p53 acetylation: regulation and consequences. Cancers (Basel) 7, 30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic procedures for the preparation of intermediates 16‐25.