

Abstract
Abstract. Iron deprivation induces apoptosis in some sensitive cultured tumour cells, while other cells are resistant. In order to elucidate the mechanisms involved in apoptosis induction by iron deprivation, we studied the expression of p53 and the expression of selected p53‐regulated genes. To discriminate between changes coupled only with iron deprivation and changes involved in apoptosis induction by iron deprivation, we compared the expression of the genes in sensitive (human Raji, mouse 38C13) versus resistant (human HeLa, mouse EL4) cells under iron deprivation. Iron deprivation was achieved by incubation in a defined iron‐free medium. The level of p53 mRNA decreased significantly under iron deprivation in sensitive cells, but it did not change in resistant cells. On the contrary, the level of the p53 protein under iron deprivation was slightly increased in sensitive cells while it was not changed in resistant cells. The activity of p53 was assessed by the expression of selected p53‐regulated targets, i.e. p21WAF1/CIP1 gene, mdm2, bcl‐2 and bax. We did not detect any relevant change in mRNA levels as well as in protein levels of these genes under iron deprivation with the exception of p21WAF1/CIP1. We detected a significant increase in the level of p21 mRNA in both (sensitive and resistant) mouse cell lines tested, however, we did not find any change in both (sensitive and resistant) human cell lines. Moreover, the p21WAF1/CIP1 protein was accumulated in mouse‐sensitive 38C13 cells under iron deprivation while all other cell lines tested, including human‐sensitive cell line Raji, did not show any accumulation of p21WAF1/CIP1 protein. It seems that the p21WAF1/CIP1 mRNA, as well as protein accumulation, is not specifically coupled with apoptosis induction by iron deprivation and that it is rather cell‐line specific. Taken together, we suggest that iron deprivation induces apoptosis at least in some cell types independently of the p53 pathway.
INTRODUCTION
Apoptosis is an evolutionarily conserved programme of cell self‐destruction, which plays a significant role in many physiological processes (Jacobson et al. 1997; Raff 1998; Fiers et al. 1999; Thomas et al. 2000). Decreased sensitivity to apoptosis induction is a feature of some tumour cells and it is connected with a weak response of tumour cells to therapeutic treatment (Hoffman & Liebermann 1994; Thompson 1995).
Apoptosis can be induced by various stimuli and many of them activate the p53 pathway of apoptosis induction. Protein p53 plays an important role in the control of the genome integrity, in the control of the cell cycle and also in the control of apoptosis induction (Levine 1997; Janus et al. 1999). It has been shown that activated p53 fulfils its function via transcriptional regulation of the expression of relevant genes. The activation of p53 can turn on apoptosis by modulation of the expression of apoptotic genes, including some members of the bcl‐2 family (El‐Deiry et al. 1993; Haldar et al. 1994; Selvakumaran et al. 1994; Miyashita & Reed 1995). Proteins of the Bcl‐2 family represent key regulators of apoptosis (Antonsson et al. 1997; Jurgensmeier et al. 1998; Marzo et al. 1998). However, it has also been shown that apoptosis onset caused by p53 may not necessarily require its transcription‐modulating activity (Haupt et al. 1995; Yan et al. 1997).
There is an apparent interest in using iron deprivation as a tool for cancer therapy (Taetle et al. 1989; Donfrancesco et al. 1996; Kemp 1997; Kovar et al. 1997). Iron represents an irreplaceable enzyme co‐factor and it is required for the catalytic function of some key enzymes, including ribonucleotide reductase and proteins of the mitochondrial respiratory chain containing Fe/S clusters (Reichard 1993; Lill et al. 1999). Iron defficiency could result in DNA damage due to the decreased activity of ribonucleotide reductase or the impaired mitochondrial respiratory chain could produce ROS (reactive oxygen species). DNA damage as well as ROS are able to activate p53 (Nelson & Kastan 1994; Yin et al. 1999; Huang et al. 2000; Smith et al. 2000; Tanaka et al. 2000). Thus, iron deprivation could result in p53 activation and cause apoptosis onset by various pathways.
We have shown previously that iron deprivation can specifically induce apoptosis (Kovar et al. 1997) and that some tumour cells are sensitive to apoptosis induction by iron deprivation while other tumour cells are resistant (Kovar et al. 2001). In the present study, we tested the role of the p53 pathway, including p53 activation and subsequent regulation of the expression of relevant genes of the bcl‐2 family such as bax and bcl‐2 in apoptosis induction by iron deprivation. We employed an experimental model based on the comparison of the expression of relevant genes in sensitive (human Raji, mouse 38C13) versus resistant (human HeLa, mouse EL4) cells under iron deprivation. We demonstrate that p53 expression and activation, as well as the expression of pro‐apoptotic Bax and anti‐apoptotic Bcl‐2 do not seem to play a substantial role in apoptosis induction by iron deprivation.
MATERIALS AND METHODS
Materials
Human transferrin (apo‐transferrin) from Sigma (St Louis, MO, USA) was rendered iron‐saturated as described previously (Kovar & Franek 1989). Mouse monoclonal antibody Pab 240 against human and mouse p53 from Calbiochem (La Jolla, CA, USA), mouse monoclonal antibody Pab 122 against human and mouse p53, mouse monoclonal antibody HZ52 against human and mouse p21CIP1/WAF1 from Neo Markers (Fremont, CA, USA), mouse monoclonal antibody F‐5 against human and mouse p21CIP1/WAF1 from Santa Cruz Biotechnology (Santa Cruz, CA, USA), mouse monoclonal antibody Bcl‐2–100 against human Bcl‐2 from Sigma, Syrian hamster monoclonal antibody 3F11 against mouse Bcl‐2 from Pharmingen (San Diego, CA, USA), rabbit polyclonal antibody P‐19 against human and mouse Bax from Santa Cruz Biotechnology and mouse monoclonal antibody Ab‐4 against human and mouse α,α‐tubulin (Neomarkers, CA, USA) were used.
Cell lines
The mouse B‐cell lymphoma 38C13 and the mouse T‐cell lymphoma EL4 were obtained from Professor J. Kemp (University of Iowa, Iowa City, IA, USA). The human Burkitt lymphoma Raji was provided by Professor G. Klein (Karolinska Institute, Stockholm, Sweden) and the human cervix carcinoma HeLa, growing as a suspension culture, by Dr A. Cvekl (Institute of Organic Chemistry and Biochemistry, Prague, Czech Republic). We have found previously that human Raji cells and mouse 38C13 cells are sensitive and human HeLa cells and mouse EL4 cells are resistant to apoptosis induction by iron deprivation (Kovar et al. 2001). Cell lines were routinely tested for mycoplasma contamination using the fluorescent Hoechst 33258 staining method (Chen 1977).
Culture media and culture conditions
Defined serum‐free media were used. Transferrin medium was a basic medium supplemented with 5 µg/ml of iron‐saturated human transferrin and iron‐free medium was a basic medium without any iron compound added. The basic medium was RPMI 1640 containing extra l‐glutamine (300 µg/ml), sodium pyruvate (110 µg/ml), penicillin (100 U/ml), streptomycin (100 µg/ml), Hepes (15 mm), ethanolamine (20 µm), ascorbic acid (20 µm), hydrocortisone (5 nm), 11 trace elements, as described previously (Kovar 1988; Kovar & Franek 1989), and 2‐mercaptoethanol (50 µm). Cells were routinely maintained in transferrin medium. In experiments, transferrin medium represented control culture conditions and iron‐free medium represented iron‐depriving conditions. Cells were incubated at 37 °C in a humidified atmosphere of 5% CO2.
Iron deprivation experiments
Cells previously grown in transferrin medium were harvested by low‐speed centrifugation, washed with iron‐free medium and seeded at 2 × 105 cells/ml (human cells) or 4 × 105 cells/ml (mouse cells) of iron‐free medium (iron deprivation) or transferrin medium (control) into plastic culture flasks. The effect of iron deprivation on the expression of the studied genes was determined after 12, 24 and 48 h (human cells) or 6, 12 and 24 h (mouse cells). It was shown previously that a significant fraction (up to 50%) of human‐sensitive Raji cells and mouse‐sensitive 38C13 cells were dead due to apoptosis after 48 and 24 h of incubation in iron‐free medium, respectively (Kovar et al. 2001).
Flow cytometric analysis
Indirect immunofluorescence according to the modified method of Pollice et al. (1992) was employed. The cells harvested by low‐speed centrifugation (approximately 4 × 106 cells per sample) were washed with phospate‐buffered saline (PBS) and then fixed in 2 ml of 0.25% paraformaldehyde in the dark for 15 min at room temperature. The cells were spun, washed with PBS and then fixed in 2 ml of 70% methanol for 1 h at 4 °C. Fixed cells were centrifuged and washed with PBS. The cell pellet (approximately 1 × 106 cells per parallel) was resuspended and incubated in 50 µl of primary antibody (5 µg/ml of PBS) or in 50 µl of relevant non‐specific IgG (5 µg/ml of PBS) as a negative control. Mouse monoclonal antibody (IgG) Pab 240 against human and mouse p53, mouse monoclonal antibody (IgG) HZ52 against human and mouse p21WAF1/CIP1, mouse monoclonal antibody (IgG) Bcl‐2–100 against human Bcl‐2, hamster monoclonal antibody (IgG) 3F11 against mouse Bcl‐2, and rabbit polyclonal antibody (IgG) P‐19 against human and mouse Bax were used as primary antibodies. After 30 min of incubation on ice, 400 µl of PBS were added and cells were resuspended. The sample was underlain with 100 µl of fetal bovine serum and spun. The cell pellet was resuspended and incubated in 50 µl of secondary staining reagent (10 µg/ml of PBS). Corresponding (anti‐mouse, anti‐hamster and anti‐rabbit) FITC‐conjugated goat antibodies (Sigma, Pharmingen) were used as the secondary staining reagents. After 30 min of incubation on ice, 400 µl of PBS were added and cells were resuspended. The sample was again underlain with 100 µl of fetal bovine serum and spun. The stained cells were resuspended in 300 µl of PBS and analysed on a FACScan flow cytometer (Becton Dickinson).
Western blot analysis
The cells (approximately 107 cells per sample) were harvested by low‐speed centrifugation and washed twice with PBS. The cell pellet was lysed in 100 µl of lysis buffer (62.5 mm Tris, 10% glycerol, 2% SDS, 20 µm leupeptin, 1 µm pepstatin A, 1 mm PMSF, pH 6.8) and crude cell lysate was sonicated discontinuously (3 s on and 1 s off, 3 × 30 s) by a ultrasonic homogenizer (model CP501, Cole Parmer). Sonicated lysate was centrifuged (20 000 g, 15 min, 4 °C). The supernatant was collected into a new Eppendorf tube and frozen at −70 °C until further analysis. Total protein content was determined by the bicinchoninic acid assay (Smith et al. 1985). Protein samples (200 µg) were heated for 5 min at 100 °C in the sample loading buffer (lysis buffer containing 5% 2‐mercaptoethanol and 0.05% bromphenol blue) and then quickly cooled on ice. Sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS–PAGE) was performed according to Laemmli (1970). Samples were run at a constant voltage of 150 V for 90 min, employing mini‐Protean II (Bio‐Rad), on 12% polyacrylamide gel (4% polyacrylamide stacking gel). Proteins separated by SDS–PAGE were blotted, according to the modified method of Kyhse‐Andersen (Kyhse‐Andersen 1984) onto 0.2 µm nitrocellulose membrane (Protran, Schleicher‐Schuell) for 1 h at a constant current of 0.2 A using a semidry blotting apparatus (Bio‐Rad). The membrane was blocked with 5% non‐fat milk in PBS for 2 h and then washed with 0.1% Tween/PBS three times. The washed membrane was incubated overnight at 4 °C with the primary antibody (0.5 µg/ml of PBS containing 1% non‐fat milk and 0.3% Tween). Mouse monoclonal antibody Pab 122 against human and mouse p53, mouse monoclonal antibody F‐5 against human and mouse p21WAF1/CIP1, mouse monoclonal antibody Bcl‐2–100 against human Bcl‐2, hamster monoclonal antibody 3F11 against mouse Bcl‐2, rabbit polyclonal antibody P‐19 against human and mouse Bax and mouse monoclonal antibody Ab‐4 against human and mouse α,α‐tubulin (loading control) were used as primary antibodies. After the incubation the membrane was washed with 0.1% Tween/PBS six times. The washed membrane was incubated for 1 hour with corresponding horseradish peroxidase‐conjugated secondary antibody (Bio‐Rad, 1 : 10 000 in PBS containing 1% non‐fat milk and 0.3% Tween). Then, the membrane was washed with 0.1% Tween/PBS six times. The horseradish peroxidase‐conjugated antibody was detected by enhanced chemiluminescence using the ECL chemiluminescence reagent (Pierce, Rockford, IL, USA) and XAR X‐ray film (Kodak).
RT‐PCR analysis
The cells were harvested by low‐speed centrifugation (approximately 106 cells per sample) and washed twice with PBS. The cell pellet was lysed in 500 µl of lysis buffer (4 m guadinium isothiocyanate, 0.5% sarcosyl, 25 mm sodium citrate, pH 7.0) on ice. Lysate was frozen at −70 °C for further analysis. Total RNA was isolated by the modified acid phenol method (Chomczynski & Sacchi 1987). The lysate (500 µl) was mixed with 50 µl of 2 m sodium acetate (pH 4.0) and shortly spun. Then, the sample was mixed with 500 µl of phenol (MERCK) and 125 µl of chloroform/isoamylalcohol (49 : 1), vortexed for 1 min and incubated for 10 min on ice. The sample was centrifuged (10 000 g, 15 min, 4 °C) and the aqueous phase was mixed with pure isopropanol (1 : 1) and incubated for 40 min/at20 °C. Precipitated RNA was centrifuged (10 000 g, 15 min), washed twice with 75% ethanol, dried and solubilized in 20 µl of RNase‐free water. Isolated RNA was stored at −70 °C. RNA integrity was checked by agarose gel electrophoresis. RNA (2 µl) was reversely transcribed by 120 U of M‐MLV (Moloney murine leukaemia virus) reverse transcriptase (Perkin Elmer) per sample according to manufacturer's instructions using random hexamer priming. Prepared cDNA (1/10 of prepared amount, 1 µl) was used for further PCR amplification.
For the PCR amplification of the human p53, p21WAF1/CIP1, mdm2 and housekeeping (β2‐microglobulin) genes, primers provided by Life Technologies (Rockville, MD, USA) were used. PCR was carried out in a 25‐µl volume using 0.625 U of Ampli‐Taq Gold polymerase according to manufacturer's instructions (Perkin Elmer). The sequences of the primers used were published previously (Seliger et al. 1996; Pallisgaard et al. 1999; Taniguchi et al. 1999). PCR reaction was run in a Biometra thermal cycler (Biometra) using a 10‐min initial denaturation at 95 °C followed by a 1‐min denaturation at 94 °C, 1‐min annealing at 50 °C (p53), 62 °C (p21WAF1/CIP1), 58 °C (mdm2), 60 °C (β2‐microglubulin), 1‐min extension at 72 °C for 39 (p53), 27 (p21WAF1/CIP1), 26 (mdm2) or 34 (β2‐microglubulin) cycles and accompanied by a 10‐min final extension at 72 °C. For the PCR amplification of the mouse p53, p21WAF1/CIP1, mdm2 and housekeeping (18S rRNA) genes, primers provided by Life Technologies were also used. PCR was carried out in a 25‐µl volume using 0.625 U of Taq polymerase according to manufacturer's instructions (Promega, Madison, WI, USA). The sequence of the primers used were: mouse 18S rRNA (5′‐CGCGGTTCTATTTTGTTGGT‐3′ and 5′‐AGTCGGCATCGTTTATGGTC‐3′), mouse p53 (5′‐GATGACTGCCATGGAGGAGT‐3′ and 5′‐CTCGGGTGGCTCATAAGGTA‐3′), mouse p21 (5′‐CGGTGGAACTTTGACTTCGT‐3′ and 5′‐CACAGAGTGAGGGCTAAG‐GC‐3′), mouse mdm2 (5′‐AAGGGCACGAGCTCTCAGAT‐3′ and 5′‐CCTCCTCAGCACA‐TGGCTC‐3′). PCR reaction was run in a Techne thermal cycler (Techne) using a 2‐min initial denaturation at 95 °C followed by a 1‐min denaturation at 94 °C, 1‐min annealing at 62 °C (p53, mdm2), 61 °C (p21WAF1/CIP1), 58 °C (18S rRNA), 1‐min extension at 72 °C for 21 (p53), 25 (p21WAF1/CIP1), 26 (mdm2) or 17 (18S rRNA) cycles and accompanied by a 10‐min final extension at 72 °C.
PCR products were analysed by electrophoresis in 1.8% agarose (Amersham Pharmacia Biotech, Piscataway, NJ. USA) gel at a constant voltage setting 8 V/cm using TAE buffer (40 mm Tris, 20 mm acetic acid, 1 mm EDTA). DNA was visualized by ethidium bromide fluorescence and photographed using the Gel 2000 Doc documentation system (Bio‐Rad) or LAS 1000 (Fuji).
RNase protection assay
The cells were harvested by low‐speed centrifugation (approximately 107 cells per sample) and lysed in 1 ml of TRIzol reagent (Life Technologies) for 5 min. Chloroform (0.2 ml) was added, the sample was vigorously mixed for 15 s and incubated for 3 min at room temperature. The mixture was centrifuged (10 000 g, 15 min, 4 °C). The aqueous phase was transferred into a new Eppendorf tube, mixed with 0.5 ml of isopropanol and incubated for 10 min at room temperature. Precipitated RNA was centrifuged (10 000 g, 10 min, 4 °C). The pellet was washed with 1 ml of 75% ethanol, spun, briefly dried, dissolved in 20 µl of RNase‐free water and stored at −70 °C. RNase Protection Assay was carried out according to manufacturer's instructions using RiboQuant‘. Starter Kit (#45024K) involving the mAPO‐2 template set (#45354P), both provided by Pharmingen (San Diego, CA, USA).
RESULTS
Expression of p53
As the first step, we assessed the effect of iron deprivation on p53 protein and mRNA levels. Sensitive human Raji cells under control conditions (transferrin medium) displayed a significantly higher level of p53 than resistant human HeLa cells. Such a difference under control conditions was not seen for tested mouse cell lines, i.e. sensitive 38C13 and resistant EL4 cells (Table 1). Flow cytometric analysis (Table 1) and western blot analysis (data not shown) showed that the p53 level was not changed by iron deprivation in both lines of resistant cells. Employing flow cytometry, a slight but reproducible increase in the p53 level could be seen in both sensitive cell lines after 48 h (Raji) or 24 h (38C13) of incubation in iron‐free medium (Table 1). However, such increase was not seen when western blot was used (data not shown). Employing RT‐PCR analysis, we surprisingly detected a reproducible decrease in the level of p53 mRNA due to iron deprivation in both sensitive cell lines while the resistant cells did not display any reproducible change (Fig. 1).
Table 1.
Effect of iron deprivation on the level of p53 in sensitive cells (human Raji, mouse 38C13) and in resistant cells (human HeLa, mouse EL4)
| Cells | Species | p53 level | |
|---|---|---|---|
| Control | –Fe | ||
| Raji | Human | 67.5 ± 4.2 (22.4 ± 3.2) | 85.0 ± 5.9 (18.6 ± 0.7) |
| HeLa | Human | 48.0 ± 1.4 (18.0 ± 0.3) | 45.3 ± 0.4 (18.0 ± 0.6) |
| 38C13 | Mouse | 32.3 ± 2.3 (16.7 ± 3.6) | 46.0 ± 5.8 (19.1 ± 3.5) |
| EL4 | Mouse | 28.7 ± 5.3 (19.2 ± 2.8) | 29.3 ± 4.8 (18.6 ± 2.8) |
Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The protein level was determined by indirect immunofluorescence analysis, employing specific monoclonal anti‐p53 antibody Pab 240, after 48 h (Raji, HeLa) or 24 h (38C13, EL4) of incubation. Stained cells were analysed by flow cytometry. Fluorescence intensities shown represent the mean ± SD of two independent experiments. Values in brackets represent control staining with relevant control non‐specific immunoglobulin.
Figure 1.
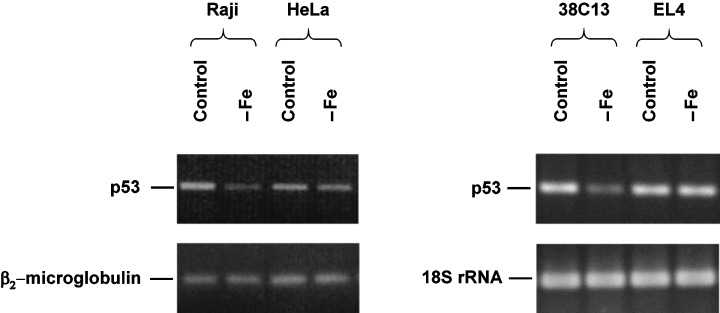
Effect of iron deprivation on the level of p53 mRNA in sensitive cells (human Raji, mouse 38C13) and in resistant cells (human HeLa, mouse EL4). Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The mRNA level was determined by RT‐PCR analysis (see Materials and methods) after 48 h (human cells) or 24 h (mouse cells) of incubation. The mRNA level of a housekeeping gene (β2‐microglobulin for human cells, 18S rRNA for mouse cells) is also shown. The data shown were obtained in one representative experiment of three independent experiments.
Expression of p21WAF1/CIP1 and Mdm2
In the next step, we assessed the effect of iron deprivation on p53 activity measured by the induction of p21WAF1/CIP1 and mdm2 expression. We did not find any change in the p21WAF1/CIP1 protein level in both resistant cell lines (human HeLa, mouse EL4) due to iron deprivation (data not shown). The level of p21WAF1/CIP1 detected by flow cytometry in sensitive mouse 38C13 cells was found to be slightly increased (20–30%) after 24‐h incubation in iron‐free medium. However, this slight but reproducible increase of the p21WAF1/CIP1 level was not detected in sensitive human Raji cells for any incubation time (Fig. 2). A confirmatory western blot showed a significant increase of the p21WAF1/CIP1 level in 38C13 cells but not in Raji cells (Fig. 3). The level of p21WAF1/CIP1 mRNA, assessed by RT‐PCR, increased significantly in both mouse cell lines (sensitive 38C13 and resistant EL4) due to iron deprivation. However, we did not detect similar changes in human tested cells, i.e. sensitive Raji and resistant HeLa (Fig. 4).
Figure 2.
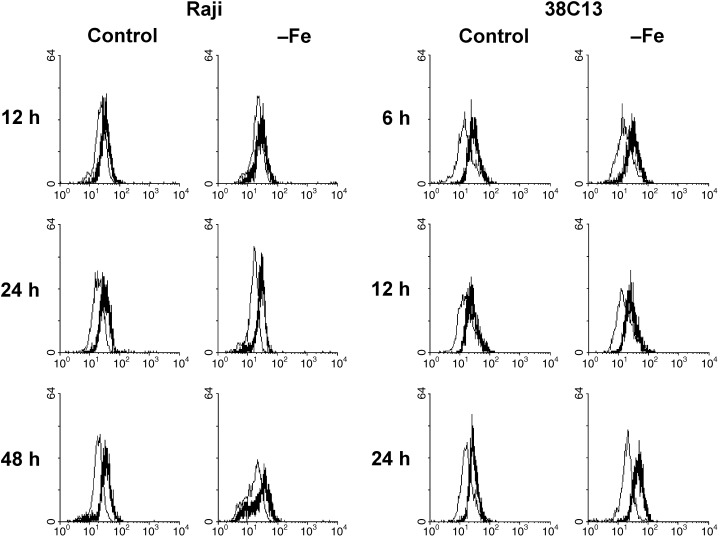
Effect of iron deprivation on the level of p21WAF1/CIP1 in sensitive cells (human Raji, mouse 38C13). Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The protein level was determined by indirect immunofluorescence analysis, employing specific monoclonal anti‐p21 antibody HZ52, after 12, 24 and 48 h (Raji) or 6, 12 and 24 h (38C13) of incubation. Stained cells were analysed by flow cytometry. The bold line represents staining with anti‐p21 antibody and the fine line represents staining with relevant control non‐specific immunoglobulin. The data shown were obtained in one representative experiment of three independent experiments.
Figure 3.
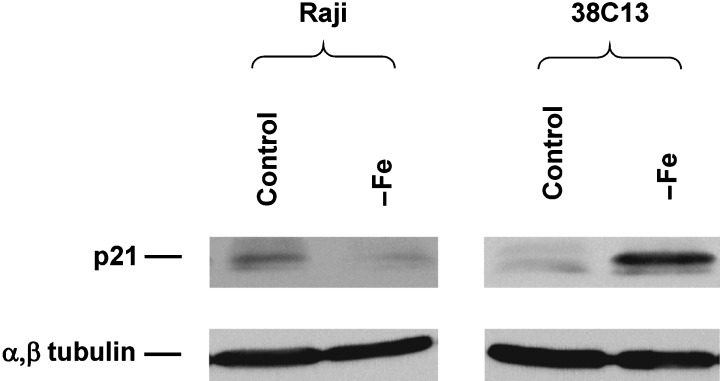
Effect of iron deprivation on the protein level of p21WAF1/CIP1 in sensitive human Raji and sensitive mouse 38C13 cells. Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The protein level was determined by western blot analysis, employing specific monoclonal anti‐p21 antibody F‐5, after 48 h (Raji) or 24 h (38C13) of incubation. Specific mouse monoclonal antibody Ab‐4 against α,α‐tubulin was used to confirm equal protein loading. The data shown were obtained in one representative experiment of five independent experiments.
Figure 4.
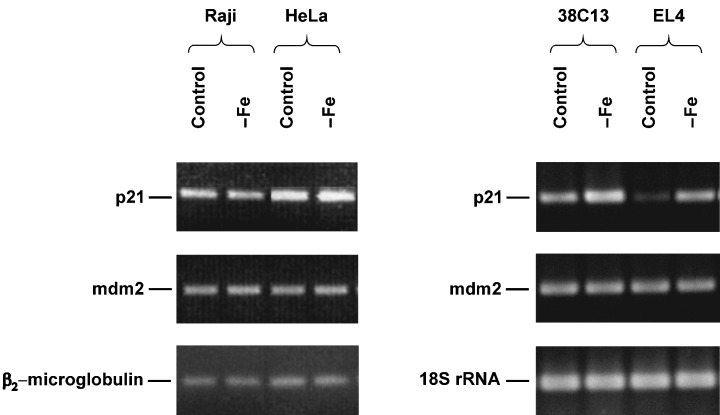
Effect of iron deprivation on the level of p21 mRNA and mdm2 mRNA in sensitive cells (human Raji, mouse 38C13) and in resistant cells (human HeLa, mouse EL4). Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The mRNA level was determined by RT‐PCR analysis (see MATERIALS AND METHODS) after 48 h (human cells) or 24 h (mouse cells) of incubation. The mRNA level of a housekeeping gene (β2‐microglobulin for human cells, 18S rRNA for mouse cells) is also shown. The data shown were obtained in one representative experiment of three independent experiments.
The expression of another p53‐controlled gene mdm2 was also assessed by RT‐PCR. The level of mdm2 mRNA was not significantly affected by iron deprivation in sensitive as well as in resistant cells independently of their species origin (Fig. 4).
Expression of Bcl‐2, Bax and other members of the Bcl‐2 family
Finally, we assessed the expression of members of the bcl‐2 family representing p53‐regulated downstream targets and apoptosis regulators. Flow cytometry and western blot revealed that sensitive cells (human Raji, mouse 38C13), incubated in control transferrin medium, had the cellular level of anti‐apoptotic Bcl‐2 protein comparable with the level of Bcl‐2 in their resistant counterparts (human HeLa, mouse EL4). However, it seems that both mouse cell lines express a higher level of the Bcl‐2 than the human cell lines. Flow cytometric data are shown in Fig. 5. Iron deprivation did not lead to any change in the Bcl‐2 level in all cells lines tested.
Figure 5.
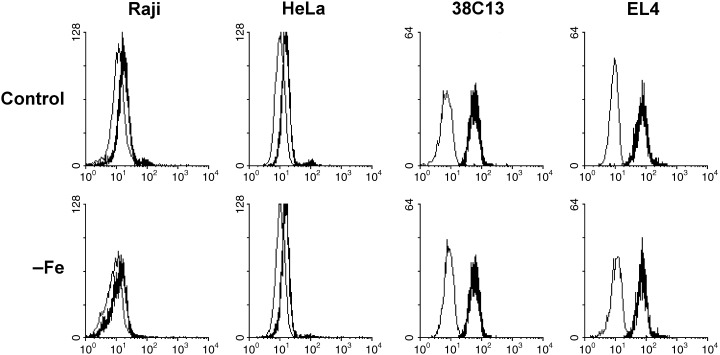
Effect of iron deprivation on the level of Bcl‐2 protein in sensitive cells (human Raji, mouse 38C13) and in resistant cells (human HeLa, mouse EL4). Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The protein level was determined by indirect immunofluorescence analysis, employing specific monoclonal anti Bcl‐2 antibody Bcl‐2–100 (human cells) and specific monoclonal anti‐Bcl‐2 antibody 3F11 (mouse cells), after 48 h (human cells) or 24 h (mouse cells) of incubation. Stained cells were analysed by flow cytometry. The bold line represents staining with anti‐Bcl‐2 antibody and the fine line represents staining with relevant control non‐specific immunoglobulin. The data shown were obtained in one representative experiment of three independent experiments.
Concerning the pro‐apoptotic Bax protein, sensitive cells under control conditions again displayed the cellular level comparable with the level in resistant cells. Principally, no significant change in the level of Bax was found in sensitive as well as in resistant cells during the incubation in iron‐free medium. Flow‐cytometric data are shown in Fig. 6. However, sometimes a slight non‐reproducible increase of the Bax level due to iron deprivation could be seen in both sensitive and resistant cells.
Figure 6.
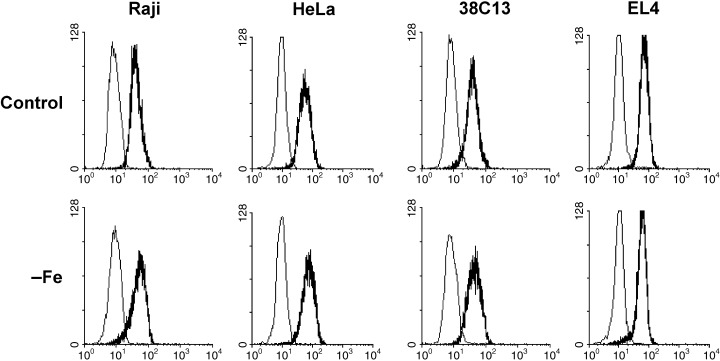
Effect of iron deprivation on the level of Bax protein in sensitive cells (human Raji, mouse 38C13) and in resistant cells (human HeLa, mouse EL4). Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The protein level was determined by indirect immunofluorescence analysis, employing specific polyclonal anti‐Bax P‐19 antibody, after 48 h (human cells) or 24 h (mouse cells) of incubation. Stained cells were analysed by flow cytometry. The bold line represents staining with anti‐Bax antibody and the fine line represents staining with relevant control non‐specific immunoglobulin. The data shown were obtained in one representative experiment of three independent experiments.
The expression of several genes (anti‐apoptotic bcl‐2, bcl‐xL, bcl‐w, bfl‐1 and pro‐apoptotic bax, bak, bad) of the bcl‐2 family was assessed at the level of mRNA by a RNase protection assay. We tested sensitive 38C13 and resistant EL4 cells. The level of bcl‐2 mRNA and the level of Bax mRNA was similar in both cell types. Sensitive 38C13 cells expressed anti‐apoptotic bfl‐1, but they did not express anti‐apoptotic bcl‐w. On the contrary, resistant EL4 cells expressed bcl‐w and they did not express bfl‐1. Furthemore, sensitive 38C13 cells displayed a significantly higher level of mRNA for anti‐apoptotic bcl‐xL. However, we did not find any change in the RNA levels coupled specifically with iron deprivation in sensitive 38C13 as well as in resistant EL4 cells (Fig. 7).
Figure 7.
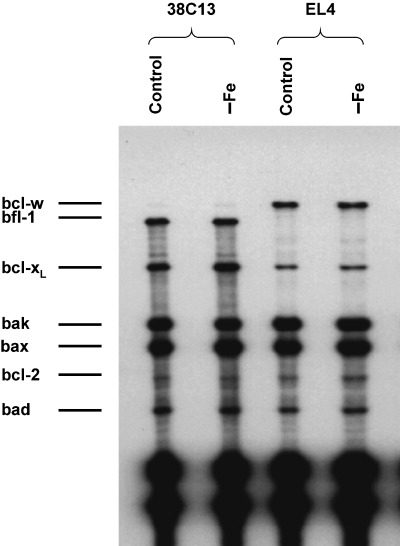
Effect of iron deprivation on the mRNA level of genes of the bcl‐2 family in sensitive mouse 38C13 and resistant mouse EL4 cells. Cells were incubated in iron‐free (–Fe) medium and cells of the control were incubated in transferrin (5 µg/ml) medium. The level of mRNAs was determined by the RNase protection assay (see MATERIALS AND METHODS) after 24 h of incubation. The data shown were obtained in one representative experiment of two independent experiments.
DISCUSSION
We have shown previously that some tumour cells are sensitive to apoptosis induction by iron deprivation while other tumour cells are resistant (Kovar et al. 1997; Kovar et al. 2001). Cells resistant to iron deprivation (human Hela, mouse EL4) do not show any sign of apoptosis during 96 h (human cells) or 48 h (mouse cells) of cultivation under iron‐depriving conditions while 30–50% of sensitive cells (human Raji, mouse 38C13) display apoptotic features after 48 h (human cells) or 24 h (mouse cells) under iron deprivation and they die completely during 96 or 48 h of cultivation, respectively. The comparison of the sensitive versus resistant cells under iron deprivation provides a tool for elucidating the unknown mechanism of apoptosis induction by iron deprivation because we can distinguish between changes accompanying just iron deprivation and changes coupled with apoptosis induction due to iron deprivation. We have established an easily controlled experimental arrangement for iron deprivation employing a defined serum‐free culture medium. The only difference between control and iron‐depriving conditions in this arrangement is the presence of an iron source (iron‐saturated transferrin). Thus, we eliminated potential non‐specific effects of compounds employed for producing iron deprivation, such as widely used iron chelators (Kovar et al. 1997; Darnell & Richardson 1999; Ido et al. 1999; Krude 1999).
Resistant human HeLa cells possess a wild‐type p53. The cells are supposed to be HPV E6 positive and thus with efficacious p53 degradation and dysfunctional p53 (Scheffner et al. 1990). Sensitive human Raji cells are supposed to be EBNA positive and carry one mutated p53 allele (Arg213?Gln) (Farrell et al. 1991; Frade et al. 1992). However, neither formation of p53‐viral protein complexes nor p53 degradation seems to be coupled with EBV infection (Farrell et al. 1991). Moreover, not all p53 mutations necessarily bring about the loss of function and it has even been shown that a particular type of mutant p53 is able to retain its pro‐apoptotic function (Bhatia et al. 1993; He et al. 2002; Shi et al. 2002). Therefore, it was not clear whether p53, even if mutated, may not play a role in apoptosis induction by iron deprivation. To our knowledge, no data concerning the p53 status of mouse‐resistant EL4 and ‐sensitive 38C13 cells have been published. Our data support the idea that Raji cells carry a mutated allele (the highest detected level of p53) and the other employed cell lines carry the wild‐type p53. Thus, it seems that the p53 status does not correlate with sensitivity or resistance to iron deprivation.
Iron plays an important role in cellular metabolism, including the function of ribonucleotide reductase and the function of the mitochondrial respiratory chain. Insufficient function in both cases may lead to p53 activation (Nelson & Kastan 1994; Yin et al. 1999; Huang et al. 2000; Smith et al. 2000; Tanaka et al. 2000), and thus p53 activation as a result of iron deprivation seemed to be a relevant hypothesis concerning the mechanism of apoptosis induction by iron deprivation.
Under our experimental arrangement, we found a significant decrease of the p53 mRNA level due to iron deprivation in both sensitive cell lines. In contrast, we detected a slight increase of the p53 protein level in both sensitive cell lines due to iron deprivation. Thus, it seems that the slight elevation of the p53 protein level might be caused by a post‐transcriptional mechanism. However, the observed changes in the p53 level were only detected by flow cytometric analysis while western blot analysis did not confirm this finding. Concerning p53 activation assessed by the induction of p21WAF1/CIP1 expression, we found that iron deprivation resulted in a significant increase of p21WAF1/CIP1 mRNA in tested mouse cells (both sensitive and resistant), but the tested human sensitive as well as resistant cells did not show such an increase. We detected a slight increase of the p21WAF1/CIP1 protein level in mouse‐sensitive 38C13 cells under iron deprivation by flow cytometry and a significant increase by western blotting. However, the human‐sensitive cell line Raji did not show such behaviour. The expression of mdm2, which represents another gene transcriptionally regulated by p53, was not affected by iron deprivation. This confirms that p53 is not regularly activated under iron deprivation. The possible slight elevation of the p53 level in both sensitive cell lines after iron deprivation seems to be likely a ‘side‐effect’ unrelated to apoptosis onset. Similarly, the change in the p21WAF1/CIP1 level found in 38C13 cells seems to be cell‐line specific and unrelated to apoptosis induction.
Other potential targets of activated p53, i.e. proteins of the Bcl‐2 family, were also studied because they play a key role in the control of apoptosis induction and, thus, they could be responsible for p53‐mediated apoptosis. We tested particularly the role of pro‐apoptotic Bax and anti‐apoptotic Bcl‐2 because both of them are p53‐regulated (Haldar et al. 1994; Selvakumaran et al. 1994). There were no relevant changes in the Bcl‐2 as well as Bax level as assesed by western blot and flow cytometric analysis due to iron deprivation. We also tested the expression of other members of the Bcl‐2 family (see Fig. 7), however, we did not find any change coupled specifically with iron deprivation.
There are some contradictory data in the literature concerning the role of p53 in apoptosis induced by iron deprivation. Fukuchi et al. (1995) have shown, employing iron chelator desferoxamine, that iron deprivation results in an increase of the p53 protein level caused by a post‐transcriptional mechanism. A cell‐line specific increase of the p21WAF1/CIP1 mRNA level was also detected. In contrast, Darnell & Richardson (1999) have not shown any involvement of p53 in the response to iron deprivation employing new iron chelator 3F11. The cell lines used in their study had mutated p53 and, despite this fact, the cells underwent apoptosis when iron deprivation was applied. However, iron deprivation in this system resulted in an increase of GADD45 and p21WAF1/CIP1 mRNA levels. The study of Abeysinghe et al. (2001) using iron chelator tachpyridine and the study of MacLean & Porter (1996) with p53 knockout mice using iron chelator deferoxamine support the idea of a p53‐independent mechanism of apoptosis induction by iron deprivation. However, all these studies employed iron chelators to produce iron deprivation.
Taken together, we can conclude that apoptosis induction by iron deprivation in the studied cells does not involve the activation of p53, including subsequent changes in the expression of Bax and Bcl‐2. Thus, the mechanism responsible for apoptosis induction by iron deprivation is independent of the classical p53 pathway. Further studies concerning mitochondrial events and the activation of caspases could provide an insight into the mechanisms responsible for apoptosis induction in tumour cells under iron deprivation. These studies are under way.
ACKNOWLEDGEMENTS
This work was supported by grant 301/01/0041 from the Grant Agency of the Czech Republic. We thank Dr C. Haskovec and Professor J. Kemp for their assistance.
REFERENCES
- Abeysinghe RD, Greene BT, Haynes R, Willingham MC, Turner J, Planalp RP, Brechbiel MW, Torti FM, Torti SV (2001) p53‐independent apoptosis mediated by tachpyridine, an anti‐cancer iron chelator. Carcinogenesis 22, 1607. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC (1997) Inhibition of Bax channel‐forming activity by Bcl‐2. Science 277, 370. [DOI] [PubMed] [Google Scholar]
- Bhatia K, Goldschmidts W, Gutierrez. M, Gaidano G, Dalla Favera R, Magrath I (1993) Hemi‐ or homozygosity: a requirement for some but not other p53 mutant proteins to accumulate and exert a pathogenic effect. FASEB J. 7, 951. [DOI] [PubMed] [Google Scholar]
- Chen TR (1977) In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 104, 255. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal. Biochem. 162, 156. [DOI] [PubMed] [Google Scholar]
- Darnell G, Richardson DR (1999) The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents III: the effect of the ligands on molecular targets involved in proliferation. Blood 94, 781. [PubMed] [Google Scholar]
- Donfrancesco A, Deb G, De Sio L, Cozza R, Castellano A (1996) Role of deferoxamine in tumor therapy. Acta Haematol. 95, 66. [DOI] [PubMed] [Google Scholar]
- El‐Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell. 75, 817. [DOI] [PubMed] [Google Scholar]
- Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T (1991) p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 10, 2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Declerq W, Vandenabeele P (1999) More than one way to die: apoptosis, necrosis, and reactive oxygen damage. Oncogene 18, 7719. [DOI] [PubMed] [Google Scholar]
- Frade R, Gauffre A, Hermann J, Barel M (1992) EBV/C3d receptor (CR2) interacts by its intracytoplasmic carboxy‐terminal domain and two distinct binding sites with the p53 anti‐oncoprotein and the p68 calcium‐binding protein. J. Immunol. 149, 3232. [PubMed] [Google Scholar]
- Fukuchi K, Tomoyasu S, Watanabe H, Kaetsu S, Tsuruoka N, Gomi K (1995) Iron deprivation results in an increase in p53 expression. Biol. Chem. Hoppe Seyler 376, 627. [DOI] [PubMed] [Google Scholar]
- Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM (1994) Down‐regulation of bcl‐2 by p53 in breast cancer cells. Cancer Res. 54, 2095. [PubMed] [Google Scholar]
- Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M (1995) Induction of apoptosis in HeLa cells by trans‐activation‐deficient p53. Genes Dev. 9, 2170. [DOI] [PubMed] [Google Scholar]
- He M, Rennie PS, Dragowska V, Nelson CC, Jia W (2002) A mutant P53 can activate apoptosis through a mechanism distinct from those induced by wild type P53. FEBS Lett. 517, 151. [DOI] [PubMed] [Google Scholar]
- Hoffman B, Liebermann DA (1994) Molecular controls of apoptosis: differentiation/growth arrest primary gene response genes, proto‐oncogenes, and tumor supressor genes as positive and negative modulators. Oncogene 9, 1807. [PubMed] [Google Scholar]
- Huang C, Zhang Z, Ding M, Li J, Ye J, Leonard SS, Shen HM, Butterworth L, Lu Y, Costa M, Rojanasakul Y, Castranova V, Vallyathan V, Shi X (2000) Vanadate induces p53 transactivation through hydrogen peroxide and causes apoptosis. J. Biol. Chem. 275, 32516. [DOI] [PubMed] [Google Scholar]
- Ido Y, Muto N, Inada A, Kohroki J, Mano M, Odani T, Itoh N, Yamamoto K, Tanaka K (1999) Induction of apoptosis by hinokitiol, a potent iron chelator, in teratocarcinoma F9 cells is mediated through the activation of caspase‐3. Cell Prolif. 32, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 88, 347. [DOI] [PubMed] [Google Scholar]
- Janus F, Albrechtsen N, Dornreiter I, Wiesmuller L, Grosse F, Deppert W (1999) The dual role model for p53 in maintaining genomic integrity. Cell. Mol. Life Sci. 55, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA 95, 4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JD (1997) Iron deprivation and cancer: a view beginning with studies of monoclonal antibodies against the transferrin receptor. Histol. Histopathol. 12, 291. [PubMed] [Google Scholar]
- Kovar J (1988) Growth‐stimulating effect of ferric citrate on hybridoma cells: characterization and relation to transferrin function. Hybridoma 7, 255. [DOI] [PubMed] [Google Scholar]
- Kovar J, Franek F (1989) Growth‐stimulating effect of transferrin on a hybridoma cell line: relation to transferrin iron‐transporting function. Exp. Cell Res. 182, 358. [DOI] [PubMed] [Google Scholar]
- Kovar J, Stunz. LL, Stewart BC, Kriegerbeckova K, Ashman RF, Kemp JD (1997) Direct evidence that iron deprivation induces apoptosis in murine lymphoma 38C13. Pathobiology 65, 61. [DOI] [PubMed] [Google Scholar]
- Kovar J, Valenta T, Stybrova H (2001) Differing sensitivity of tumor cells to apoptosis induced by iron deprivation in vitro . In Vitro Cell Dev. Biol. Anim. 37, 450. [DOI] [PubMed] [Google Scholar]
- Krude T (1999) Mimosine arrests proliferating human cells before onset of DNA replication in a dose‐dependent manner. Exp. Cell Res. 247, 148. [DOI] [PubMed] [Google Scholar]
- Kyhse‐Andersen J (1984) Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J. Biochem. Biophys. Meth 10, 203. [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680. [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323. [DOI] [PubMed] [Google Scholar]
- Lill R, Diekert K, Kaut A, Lange H, Pelzer W, Prohl C, Kispal G (1999) The essential role of mitochondria in the biogenesis of cellular iron‐sulfur proteins. Biol. Chem. 380, 1157. [DOI] [PubMed] [Google Scholar]
- Maclean K, Porter JB (1996) Iron chelators induce apoptosis in thymocytes by mechanisms independent of p53 and DNA synthesis inhibition. European Iron Club Meeting, Jaca; proffered paper.
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293. [DOI] [PubMed] [Google Scholar]
- Nelson WG, Kastan MB (1994) DNA strand breaks: the DNA template alterations that trigger p53‐dependent DNA damage response pathways. Mol. Cell Biol. 14, 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallisgaard N, Clausen N, Schroder H, Hokland P (1999) Rapid and sensitive minimal residual disease detection in acute leukemia by quantitative real‐time RT‐PCR exemplified by t (12; 21) TEL‐AML1 fusion transcript. Genes Chromosomes Cancer 26, 355. [DOI] [PubMed] [Google Scholar]
- Pollice AA, McCoy JP Jr, Shackney SE, Smith CA, Agarwal J, Burholt DR, Janocko LE, Hornicek FJ, Singh SG, Hartsock RJ (1992) Sequential paraformaldehyde and methanol fixation for simultaneous flow cytometric analysis of DNA, cell surface proteins, and intracellular proteins. Cytometry 13, 432. [DOI] [PubMed] [Google Scholar]
- Raff M (1998) Cell suicide for beginners. Nature 396, 119. [DOI] [PubMed] [Google Scholar]
- Reichard P (1993) From RNA to DNA, why so many ribonucleotide reductases? Science 260, 1773. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129. [DOI] [PubMed] [Google Scholar]
- Seliger B, Papadileris S, Vogel D, Hess G, Brendel C, Storkel S, Ortel J, Kolbe K, Huber C, Huhn D, Neubauer A (1996) Analysis of the p53 and MDM‐2 gene in acute myeloid leukemia. Eur. J. Haematol. 57, 230. [DOI] [PubMed] [Google Scholar]
- Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, Hoffman B, Liebermann D (1994) Immediate early up‐regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways. Oncogene 9, 1791. [PubMed] [Google Scholar]
- Shi XB, Nesslinger NJ, Deitch AD, Gumerlock PH, Devere White RW (2002) Complex functions of mutant p53 alleles from human prostate cancer. Prostate 51, 59. [DOI] [PubMed] [Google Scholar]
- Smith ML, Ford JM, Hollander MC, Bortnick RA, Amundson SA, Seo YR, Deng CX, Hanawalt PC, Fornace AJ Jr (2000) p53‐mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, 21, and/or gadd45 genes. Mol. Cell Biol. 20, 3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76. [DOI] [PubMed] [Google Scholar]
- Taetle R, Honeysett JM, Bergeron R (1989) Combination iron depletion therapy. J. Natl Cancer Inst 81, 1229. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y (2000) A ribonucleotide reductase gene involved in a p53‐dependent cell‐cycle checkpoint for DNA damage. Nature 404, 42. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Endo H, Chikatsu N, Uchimaru K, Asano S, Fujita T, Nakahata T, Motokura T (1999) Expression of p21 (Cip1/Waf1/Sdi1) and p27 (Kip1) cyclin‐dependent kinase inhibitors during human hematopoiesis. Blood 93, 4167. [PubMed] [Google Scholar]
- Thomas DA, Du C, Xu M, Wang X, Ley TJ (2000) DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 12, 621. [DOI] [PubMed] [Google Scholar]
- Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456. [DOI] [PubMed] [Google Scholar]
- Yan Y, Shay JW, Wright WE, Mumby MC (1997) Inhibition of protein phosphatase activity induces p53‐dependent apoptosis in the absence of p53 transactivation. J. Biol. Chem. 272, 15220. [DOI] [PubMed] [Google Scholar]
- Yin Y, Solomon G, Deng C, Barrett JC (1999) Differential regulation of p21 by p53 and Rb in cellular response to oxidative stress. Mol. Carcinog. 24, 15. [PubMed] [Google Scholar]