

Abstract
Objectives: Schwann cell (SC) transplantation is a promising therapy for peripheral nerve transaction, however, clinical use of SCs is limited due to their very limited availability. Adipose‐derived stem cells (ADSCs) have been identified as an alternative source of adult stem cells in recent years. The aim of this study was to evaluate the feasibility of using ADSCs as a source of stem cells for differentiation into Schwann‐like cells by an indirect co‐culture approach, in vitro.
Materials and methods: Multilineage differentiation potential of the obtained ADSCs was assayed by testing their ability to differentiate into osteoblasts and adipocytes. The ADSCs were co‐cultured with SCs to be induced into Schwann‐like cells through proximity, using a Millicell system. Expression of typical SC markers S‐100, GFAP and P75NTR of the treated ADSCs was determined by immunocytochemical staining, western blotting and RT‐PCR. Myelination capacity of the differentiated ADSCs (dADSCs) was evaluated in dADSC/dorsal root ganglia neuron (DRGN) co‐cultures.
Results: The treated ADSCs adopted a spindle shaped‐like morphology after co‐cultured with SCs for 6 days. All results of immunocytochemical staining, western blotting and RT‐PCR showed that the treated cells expressed S‐100, GFAP and P75NTR, indications of differentiation. dADSCs could form Schwann‐like cell myelin in co‐culture with DRGNs. Undifferentiated ADSCs (uADSCs) did not form myelin compared to DRGNs cultured alone, but could produce neurite extension.
Conclusions: These results demonstrate that this indirect co‐culture microenvironment could induce ADSCs to differentiate into Schwann‐like cells in vitro, which may be beneficial for treatment of peripheral nerve injuries in the near future.
Introduction
Schwann cells (SCs) play a critical role in regeneration of peripheral nerves and are thus, the most promising candidate cell type as healing treatment for transected peripheral nerves (1). In animal models, SCs transplanted into nerve bridges in injured sciatic nerves have been shown to promote re‐growth of injured axons and of functional recovery (2). However, it is difficult to obtain sufficiently large numbers of functional SCs for in vitro work within a short span of time, which greatly limits their clinical application.
In the past few decades, bone marrow stromal cells (BMSCs) have been considered as an alternative cell source for tissue regeneration after peripheral nerve injury as they are capable of selectively differentiating into SCs and enhancing axonal regeneration (3). However, BMSC procurement procedures are painful for the donor and frequently require general or spinal anaesthesia and yet may yield low numbers of BMSCs upon harvest (4). For these reasons, alternative stem cell types need to be investigated.
The ideal transplantable source cell should be easily accessible, proliferate rapidly in vitro, possess multipotental differentiation properties and successfully integrate into host tissue, with non‐ or hypo‐immunogenicity (5). Cells isolated from adipose tissue, termed adipose‐derived stem cells (ADSCs), can satisfy the above requirements. Both ADSCs and BMSCs are originally derived from embryonic mesoderm, they have similar phenotype and gene expression profiles (6, 7); both can differentiate into several mesenchymal tissue lineages, including adipocytes (8), osteoblasts (9), myocytes (10), chondrocytes (11), endothelial cells (12) and cardiomyocytes (13). They can also be induced into neural lineages in vitro (14, 15, 16). Subcutaneous adipose tissue is abundant and possesses more mesenchymal stem cells (MSCs; 2 in 100 cells) than does the bone marrow (1 in 25 000 or 100 000 cells) (17, 18, 19, 20). ADSCs can be easily isolated by conventional liposuction procedures, which overcome tissue morbidity compared to bone marrow aspiration, and proliferate rapidly in culture. Thus, ADSCs may be an ideal alternative cell source for SC transplantation in regeneration of peripheral nerve.
Previous studies have shown that ADSCs can be induced into SC‐like cells using a protocol similar to the method used to induce BMSCs to differentiate into SC‐like cells (21, 22). Reports have documented that SC‐like cells from ADSCs are spindle‐like in shape and express SC markers such as S‐100. In our study, we have aimed to investigate whether ADSCs could be induced into Schwann‐like cells by indirect co‐culture with SCs in vitro, and whether the differentiated ADSCs (dADSCs) would exhibit Schwann‐like cell behaviour when co‐cultured with dorsal root ganglia neurons (DRGNs) in vitro, which would provide a foundation for transplantation of dADSCs in the vicinity of transected peripheral nerves.
Materials and methods
Materials
Trypsin and Dulbecco’s modified Eagle’s medium/F‐12 Ham nutrient mixture (DMEM/F‐12) were purchased from Invitrogen, Carlsbad, CA, USA. Collagenase type α was purchased from Merck. Recombinant human beta‐NGF was from Peprotech (Rocky Hill, Connecticut, USA). Cytosine‐B‐arabinoside hydrochloride (Ara‐C), 3‐isobutyl‐1‐methylxanthine, ascorbate, dexamethasone, β‐glycerophosphate, insulin, penicillin and streptomycin were purchased from Sigma, St. Louis, MO, USA. Human heregulin1‐β1 extracellular domain (HRG1‐β1 ECD) was from R&D (Minneapolis, USA). Foetal bovine serum (FBS) was obtained from Hyclone, Logan, UT, USA. 12‐well Millicell cell culture dishes with inserted hanging geometry (1.0 μm PET transparent) were purchased from Millipore (Boston, MA, USA). All culture plates were from Costar (IL, USA). All other reagents were local products of analytical grade.
Primary antibody employed for immunocytochemistry, rabbit anti‐S‐100 protein monoclonal antibody, was from Sigma. Mouse anti‐tubulin, beta isoform monoclonal antibody, rabbit anti‐P75 neurotrophin receptor polyclonal antibody and rabbit anti‐glial fibrillary acidic protein (GFAP) polyclonal antibody were from Millipore. Secondary antibodies including FITC goat anti‐rabbit IgG (H+L), FITC goat anti‐mouse IgG (H+L), TRITC goat anti‐rabbit IgG (H+L) and goat anti‐rabbit IgG/HRP were purchased from Invitrogen.
Neonatal Sprague–Dawley rats (1–3 days old) and adult Sprague–Dawley rats (male, 6–8 weeks old) were obtained from Beijing Haidian Experimental Animal Center. All animal experiments were carried out in accordance with the US National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80‐23) revised 1996 and approved by the Beijing Administration Committee for Experimental Animals.
Isolation and culture of ADSCs
Adult Sprague–Dawley rats were deeply anaesthetized using chloral hydrate, and adipose tissue was obtained from testicular fat pads. Isolation of ADSCs was accomplished using a modification of a previously published method (22). Adipose tissue samples were washed in cold DMEM/F‐12 medium (containing 1% penicillin/streptomycin) and were mechanically dissociated carefully after digestion in collagenase type II for 40 min at 37 °C. Suspensions were filtered through 80‐μm metal mesh and centrifuged to separate floating adipocytes from the stromal vascular fraction. Supernatant was discarded and cells in the stromal vascular fraction were resuspended in DMEM/F‐12 medium (containing 1% penicillin/streptomycin and 10% FBS) and seeded on culture plates. Twenty‐four hours later, culture medium was changed to eliminate non‐adherent cells. Cells used in our experiments were passaged 3–5 times.
Isolation and culture of SC
Schwann cells were enriched from sciatic nerves of neonatal Sprague–Dawley rats as described in our previous study (23). The protocol is based on different rates of cell proliferation and attachment properties between cultured fibroblasts and SCs in vitro. In brief, cells were first treated with Ara‐C for 24 h. Then medium was replaced with growth medium containing 20 ng/ml human heregulin1‐β1 extracellular domain (HRG1‐β1 ECD). After 48 h further incubation, cells were treated with 0.05% trypsin, which resulted in detachment of SCs from the dishes but not fibroblasts. In each category, suspended cells were collected into conical tubes and centrifuged. After removal of supernatant, pellets were resuspended in DMEM/F‐12 medium (containing 1% penicillin/streptomycin, 20 ng/ml HRG1‐β1 ECD and 10% FBS) and plated on culture plates at density of 5 × 104 cells/ml. Cells were grown to confluence, and then passaged by splitting them 1:3.
Proliferation assay of ADSCs
ADSCs at passages 3, 5 and 10, were seeded 1 × 104 cells/ml on 24‐well plates to determine proliferation activity of the cells. After growing ADSCs for 1, 3, or 5 days, cell proliferation was determined by MTT assay, which detects mitochondrial dehydrogenase activity of viable cell, s spectrophotometrically. Briefly, cells were cultured for 1, 3, and 5 days, and 100 μl of MTT (3‐[4, 5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl tetrasodium bromide, 5 mg/ml), was added to each well and incubated at 37 °C for 4 h. Then, MTT solution was carefully removed and insoluble blue formazan crystals were dissolved in 1 ml dimethyl sulphoxide. One hundred microlitres of solution was aspirated from each well and poured into 96‐well plates. Absorbance of each well was measured by ELISA at 570 nm. In addition, MTT assay was performed on directly counted SCs (0.5 × 105, 1 × 105, 2 × 105, 4 × 105, 8 × 105, 1.6 × 106, 3.2 × 106), and absorbance values were plotted against counted cell numbers to establish a standard calibration curve. Viable cell numbers were then determined from the standard curve based on their MTT absorbance values. Cell numbers are represented as mean ± standard deviation (SD).
Adipogenic and osteogenic differentiation of rat ADSCs
Multilineage differentiation potential was assayed by testing ability of ADSCs to differentiate into osteoblasts and adipocytes, as described previously (24). Briefly, osteoblast differentiation was achieved after cells were treated with 50 μg/ml ascorbate, 0.1 μm dexamethasone and 10 mmβ‐glycerophosphate for 3 weeks. For adipocyte differentiation, cultures were treated with 1 μm dexamethasone, 10 μg/ml insulin and 100 μg/ml 3‐isobutyl‐1‐methylxanthine for 3 weeks. von Kossa and oil red O staining methods were employed to identify osteoblasts and adipocytes respectively.
Differentiation of ADSCs by co‐culture with SCs
For co‐culture experiments, 5 × 103 ADSCs/well were seeded on 12‐well plates with DMEM/F‐12 medium containing 10% FBS overnight and replaced with fresh medium to eliminate non‐adherent cells. As shown in Fig. 1 (III), SCs were seeded on Millicell and co‐cultured with ADSCs in 12‐well plates with replacement of 1/2 volume co‐culture medium every 3 days. Millicell plates had porous polystyrene membranes with pore size of 1 μm, allowing free exchange of proteins in the medium while preventing cell migration between the two compartments. ADSCs without co‐culturing with SCs under identical conditions were negative controls and SCs alone were positive controls.
Figure 1.
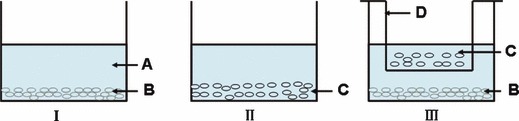
Model of ADSCs co‐cultured with SCs. (I) ADSCs cultured alone; (II) Schwann cells; (III) ADSCs co‐cultured with Schwann cells. A: DMEM/F‐12 medium; B: ADSCs; C: SCs; D: Millicell.
Immunofluorescence staining
For immunophenotype characterization of dADSCs, SC markers, including S‐100, GFAP and P75NTR, were used. Immunostaining was performed according to protocols published previously (25). Briefly, cells cultured on coverslips were fixed in 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS) for 10 min, and then permeabilized with 0.05% Triton X‐100 for 10 min. Non‐specific binding sites were blocked using 5% goat serum for 1 h. Cells were then incubated with primary monoclonal antibody (1:200) at room temperature for 1 h. Cells were washed three times in PBS and incubated with corresponding secondary antibodies (1:200) for 2 h at room temperature. After second rinsing in PBS, cells were incubated in 4′, 6′‐diamidino‐2‐phenylindole dihydrochloride (DAPI, 1:1000) to counterstain cell nuclei, for 5 min at room temperature. Finally, specimens were washed three times in PBS, each for 5 min, and were then mounted with antifade solution. Labelled cells were examined by fluorescence microscopy. Images were digitally recorded and processed using Image‐Pro Plus (Media Cybernetics, USA). Cultures of SCs and uADSCs were similarly stained as positive and negative controls accordingly, with antibodies above.
Cultures to be stained for β‐tubulin were fixed in 4% PFA for 30 min at room temperature. Cultures were permeabilized with 0.1% Triton X‐100 for 10 min. Cultures were then treated with ice‐cold 50% acetone for 2 min, ice‐cold 100% acetone for 2 min and 50% acetone for 2 min. They were then washed twice in PBS for 3 min each and incubated with mouse anti‐tubulin beta β isoform, monoclonal antibody for 2 h at room temperature. Rinsing three times in PBS was followed by incubation with corresponding secondary antibodies (1:100) for 1 h at room temperature. After the second rinsing step in PBS, cells were incubated in DAPI (1:1000) for 5 min at room temperature to counterstain cell nuclei. Finally, specimens were washed three times in PBS, each for 5 min, and then were mounted with antifade solution. Labelled cells were examined by fluorescence microscopy.
Western blot analysis
Cells were washed three times in cold PBS, scraped into ice‐cold lysis buffer (20 mm Tris–HCl, pH 7.4, 2 mm EDTA, 25 mm NaF, 1% Triton X‐100 plus protease inhibitors) and incubated at 4 °C for 30 min. After incubation, cell lysates were sonicated for 15 s and then centrifuged at 12 520 g at 4 °C for 15 min. Supernatants were collected and their total protein concentration was determined using BCA Protein Assay Kit (Pierce, Rockford, USA). Protein samples were completely denatured by boiling in bromophenol blue sample buffer for 5 min, separated by SDS–PAGE (10%), and then transferred electrophoretically on to polyvinylidenedifluoride (PVDF) membranes. Non‐specific antibody binding was blocked with 5% non‐fat dry milk in Tris‐buffered saline containing 0.1% Tween 20 (TBST) for 2 h at room temperature. Immunoblotting was performed with specific primary antibody and secondary antibodies conjugated to horseradish peroxidase. Protein blots were developed by enhanced chemiluminescence method. β‐actin was used as loading control, and results were analysed as detected protein/β‐actin ratio.
RT‐PCR analysis
RT‐PCR was used to evaluate expression of genes encoding S‐100, GFAP and P75NTR in ADSCs co‐cultured with SCs. Total RNA was isolated using Trizol reagent (Invitrogen) and genomic DNA was removed using DNase (Takara, Japan). Two micrograms of RNA was reverse transcribed at 42 °C for 45 min in 20 μl reaction mixture using Reverse Transcription System (Promega, Madison, USA). Total RNA concentration was determined spectrophotometrically at 260 nm. Primers of detected genes are listed in Table 1. The reaction system and procession were employed as described previously (26). GAPDH was used to normalize the amount of template for each sample. ADSCs not co‐cultured with SCs under identical conditions were negative controls and SCs alone were positive controls.
Table 1.
Primers for RT‐PCR
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) | PCR product (bp) |
|---|---|---|---|
| S‐100 | GAGAGAGGGTGACAAGCACAA | GGCCATAAACTCCTGGAAGTC | 169 |
| P75NTR | AGGGCACATACTCAGACGAA | CAAGATGGAGCAATAGAGAG | 327 |
| GFAP | CTTCCCGCAACGCAGAG | GAGCCGTGGGCACTAAA | 631 |
| GAPDH | GCCACCCAGAAGACTGTGGAT | TGGTCCAGGGTTTCTTACTCC | 500 |
Functional assay in dADSC/DRGN co‐cultures
DRGN culture was prepared according to a previous study with minor modification (27). In brief, dorsal root ganglia (DRG) neurons were removed from neonatal Sprague–Dawley rats, cleaned of connective tissue and chemically dissociated in trypsin. Trypsin activity was stopped by addition of DMEM/F‐12 medium containing foetal bovine serum. Ganglia were then washed in DMEM/F‐12, before mechanical dissociation by gentle trituration using a pipette. Dissociated neurons were passed though a 70 μm mesh to remove non‐dissociated tissues and myelin debris. Suspensions were centrifuged, and pellets were resuspended in NLA negative (neurobasal medium plus B27, supplemented with nerve growth factor at 10 ng/ml). Approximately 5000–7000 DRG cells were plated per well (24‐well plate). After seeding and overnight incubation, cultures were flooded and subsequently treated with NLA negative medium containing 5′‐fluoro‐2′‐deoxyuridine (FUDR) at 20 μg/ml and uridine at 10 μg/ml 2 days to kill any remaining non‐neuronal cells. After this antimitotic treatment, resulting DRGN cultures were maintained on NLA− medium for at least 1 week to ensure that residual FUDR was removed when SCs or dADSCs were added.
For myelination experiments, co‐cultures were prepared by addition of 50 000 dADSCs, SCs and uADSCs, to purified DRGN cultures. DRGN cultures and co‐cultures were fed every 2–3 days with NLA negative medium for 2 weeks. For myelination induction, each culture of DRGN, SC/DRGN, dADSC/DRGN and uADSC/DRGN was switched to NLA+ medium. NLA+ medium was the same as NLA− medium but with addition of 50 μg/ml ascorbic acid. All cultures were then maintained for additional 2 weeks, with replacement of appropriate medium every 2–3 days.
Sudan black staining and cell counting
Cultures were fixed for 10 min in 4% PFA, rinsed three times in 0.1 m PO4 and further fixed in 0.1% OsO4 for 1 h at 4 °C. Cultures were then washed four times in 0.1 m PO4 followed by dehydration in 25%, 50% and 70% ethanol, each for 5 min; 0.5% Sudan black/70% ethanol solution was filtered before staining for 1 h. Cultures were rinsed in 0.1 m PO4 and mounted on glycerine jelly after trimming the dish. Images of myelinated axons were captured using a CCD video camera (CCD colour camera; Hitachi, Japan). All experiments were performed in triplicate and repeated at least twice.
Statistical analysis
Data are expressed as mean ± SD. For comparison of quantitative measures, the values were subjected to statistical analysis using Student’s t‐test, considered significant at P < 0.05.
Results
Characterization of rat ADSCs and SCs
Rat testicular adipose tissue was enzymatically digested and then centrifuged to isolate the stromal cell fraction from mature adipocytes. After two passages in culture, cells from the stromal fraction morphologically exhibited a homogenous population of fibroblast‐like cells (Fig. 2a). Cells were then trypsinized and plated for MTT proliferation assay (Fig. 3). There was an apparent stationary phase in population growth of cells during the first 24 h. Rate of cell proliferation then expanded more rapidly. Although overall cell population growth rate of ADSCs at passages 5 and 10 decreased, they still proliferated very rapidly. These rat ADSCs could be passaged at least 10 times, and did not spontaneously differentiate during culture expansion (data not shown).
Figure 2.
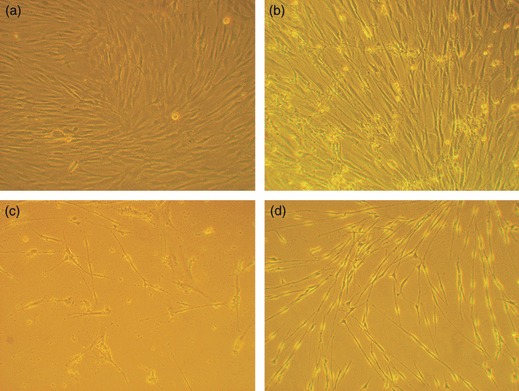
Morphology of ADSCs, dADSCs and SCs by phase‐contrast microscopy. (a) ADSCs cultured alone showed flattened fibroblast‐like morphology; (b) ADSCs co‐cultured with Schwann cells for 6 days differentiated to a Schwann cell phenotype; (c) Morphology of treated cells was exactly the same as SCs, when co‐cultured for 21 days; (d) Images of purified Schwann cells (magnification: ×100).
Figure 3.
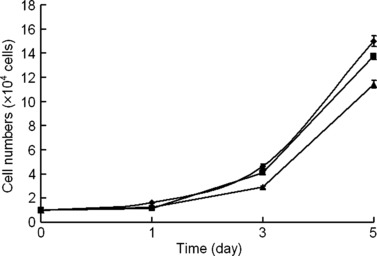
Cell proliferation of ADSCs isolated from adipose tissue. MTT assay was used to determine growth rate of cell populations obtained from adipose tissue at passage 3( ), 5(
), 5( ) and 10(
) and 10( ). Values represent mean ± SEM, corresponding to five replicates.
). Values represent mean ± SEM, corresponding to five replicates.
As Fig. 2d shows, SCs were typically spindle‐shaped and were either bipolar or occasionally multipolar, often aligning in fascicles; purity of SCs was more than 99%.
To determine whether cells isolated from adipose tissue exhibited multipotential properties of mesenchymal stem cells, they were treated with agents known to induce differentiation in cells originating from the mesoderm. Adipocyte differentiation was confirmed by lipid droplets with oil red O (Fig. 4a) and osteoblast differentiation was assessed by production of calcium deposits detected by staining with the von Kossa reagent (Fig. 4b).
Figure 4.
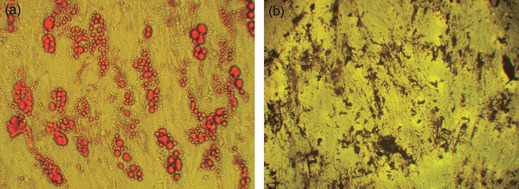
ADSCs exhibit properties of stem cells. Cultures were treated with agents to induce differentiation to adipogenic and osteogenic lineages. Cells were then stained with oil red O (a) and von Kossa reagent (b) respectively (magnification: ×100).
Morphology of dADSCs
At co‐culture days 6 and 21, ADSCs were visualized using a phase‐contrast microscope. As Fig. 2 shows, ADSCs co‐cultured with SCs exhibited striking morphological changes compared to controls. ADSCs cultured alone appeared as a monolayer of large, fibroblast‐like flattened cells. When ADSCs were co‐cultured with SCs for 21 days, cells showed spindle shaped morphology and were bi‐ or tripolar, resembling the appearance of SCs (Fig. 2c). Unfortunately, almost all control ADSCs detached from the plates and died after 21 days of culture without passage.
Expression of SC markers
dADSCs were assessed for expression of SC markers: S‐100, GFAP and P75NTR, to determine evidence of phenotypic progression along an SC lineage. Control SC cultures were immunoreactive for all three makers, S‐100, GFAP and P75NTR. The cells demonstrated typical bipolar or tripolar morphology, had oval nuclei, and brightly stained for the above three protein markers, as shown in Fig. 5a–c respectively. Positive expression of S‐100, GFAP and P75NTR was observed in treated groups on days 6 and 21 (Fig. 5d–i). ADSCs cultured alone were negative for S‐100, GFAP and P75NTR (Fig. 5j–l); however, if ADSCs were cultured up to passage 8–10, there was occasional, faint expression of S‐100, but not of GFAP or P75NTR (Fig. 6).
Figure 5.
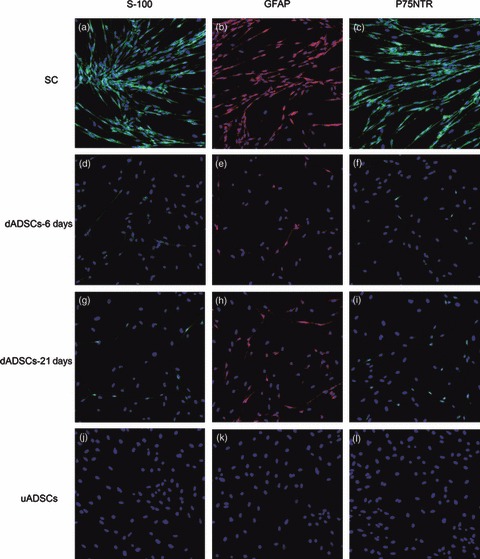
Immunofluorescence staining in Schwann cells, dADSCs and uADSCs. (a, d, g, j) S‐100; (b, e, h, k) GFAP and (c, f, i, l) P75NTR. Nuclei labelled blue with DAPI. Morphology of dADSCs: purity of isolated Schwann cells was more than 99% (magnification: ×100).
Figure 6.

S‐100 immunostaining in ADSCs. (a) DAPI; (b) S‐100 and (c) merged. There was occasional, faint expression of S‐100 when ADSCs were cultured up to passage 8–10 (magnification: ×200).
To confirm that results obtained by immunocytochemical staining were not due to artefacts of cell shrinkage, western blotting and RT‐PCR were performed. Lysates of the treated ADSCs but not ADSCs cultured alone showed S‐100, GFAP and P75NTR‐immunoreactive bands corresponding to a molecular weight of 12, 52 and 75 kDa respectively. This was present in SC lysates as well (Fig. 7). Results of RT‐PCR of the treated ADSCs mRNA probed with S‐100, GFAP and P75NTR primers also supported the results found on immunocytochemical staining. As shown in Fig. 8, S‐100, GFAP and P75NTR gene expressions were detected in dADSCs and SCs, but not in uADSCs cultures. All the above results supported the results found on immunocytochemical staining.
Figure 7.
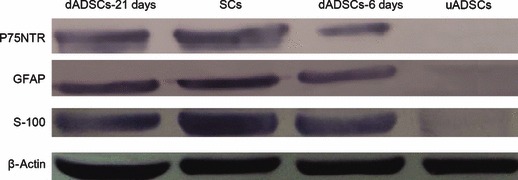
Western blotting analysis of S‐100, GFAP and P75NTR in Schwann cells, dADSCs and uADSCs. SCs, dADSCs and uADSCs lysates were blotted for S‐100, GFAP and P75NTR proteins. β‐actin was used as a loading control.
Figure 8.
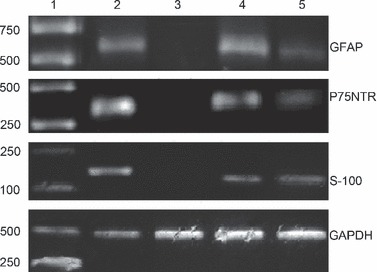
Detection of S‐100, P75NTR and GFAP expression in Schwann cells, dADSCs and uADSCs. Lane 1: DNA markers; Lane 2: Schwann cells; Lane 3: ADSCs cultured alone; Lane 4: ADSCs co‐cultured with Schwann cells for 6 days; Lane 5: ADSCs co‐cultured with Schwann cells for 21 days.
To investigate whether the cells could maintain their differentiated state after removing SCs from the co‐culture system, dADSCs were transferred to normal medium. Absence of SCs induced morphological changes in dADSCs during the first 2 weeks, which though maintained expression of SC markers, thus suggesting an indication of long‐lasting differentiation (data not shown). After that, cells gradually regained basal morphology and phenotype. However, if ADSCs was kept in co‐culture with SCs, their differentiated state could last for 21 days (Fig. 5g–i).
Functional assay of dADSC/DRGN co‐cultures
To evaluate ability of dADSCs to produce myelin under conditions that enabled SCs to produce myelin, dADSC/DRGN, SC/DRGN and uADSC/DRGN co‐cultures were prepared as described previously (27). After addition of dADSCs, SCs and uADSCs, cultures were maintained for a 2‐week period in NLA− medium to allow for cell proliferation. Cultures were then switched to NLA+ medium and incubated for an additional 2‐week period to allow myelination to occur. In both dADSC/DRGN and SC/DRGN co‐cultures, extensive myelin was formed during the final weeks (Fig. 9a,b). No myelin was formed in uADSC/DRGN co‐cultures over the same period (Fig. 9c); identical results were obtained in repeat experiments. Myelin was not formed in control DRGN cultures to which no exogenous cells were added (Fig. 9d).
Figure 9.
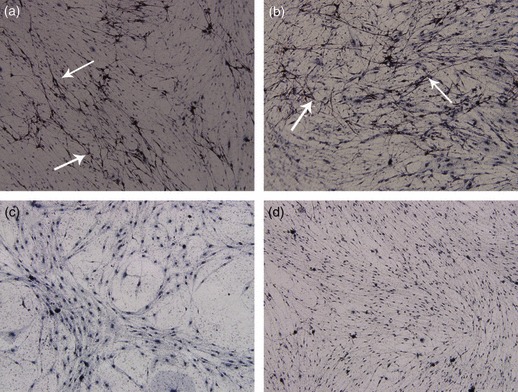
dADSCs could form myelin under conditions that enabled SCs to produce myelin. (a) SC/DRGN co‐cultures; (b) dADSCs/DRGN co‐cultures; (c) uADSCs/DRGN co‐cultures; (d) DRGN cultures. Myelin sheaths (arrows in a and b) are found in both SC/DRGN and dADSCs/DRGN co‐cultures (magnification: ×100).
One week after addition of dADSCs, SC and uADSC, cultures were further studied by immunocytochemical staining to better characterize axon–glial relationships in SC/DRGN, dADSC/DRGN and uADSC/DRGN co‐cultures. At termination of cultures for analysis, dADSCs were demonstrated to have greatly enhanced neurite outgrowth (Fig. 10b), which was similar to the level achieved by SCs (Fig. 10a). uADSCs produced neurite extensions superior to those seen with DRGNs cultured alone (Fig. 10c,d), but inferior to those seen with SCs and dADSCs.
Figure 10.
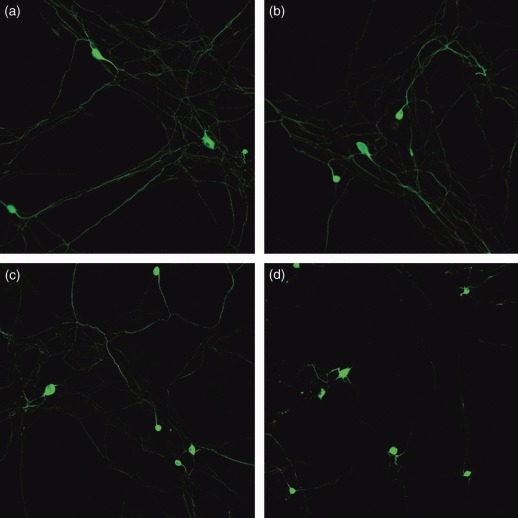
DRGNs stained for β‐tubulin to show neurite outgrowth. (a) SC/DRGN co‐cultures; (b) dADSC/DRGN co‐cultures; (c) uADSC/DRGN co‐cultures; (d) DRGN cultures. Differentiated ADSCs enhanced neurite outgrowth in co‐culture with neurons. Schwann cells evoked a similar response, while undifferentiated ADSCs had a definite effect compared to the neurons cultured alone (magnification: ×200).
Discussion
After transaction of peripheral nerves, SCs play a crucial part in functional recovery of injured peripheral nerves by forming Büngner bands, to help regrowing axons elongate, secrete surface cell adhesion molecules, cell matrix protein and neurotrophic factors, and remyelinating regenerative axons (28, 29). Therefore, reduced time periods between injury and SC transplantation would facilitate tissue integration in clinical processes. This would enhance probability of functional recovery after reconstruction surgery, which depends definitely on a crucial time window to obtain enough population of SCs, naïve or differentiated, from stem cells.
Schwann‐like cell differentiation of ADSCs, induced either with chemical factors or by co‐culture with SCs – by cell–cell contact or SC‐conditioned medium, has been obtained by several groups (21, 30). In our study, we aimed to evaluate the potential of ADSCs to differentiate into Schwann‐like cells by an indirect co‐culture approach through Millicell porous membranes. Millicell membranes were placed in such a way that allowed molecules secreted by SCs to permeate through the pores and reach the ADSCs beneath. There was no direct contact between SCs and ADSCs; ADSCs and SCs could communicate with each other only via secreted proteins in the medium during the co‐culture process. Our purpose was to assess whether such communication could induce Schwann‐like cell differentiation of ADSCs in vitro. Results showed that morphological changes were induced in the treated ADSCs co‐cultured with SCs, which subsequently successfully expressed SC markers, S‐100, GFAP and P75NTR, both at protein and mRNA levels, thus suggesting that some molecules secreted by SCs might be responsible for the Schwann‐like cell differentiation of ADSCs.
We focused our attention on ADSCs as they can be harvested by less invasive procedures than can BMSCs, and cultured with higher proliferation rates. Similar to BMSCs, ADSCs are non‐haematopoietic progenitor cells and possess multilineage differentiation potential into tissues of mesenchymal origin, as well as tissues of endodermal and ectodermal origin. Previous studies have reported that a mixture of glial growth factors (containing GGF/heregulin, FSK, PDGF and bFGF) can induce ADSCs to differentiate into cells that show spindle shapes with bi‐ or tripolar orientation, morphology resembling the appearance of SCs, and express S‐100, GFAP and P75NTR, similar to natural SCs (30). However, a further study has reported that such chemical factors may be a potent stimulus inducing initial, although poorly specific, changes in neural differentiation of MSCs, and that SC co‐culture may provide additional, necessary conditions to complete such a process (21). They also showed that SC‐conditioned medium could not induce morphological changes in MSCs, and their treated MSCs failed to express any myelin protein. In our co‐culture system, ADSCs were used for culture with SCs in indirect co‐culture. After 6 days co‐culture, the majority of ADSCs changed shape from a fibroblast‐like appearance to bipolar and tripolar spindle shapes, a characteristic phenotype of SCs. Most excitingly, morphology of the treated cells was exactly the same as SCs, when co‐cultured for 21 days. Immunocytochemical staining revealed that bipolar and tripolar cells were positive for S‐100, GFAP and P75NTR. To confirm that the results obtained by immunocytochemical staining were not due to any artefact of cell shrinkage, western blotting and RT‐PCR were performed. Lysates of treated ADSCs but not ADSCs cultured alone, showed S‐100, GFAP and P75NTR ‐immunoreactive bands corresponding to molecular weights of 12, 52 and 75 kDa respectively. Results of RT‐PCR of treated ADSC mRNA probed with S‐100, GFAP and P75NTR primers also supported the results found by immunocytochemical staining. S‐100, GFAP and P75NTR gene expressions were detected in ADSCs co‐cultured with SCs and SCs cultured alone, but not in ADSCs cultured alone, arguing against the notion that staining was merely caused by disruption of the cytoskeleton (31). To investigate whether the cells could maintain their differentiated state after removing SCs from the co‐culture system, dADSCs were transferred to normal medium. Absence of SCs induced morphological changes in dADSCs during the first 2 weeks, which retained the ability to express SC markers, thus suggesting an indication for long‐lasting differentiation. In our experiments, we also found that SCs proliferated rapidly during the co‐culture process without adding any additional neurotrophic factors (data not shown), suggesting cell–cell cross‐talk between the two co‐cultured cell types by soluble secreted proteins, which has also been observed in SC/BMSC co‐culture systems in recent reports (32).
One defining property of SCs is their unique affinity for axons. The manner in which SCs are associated with axons is an easily recognizable feature of SC/DRGN cultures. Thus, it is a well‐established method to evaluate myelinating capacity of dADSCs. When presented with DRGN axons in vitro, extensive myelin was formed in both dADSC/DRGN and SC/DRGN co‐cultures; also, no myelin was formed in uADSC/DRGN co‐cultures during the same period. Myelin was not formed in control DRGN cultures. These results demonstrate that dADSCs demonstrate the same property as SCs. Immunocytochemical staining demonstrated that dADSCs produced neurite extensions superior to those seen with uADSCs, and moderate neurite growth caused with the latter would suggest that some benefit can be conferred by stem cells, even prior to their differentiation.
Cell transplantation needs large numbers of cells, but in vitro expansion potential of committed SCs is limited. In vitro expansion of ADSCs is considered to be a powerful approach to overcome some practical and ethical concerns of cell transplantation. In this study, ADSCs were obtained from rat testicular adipose tissue. Primary cultures of adipose tissue are heterogeneous, containing haematopoietic cells, endothelial cells, smooth muscle cells and pericytes (4). Numbers of these contaminating cells is small though, and frequency of these cells diminishes quickly through serial passaging (33). Purified rat ADSCs exhibit typical mesenchymal stem cell characteristics: fibroblast morphology and multipotential capability. We used rat ADSCs at passages 3–5 for our experiments and observed that they could undergo osteogenic and adipogenic differentiation. They can be expanded through at least 10 passages and maintained in their undifferentiated state, except for occasional, faint expression of S‐100. Compared to previous methods, our protocol is relatively simple and easy to perform. It is similar to regular cell culture procedures, and does not require any special growth factors. Furthermore, this method is of low cost and is reproducible, which might become an alternative culture system for Schwann‐like cell differentiation of ADSCs.
In summary, our results demonstrate that ADSCs can be easily obtained from a small amount of fat tissue and expanded rapidly in culture. These ADSCs have the ability to differentiate into osteoblasts and adipocytes. They can be selectively induced to undergo morphological and phenotypic changes consistent with Schwann‐like cell differentiation in an in vitro co‐culture approach. Furthermore, the dADSCs could form myelin and enhance neurite outgrowth in a co‐culture bioassay, which clearly demonstrated functional potential of differentiated stem cells. Although a broad spectrum of phenotypic changes consistent with Schwann‐like cell differentiation was observed in treated ADSCs in vitro, further studies are needed to detect which growth factors secreted by SCs play a key role in Schwann‐like cell differentiation of ADSCs and to confirm whether dADSCs can enhance regeneration of injured peripheral nerves after they are transplanted into experimental nerve bridges.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (contract grant number: 30670528, 30700848, 30772443), National Basic Research Program of China (also called the 973 Program, contract grant number: 2005CB623905) and the National Natural Science Foundation of Beijing (contract grant number: 7082090).
References
- 1. Bunge RP (1993) Expanding roles for the Schwann cell: ensheathment, myelination, trophism and regeneration. Curr. Opin. Neurobiol. 3, 805–809. [DOI] [PubMed] [Google Scholar]
- 2. Mosahebi A, Woodward B, Wiberg M, Martin R, Terenghi G (2001) Retrovitral labeling of Schwann cells: in vitro characterization and in vivo transplantation to improve peripheral nerve regeneration. Glia 34, 8–17. [DOI] [PubMed] [Google Scholar]
- 3. Hou SY, Zhang HY, Quan DP, Liu XL, Zhu JK (2006) Tissue‐engineered peripheral nerve grafting by differentiated bone marrow stromal cells. Neuroscience 140, 101–110. [DOI] [PubMed] [Google Scholar]
- 4. Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ et al. (2001) Multilineage cells from human adipose tissue: implications for cell‐based therapies. Tissue Eng. 7, 211–228. [DOI] [PubMed] [Google Scholar]
- 5. Baksh D, Song L, Tuan RS (2004) Adult mesenchymal stem cells: characterization, differentiation and application in cell and gene therapy. J. Cell Mol. Med. 8, 301–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Ugarte DA, Alfonso Z, Zuk PA, Elbarbary A, Zhu M, Ashijian P et al. (2003) Different expression of stem cell mobilization‐associated molecules on multi‐lineage cells from adipose tissue and bone marrow. Immunol. Lett. 89, 267–270. [DOI] [PubMed] [Google Scholar]
- 7. De Ugarte DA, Morizono K, Elbarbary A, Alfonso Z, Zuk PA, Zhu M et al. (2003) Comparison of multi‐lineage cells from human adipose tissue and bone marrow. Cells Tissues Organs 174, 101–109. [DOI] [PubMed] [Google Scholar]
- 8. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. (1999) Multilineage potential of adult human mesenchymal stem cells. Science 288, 143–147. [DOI] [PubMed] [Google Scholar]
- 9. Erickson GR, Gimble JM, Franklin DM, Rice HE, Awad H, Guilak F (2006) Chondrogenic potential of adipose tissue‐derived stromal cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 290, 763–769. [DOI] [PubMed] [Google Scholar]
- 10. Lee JH, Kemp DM (2006) Human adipose‐derived stem cells display myogenic potential and perturbed function in hypoxic conditions. Biochem. Biophys. Res. Commun. 341, 882–888. [DOI] [PubMed] [Google Scholar]
- 11. Mehlhorn AT, Zwingmann J, Finkenzeller G, Niemeyer P, Dauner M, Stark B et al. (2009) Chondrogenesis of adipose‐derived adult stem cells in a poly‐lactide‐co‐glycolide scaffold. Tissue Eng. Part A 15, 1159–1167. [DOI] [PubMed] [Google Scholar]
- 12. Altman AM, Yan YS, Matthias N, Bai XW, Rios C, Mathur AB et al. (2009) IFATS collection: human adipose‐derived stem cells seeded on a silk fibroin‐chitosan scaffold enhance wound repair in a murine soft tissue injury model. Stem Cells 27, 250–258. [DOI] [PubMed] [Google Scholar]
- 13. Zhu Y, Liu T, Song K, Ning R, Ma X, Cui Z (2009) ADSCs differentiated into cardiomyocytes in cardiac microenvironment. Mol. Cell. Biochem. 324, 117–129. [DOI] [PubMed] [Google Scholar]
- 14. Wang B, Han J, Gao Y, Xiao ZF, Chen B, Wang X et al. (2007) The differentiation of rat adipose‐derived stem cells into OEC‐like cells on collagen scaffolds by co‐culturing with OECs. Neurosci. Lett. 421, 191–196. [DOI] [PubMed] [Google Scholar]
- 15. Franco Lambert AP, Fraga Zandonai A, Bonatto D, Cantarelli Machado D, Pegas Henriques JA (2009) Differentiation of human adipose‐derived adult stem cells into neuronal tissue: does it work? Differentiation 77, 221–228. [DOI] [PubMed] [Google Scholar]
- 16. Caddick J, Kingham PJ, Gardiner NJ, Wiberg M, Terenghi G (2006) Phenotypic and functional characteristics of mesenchymal stem cells differentiated along a Schwann cell lineage. Glia 54, 840–849. [DOI] [PubMed] [Google Scholar]
- 17. Strem BM, Hicok KC, Zhu M, Wulur I, Alfonso Z, Schreiber RE et al. (2005) Multipotential differentiation of adipose tissue‐derived stem cells. Keio J. Med. 54, 132–141. [DOI] [PubMed] [Google Scholar]
- 18. D’Ippolito G, Schilller PC, Ricordi C, Roos BA, Howard GA (1999) Age‐related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J. Bone Miner. Res. 14, 1115–1122. [DOI] [PubMed] [Google Scholar]
- 19. Banfi A, Bianchi G, Galotto M, Cancedda R, Quarto R (2001) Bone marrow stromal damage after chemo/radiotherapy: occurrence, consequences and possibilities of treatment. Leuk. Lymphoma 42, 863–870. [DOI] [PubMed] [Google Scholar]
- 20. Muschler GF, Nitto H, Boehm CA, Easley KA (2001) Age‐ and gender‐related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J. Orthop. Res. 19, 117–125. [DOI] [PubMed] [Google Scholar]
- 21. Kingham PJ, Kalbermatten DF, Mahay D, Armstrong SJ, Wiberg M, Terenghi G (2007) Adipose‐derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp. Neurol. 207, 267–274. [DOI] [PubMed] [Google Scholar]
- 22. Xu YF, Liu L, Li Y, Zhou C, Xiong F, Liu ZS et al. (2008) Myelin‐forming ability of Schwann cell‐like cells induced from rat adipose‐derived stem cells in vitro. Brain Res. 1239, 49–55. [DOI] [PubMed] [Google Scholar]
- 23. Wei YJ, Zhou JL, Zheng ZH, Wang AJ, Ao Q, Gong YD et al. (2009) An improved method for isolating Schwann cells from postnatal rat sciatic nerves. Cell Tissue Res. 337, 361–369. [DOI] [PubMed] [Google Scholar]
- 24. Bunnell BA, Flaat M, Gagliardi C, Patel B, Ripoll C (2008) Adipose‐derived stem cells: isolation, expansion and differentiation. Methods 45, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Komiyama T, Nakao Y, Toyama Y, Asou H, Vacanti CA, Vacanti MP (2003) A novel technique to isolate adult Schwann cells for an artificial nerve conduit. J. Neurosci. Methods 122, 195–200. [DOI] [PubMed] [Google Scholar]
- 26. Keilhoff G, Goihl A, Langnäse K, Fansa H, Wlof G (2006) Transdifferentiation of mesenchymal stem cells into Schwann cell‐like myelinating cells. Eur. J. Cell Biol. 85, 11–24. [DOI] [PubMed] [Google Scholar]
- 27. Plant GW, Currier PF, Cuervo EP, Bates ML, Pressman Y, Bunge MB et al. (2002) Purified adult ensheathing glia fail to myelinate axons under culture conditions that enable Schwann cells to form myelin. J. Neurosci. 22, 6083–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dezawa M, Takahashi I, Esaki M, Takano M, Sawada H (2001) Sciatic nerve regeneration in rats induced by transplantation of in vitro differentiated bone‐marrow stromal cells. Eur. J. Neurosci. 14, 1771–1776. [DOI] [PubMed] [Google Scholar]
- 29. Ribeiro‐Resende VT, Koenig B, Nichterwitz S, Oberhoffner S, Schlosshauer B (2009) Strategies for inducing the formation of bands of Büngner in peripheral nerve regeneration. Biomaterials 30, 5251–5259. [DOI] [PubMed] [Google Scholar]
- 30. Xu YF, Liu ZS, Liu L, Zhao CP, Xiong F, Zhou C et al. (2008) Neurospheres from rat adipose‐derived stem cells could be induced into functional Schwann cell‐like cells in vitro. BMC Neurosci. 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu P, Blesch A, Tuszynski MH (2004) Induction of bone marrow stromal cells to neurons: differentiation, transdifferentiation, or artifact? J. Neurosci. Res. 77, 174–191. [DOI] [PubMed] [Google Scholar]
- 32. Wang J, Ding F, Gu Y, Liu J, Gu XS (2009) Bone marrow mesenchymal stem cells promote cell proliferation and neurotrophic function of Schwann cells in vitro and in vivo. Brain Res. 1262, 7–15. [DOI] [PubMed] [Google Scholar]
- 33. Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM (2001) Surface protein characterization of human adipose tissue‐derived stromal cells. J. Cell. Physiol. 189, 54–63. [DOI] [PubMed] [Google Scholar]