

Abstract
Abstract. Objective: The exposure of mammalian cells to extracellular stress induces the expression of immediate early genes such as c‐fos and c‐jun and activates transcription factor activator protein‐1 (AP‐1). The purpose of the current study was to investigate the role of c‐Fos and JunD in stress‐induced cell death. Materials and methods: We exposed cultured primary mouse embryonic fibroblasts (MEF) to ultraviolet light (UV‐C) or hydrogen peroxide (H2O2). Induction of c‐Fos and JunD and activation of MAPK/ERK1/2 signalling in the presence or absence of a MAPK inhibitor were analyzed by western blotting. Activation of AP‐1 transcription factors was detected by the electrophoretic mobility shift assay and immunoprecipitation. Cell death was measured by changes in caspase 3 activities and nuclear morphology. Effects of c‐Fos and JunD expression on cell death were investigated by transfection. Results: We found that the exposure of cultured primary MEF cells to UV or H2O2 caused a significant increase in c‐Fos and JunD protein levels. In addition, these two proteins formed complexes with each other and contributed to activation of AP‐1 transcription complexes. More importantly, under both stress conditions, overexpression of JunD alone or overexpression of both c‐Fos and JunD reduced caspase 3 activity and cell death. At the same time, UV irradiation activated the MAPK/ERK1/2 signalling pathway. The suppression of MEK1/ERK1/2 activation inhibited UV‐induced expression of c‐Fos and JunD and increased caspase 3 activity and cell death. Conclusion: Our results suggest that both UV and H2O2 induce the activation of c‐Fos/JunD AP‐1 complexes resulting in the prevention of cell death. Moreover, UV irradiation‐induced increases in c‐Fos/JunD expression in primary MEF cells are mediated through the activation of the MAPK/ERK1/2 signalling pathway.
INTRODUCTION
Activator protein‐1 (AP‐1) was among the first mammalian transcription factors to be identified, yet its physiological functions are still being unravelled. AP‐1 activity is induced by a plethora of physiological stimuli and environmental insults. In turn, AP‐1 regulates a wide range of cellular processes, including cell proliferation, cell death, survival and differentiation. The ability of a single transcription factor to control a collection of biological processes stems primarily from its structural and regulatory complexity. AP‐1 is a menagerie of dimeric basic region‐leucine zipper (bZIP) proteins that belong to the Jun (c‐Jun, JunB and JunD) and Fos (c‐Fos, FosB, Fra‐1 and Fra‐2) subfamilies in mammalian cells. These Jun and Fos proteins recognize either 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) response elements (5′‐TGAG/CTCA‐3′) or cAMP response elements (CRE, 5′‐TGACGTCA‐3′) to regulate transcription (Shaulian & Karin 2002; Eferl & Wagner 2003).
Previous studies of c‐fos and c‐jun suggest that c‐fos/c‐jun is up‐regulated in stress‐induced cell death, whereas exogenous inhibition of c‐fos and c‐jun mRNA synthesis attenuated cell death (Colotta et al. 1992; Moreno‐Manzano et al. 1999). FosB and JunD containing AP‐1 transcription complexes are activated in transforming growth factor (TGF)‐β1‐induced cell death. In addition, ectopic overexpression of FosB and JunD enhances apoptosis (Yamamura et al. 2000). In addition, AP‐1 composed of c‐Fos and JunD has been shown to be involved in the process of cell death (Marti et al. 1994; Hafezi et al. 1999b; Vollgraf et al. 1999). However, little is known about the effect of AP‐1 containing c‐Fos and JunD in stress‐induced cell death. Previous studies have demonstrated that jun‐D−/– primary mouse embryonic fibroblast (MEF) cells exhibited an increased p53‐dependent apoptosis in response to ultraviolet (UV)‐C irradiation (Weitzman et al. 2000). c‐fos‐Deficient fibroblasts are also hypersensitive to UV irradiation (Schreiber et al. 1995; Lackinger & Kaina 2000). It is possible that c‐Fos/JunD containing AP‐1 transcription complexes play protective functions in UV‐induced cell death. UV irradiation provokes oxidative stress in cells (Halliwell & Gutteridge 1990; Scharffetter‐Kochanek et al. 1997) and reactive oxygen species (ROS) such as H2O2 and nitric oxide are effective apoptotic inducers (Buttke & Sandstrom 1994). Thus, UV irradiation could elicit formation of cellular ROS and these ROS could, in turn, activate downstream cell death signalling pathway(s). Previous studies showed that H2O2 induces the activation of AP‐1 transcription complexes containing c‐Fos and JunD (Lakshminarayanan et al. 1998; Vollgraf et al. 1999; Mendes et al. 2003). Based on these factors, we have examined whether signalling pathways induced by UV, in our system, can be mimicked by H2O2.
One of the most explored functions of MAPK signalling modules is regulation of gene expression in response to extracellular stimuli. By and large, activation of JNK and p38 has been linked to induction of apoptosis, while ERK1/2 is linked to cell survival (Chang & Karin 2001). UV irradiation increases the activity of ERK1/2 in various cell types (Dhanwada et al. 1995; Price et al. 1996). Therefore, it is important to investigate whether, in our system, ERK1/2 would be involved in the signalling pathway in UV‐induced apoptosis. Here, we report that both UV and H2O2 induce the activation of c‐Fos/JunD AP‐1 transcription complexes, resulting in the prevention of cell death. Our results also demonstrate that it is the MEK1/ERK1/2 signalling pathway that is responsible for c‐Fos/JunD expression in response to UV irradiation in primary MEF cells.
MATERIALS AND METHODS
Cell cultures and treatments
Day 14.5 mouse embryos were isolated from maternal uteri. Heads and internal organs were removed and the remaining tissue was digested with trypsin. Tissue debris was allowed to settle and the supernatant was collected. MEF cells obtained by this method were cultured in Dulbecco's modified Eagle's medium containing 10% foetal bovine serum (PAA Laboratories, Pasching, Austria) and 100 µg/ml penicillin and 50 µg/ml streptomycin sulphate (Invitrogen, Carlsbad, CA, USA) in a humidified incubator at 37 °C with 5% CO2; medium was replaced every 3 days. Cells used in all experiments were allowed to each confluence of 50–60%.
For UV irradiation experiments, cells were exposed to UV‐C at an intensity of 300 J/m2 in a UV cross‐linker (UVP Products, Upland, CA, USA) and were allowed to recover for a selection of time periods. Cells were exposed to H2O2 at 10 mm for 15 min and were also allowed to recover for various times.
Immunoprecipitations and immunoblots
Cells harvested by scraping and centrifugation were lysed in a lysis buffer [20 mm Tris‐HCl, pH 7.5, 137 mm NaCl, 1.5 mm MgCl2, 2 mm ethylenediaminetetraacetic acid (EDTA), 10 mm sodium pyrophosphate, 25 mmβ‐glycerophosphate, 10% glycerol, 1% Triton X‐100, 1 mm sodium orthovanadate and 1 mm phenylmethylsulphonyl fluoride] supplemented with 1 × protease inhibitor cocktail and 10 µm okadaic acid, were incubated on ice for 30 min and were cleared by centrifugation at 13 000 g for 25 min at 4 °C. Cell lysates were quantified using a Bicinchoninic Acid Protein Assay kit (Pierce Biotechnology Inc., Rockford, IL, USA). For immunoprecipitation, 1 mg aliquots of each sample were incubated overnight with the appropriate antibody and Protein G PLUS‐Agarose (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Immunocomplexes were washed three times with lysis buffer, were re‐suspended in 2 × sodium dodecyl sulphate‐polyacrylamide gel electrophoresis sample buffer, boiled for 5 min and were fractionated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis followed by electro‐transferring to nitrocellulose membranes. Immunoblotting was performed by standard procedures as previously described (Li et al. 2002).
Normal rabbit immunoglobulin G and rabbit polyclonal antibodies against c‐Fos and JunD were obtained from Santa Cruz Biotechnology Inc. Rabbit polyclonal antibodies against phospho‐ERK1/2, ERK1/2 and poly(ADP‐ribose)polymerase (PARP) were from Cell Signalling Technology Inc. (Beverly, MA, USA). Mouse monoclonal antibody against c‐Fos was from Calbiochem (San Diego, CA, USA). Mouse monoclonal antibody against β‐actin was obtained from Sigma‐Aldrich Inc. (St. Louis, MO, USA). Horseradish peroxidase‐conjugated rabbit immunoglobulin G was from Amersham Biosciences (Pittsburgh, PA, USA). Specific signals were detected in an enhanced chemiluminescent procedure using a Supersignal West Pico kit (Pierce Biotechnology Inc., Rockford, IL, USA).
Nuclear extracts
Following treatment, cells were scraped gently into the medium, were pelleted, washed in ice‐cold phosphate‐buffered saline and were lysed on ice in four volumes of homogenization buffer [10 mm Hepes‐NaOH buffer (pH 7.9), 10 mm KCl, 1 mm EDTA, 1 mm ethylene glycol‐bis(2‐aminoethyl ether)N,N,N′N′‐tetraacetic acid (EGTA), 10 mm sodium fluoride, 10 mm sodium‐β‐glycerophosphate, 5 mm dithiothreitol (DTT) and the CompleteTM protease inhibitor cocktail (Sigma‐Aldrich Inc.)]. Following addition of Nonidet P‐40 at a final concentration of 0.6%, the homogenates were vortexed for 10 s constantly and were then incubated for 15 min on ice. After centrifugation at 16 430 g for 10 min at 4 °C, resulting pellets were re‐suspended with 200 mm Tris‐HCl buffer (pH 7.5) containing 400 mm KCl, 1 mm EDTA, 1 mm EGTA, 10 mm sodium fluoride, 10 mm sodium‐β‐glycerophosphate, 1 mm DTT, 0.5 mm phenylmethylsulphonyl fluoride and the above protease inhibitor cocktail. Samples were incubated for 30 min on ice and again were centrifuged at 16 430 g for 10 min at 4 °C. Protein concentrations of nuclear extracts obtained were determined by the bicinchoninic acid method and specimens were stored at –80 °C until ready for use.
Electrophoretic mobility shift assay
For electrophoretic mobility shift assay (EMSA), a double‐stranded, 21‐base‐pair oligonucleotide containing the consensus binding sequence of AP‐1 (5′‐CGC TTG ATG ACT CAG CCG GAA‐3′, Santa Cruz Biotechnology Inc.) was radiolabelled with [γ‐32P] deoxy ATP (~3.000 mCi/mmol) by T4 polynucleotide kinase (Promega, Madison, WI, USA) and was purified on a Sephadex G‐25 column according to directions provided (Sambrook & Russell 2002). Nuclear protein extracts (10 µg) were incubated in 19 µl of 10 mm Hepes‐KOH buffer (pH 7.9), containing 0.5 mg poly(dI‐dC), 0.1 mm EDTA, 75 mm NaCl, 4 mm MgCl2, 17.5% glycerol, 20% Ficoll 400 and 2 mm DTT on ice for 15 min. The binding reaction was initiated by the addition of 68.8 fmol (1 ng) of labelled probe to a total volume of 20 µl and was incubated on ice for 30 min. Bound and free oligonucleotides were electrophoretically separated on a native 4% polyacrylamide gel in 0.5 × Tris‐glycine running buffer (12.5 mm Tris, 96 mm glycine, 1.25 mm EDTA, pH 8.0) at a constant voltage of 10 V/cm for 2 h at room temperature. Gels were vacuum‐dried on filter membranes in a GD‐5040 gel‐dryer (SCIE‐PLAS, Warwickshire, UK) at 80 °C for 45 min. Dried gels were exposed in a phosphor imaging cassette overnight and then were scanned with a Storm 860 (Amersham Biosciences) laser scanner.
Competition experiments were performed by pre‐incubating 5 µg nuclear protein extracts derived from MEF cells 2 h after UV irradiation or H2O2 treatment with measured molar ratios of unlabelled AP‐1 specific oligonucleotides for 30 min on ice before adding a constant amount (0.5 ng) of the radioprobe. Furthermore, 0.5 ng of AP‐1 radioprobes containing mutant AP‐1 sequences were added to the same nuclear extracts without pre‐incubation and were loaded on the same gel.
Supershift assays were performed with specific antibodies against c‐Fos and JunD. In brief, 2 µg of antibodies and 10 µg nuclear extracts from MEF cells were pre‐incubated in the presence of binding buffer for 30 min on ice. The binding reaction was then initiated by adding 1 ng of radiolabelled AP‐1 specific oligoprobes and mutant probes.
Nuclear staining with acridine orange and ethidium bromide
Apoptotic cells were revealed by nuclear staining to detect DNA condensation. A dye mixture containing acridine orange (AO) and ethidium bromide (EB), each of which was present at 100 µg/ml (AO/EB), was added to MEF cells cultured in 35‐mm dishes at a final confluence of 50–60% after exposure to each stimulus. The cells were visualized under a UV‐fluorescence microscope. Cells with nuclei stained green had not lost membrane integrity, but in contrast, cells in which the nuclei were stained orange had damaged membranes. Apoptotic cells can be distinguished from non‐apoptotic cells on the basis of the presence or absence of nuclear condensation and DNA fragmentation (Wang et al. 2005). The extent of cell death was calculated as (number of orange) cells/(number of orange + number of green) cells × 100%. Ten visual fields (100×) were chosen randomly and a minimum of 1000 cells was scored.
Transfection
Plasmid pcMV‐c‐fos containing a c‐fos cDNA has previously been described (Zhang et al. 2002). The pcDNA3.1 junD plasmid was kindly provided by Dr Sunita K. Agarwal (National Institute of Health, Bethesda, MD, USA). MEF cells were transfected by electroporation using a Bio‐Rad Gene Pulser electroporator (Bio‐Rad Laboratories, Hercules, CA, USA). Cells (4 × 106) were washed, re‐suspended in 0.6 ml phosphate‐buffered saline and were placed in a 0.4‐cm gap electroporation cuvette (Bio‐Rad Laboratories). Plasmid DNA 10 µg was added. After brief incubation on ice, cells were subjected to a single electric pulse (975 µf, 280 V) followed by further incubation on ice for 10 min. They were then diluted to a concentration of 2 × 105 cells/ml with Dulbecco's modified Eagle's medium containing 10% foetal bovine serum and antibiotics. Thereafter, cells were incubated at 37 °C for 24 h prior to use.
MAPK inhibitor treatment
MEF cells were treated with PD98059 (Cell Signalling Technology Inc.), a specific inhibitor for MEK1, for an hour prior to UV irradiation and were allowed to recover for a period of time. Cells were collected subsequently for apoptosis and immunoblot assays.
Statistical analysis
Statistical significance of results was determined using Student's t‐test analysis.
RESULTS
Activation of c‐Fos/JunD AP‐1 transcription complexes in response to UV or H2O2
In MEF cells treated with UV or H2O2, we measured the expression and activity of c‐Fos and JunD by immunoblot and EMSA techniques, respectively. Basal levels of c‐Fos and JunD were very low but increased dramatically in response to UV irradiation or H2O2 treatment (Fig. 1a,b). Expression of both c‐Fos and JunD was time‐dependent, reaching its maximum at 2 h for c‐Fos and 2–4 h for JunD, decreasing modestly thereafter during the process of observation (Fig. 1a,b). Degree of induction was quantitatively significant, reaching at its peak 20‐fold (UV) and 6‐fold (H2O2) over untreated controls for c‐Fos and 5‐fold for JunD, respectively (Fig. 1a,b). As the components of the AP‐1 transcription complexes, c‐Fos and JunD function was via their AP‐1 binding activity. EMSA studies using nuclear extracts derived from MEF cells after exposure to UV or H2O2 showed markedly increased AP‐1 binding activities, which peaked at 2 h and decreased slowly during the following 4 h (Fig. 1c). To rule out the possibility of non‐specific binding, a labelled AP‐1 mutant oligonucleotide was used; this failed to bind to any of the AP‐1 transcription complexes (Fig. 1c). In addition, inclusion of increasing amounts of a cold AP‐1 oligonucleotide effectively competed out AP‐1 transcription complex binding to the radiolabelled probe (Fig. 1c). These results suggest that both UV and H2O2 were able to induce c‐Fos and JunD expression and the formation of new AP‐1 transcription complexes.
Figure 1.
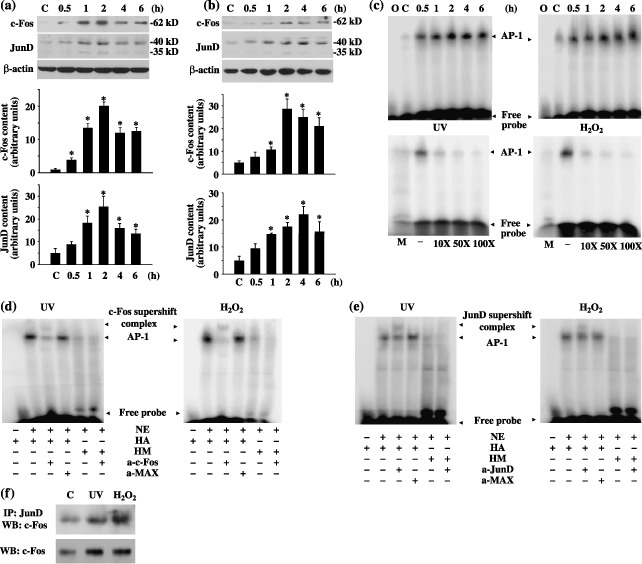
Stress‐induced c‐Fos and JunD activation involving activator protein‐1 (AP‐1) activation. (a) UV‐irradiation‐induced c‐Fos and JunD expression. (b) H2O2‐induced c‐Fos and JunD expression. For (a) and (b), mouse embryonic fibroblast (MEF) cells were treated with either ultraviolet (UV)‐C at 300 J/m2 or H2O2 at 10 mm for 15 min and harvested at various times, as indicated. Cell extracts were isolated and immunoblot was performed. Representative immunoblots and quantifications of at least three independent experiments are shown. C is untreated control sample. β‐Actin is the loading control. * statistically different (P < 0.05) compared to controls. (c) Stress‐induced AP‐1 activation. MEF cells were exposed to either UV or H2O2 and collected at various times. Nuclear extracts (10 µg) were isolated and mixed with a radioactive AP‐1 probe. O represents radiolabelled AP‐1 oligonucleotides only. C is untreated control sample. M represents nuclear extracts incubated with a radiolabelled mutant AP‐1 probe. – represents the absence of unlabelled oligonucleotides. (d) Supershift with a c‐Fos antibody. (e) Supershift with a JunD antibody. For (d) and (e), nuclear extracts (NE, 10 µg) from MEF (2 h of recovery time) was incubated in the presence (+) or absence (–) of the indicated antibodies (2 µg), followed by the binding reaction with 0.5 ng of the radiolabelled AP‐1 (HA) or mutant AP‐1 oligonucleotides (HM). MAX indicates the use of a negative control antibody. (f) Interaction of c‐Fos and JunD. MEF cells were exposed to either UV or H2O2 and collected after 2 h of recovery. c‐Fos expression was analysed in either anti‐JunD immunoprecipitates or whole cell lysates using a mouse monoclonal antic‐Fos antibody. C is untreated control sample.
To further determine the molecular identity and specificity of the AP‐1 transcription complexes, supershift assays were carried out by pre‐incubating the nuclear extracts with either the c‐Fos (Fig. 1d) or JunD (Fig. 1e) antibody. There was no change when the binding reaction was pre‐incubated with non‐specific MAX antibody. In addition, c‐Fos was detected in JunD immunocomplexes from untreated and UV‐ or H2O2‐treated samples (Fig. 1f). These results suggest that c‐Fos and JunD heterodimerized to form AP‐1 transcription complexes.
Cell death induced by UV or H2O2
To investigate the induction of cell death in response to UV and H2O2, we measured caspase 3 activities and nuclear morphology, respectively. PARP, a 116 kD protein and a cleavage target of activated caspase 3, served as a marker of apoptosis (Tewari et al. 1995). The 89kD product of PARP cleavage was detected within 4 h after UV or H2O2 treatment and the quantity of this fragment increased over a prolonged period of time (Fig. 2a). Moreover, increasing amount of the PARP cleavage product coincided with a steady decrease of the full length PARP protein (Fig. 2a). Activation of caspase 3 activity was, as expected, paralleled by development of apoptosis, documented here by the increasing aspect of nuclear condensation. Nuclear condensation was evaluated based on the extent of nuclear AO/EB staining, in which orange‐stained nuclei indicated dead cells. Exposure of MEF cells to UV or H2O2 progressively increased cell death, starting as early as 1 h post‐treatment and reaching a plateau between 8 and 24 h (Fig. 2b). Levels of apoptosis were below 20% in the first 4 h but rose sharply and dramatically after that, reaching nearly 70% (UV) or 50% (H2O2) by 24 h (Fig. 2b). These results demonstrate that both UV and H2O2 are capable of inducing apoptosis in MEF cells.
Figure 2.
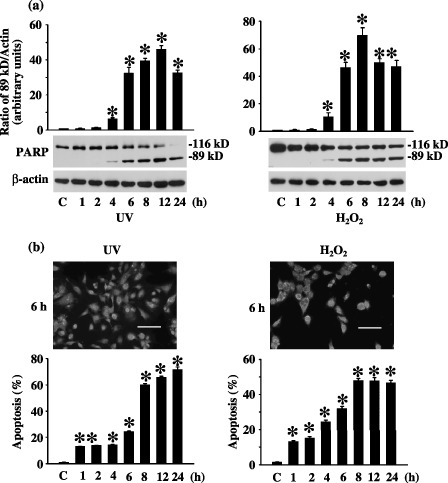
Cell death induced by ultraviolet (UV) or H2O2. (a) Stress‐induced caspase 3 activation. Cleavage of PARP was used as one measure of cell death and was performed by immunoblot of total poly(ADP‐ribose)polymerase (PARP) (116 kD) and the large cleavage fragment (89 kD) after cells were treated with UV or H2O2. Quantification of the densitometric ratio of the 89 kD PARP fragment and β‐actin is presented, as well as the representative Western blotting results. C is untreated control sample. β‐Actin is the loading control. (b) Stress‐induced nuclear condensation. Percentage of mouse embryonic fibroblast (MEF) apoptosis after UV or H2O2 treatment was measured by acridine orange (AO)/ethidium bromide (EB) nuclear staining. Cells stained in orange (apoptotic cells) and green (living cells) were scored under a fluorescence microscope. To the top are representatives of the photographs. To the bottom is the quantification. The scale bar is 100 µm. C is untreated control samples. * statistically different (P < 0.05, n = 3) compared to controls.
Effect of overexpression of JunD alone or both c‐Fos and JunD on stress‐induced cell death
To further determine the role of c‐Fos and JunD in cell death induced by stress signals, we transfected cells with a eukaryotic expression vector containing either a full length junD cDNA or cDNAs for both c‐fos and junD before exposing the cells to UV or H2O2. Overexpression of JunD alone or both c‐Fos and JunD resulted in no change in the amount of total PARP (cleaved plus uncleaved) but significant changes in cleaved PARP. As shown in Fig. 3(a), 4 h after UV stimulation, compared to non‐transfected controls, there was in the range of 65% and 80% decrease in PARP cleavage in JunD overexpressing or c‐Fos plus JunD overexpressing MEF cells, respectively. Moreover, 4 h after H2O2 treatment, PARP cleavage decreased by around 75% and 50% in JunD overexpressing and c‐Fos plus JunD overexpressing cells, compared to non‐transfected controls, respectively (Fig. 3b).
Figure 3.
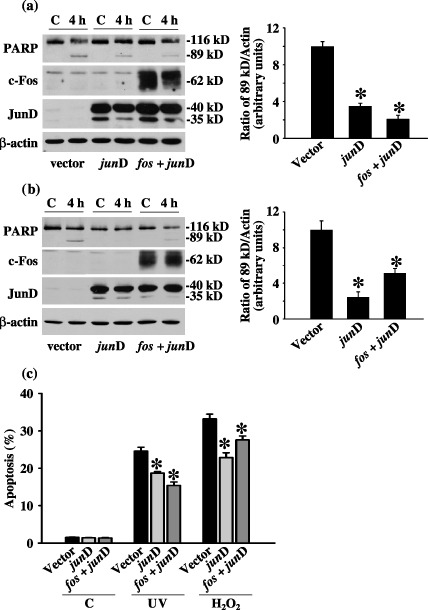
Effect of overexpressing JunD or c‐Fos plus JunD on ultraviolet (UV)‐ and H2O2‐induced cell death. (a) Effect of overexpressing JunD and c‐Fos plus JunD on UV‐induced caspase 3 activation. (b) Effect of overexpressing JunD and c‐Fos plus JunD on H2O2‐induced caspase 3 activation. Cleavage of poly(ADP‐ribose)polymerase (PARP) as well as expression of c‐Fos and JunD was detected in cells 4 h after UV or H2O2 treatment. Mouse embryonic fibroblast (MEF) cells were transfected with either 10 µg of the pcDNA4 plasmid (vector), junD plasmid (junD) alone or both c‐fos and junD plasmids (fos+junD) for 24 h before they were exposed to UV or H2O2. C is untreated control sample. Shown are the results of a typical experiment and the quantification of at least three independent experiments. (c) Effect of overexpressing JunD and c‐Fos plus JunD on stress‐induced cell death. Percentage of apoptosis 6 h after UV irradiation or H2O2 treatment was measured by AO/EB nuclear staining. C indicated untreated control samples. * statistically different (P < 0.05, n = 3) compared to controls in respective UV‐ or H2O2‐treated groups.
To further substantiate these findings, we performed AO/EB nuclear staining after cells had recovered for 6 h from either UV or H2O2 treatment (Fig. 3c). Interestingly, overexpression of JunD or c‐Fos plus JunD did not affect cell death in untreated cells, but reduced apoptosis in UV‐ or H2O2‐treated cells. The results are consistent with those of the caspase 3 activity analysis (Fig. 3a,b). Together, these results demonstrate that MEF cells overexpressing c‐Fos and JunD underwent cell death after either UV or H2O2 stimulation, as would normal cells. More importantly, our data suggest a protective role for c‐Fos/JunD against stress‐induced cell death.
Effect of suppressing ERK activation on c‐Fos and JunD expression induced by UV
To investigate whether UV irradiation induces ERK1/2 activity in MEF cells, the effect of UV irradiation on phospho‐ERK1/2 expression was measured. As shown in Fig. 4(a), basal levels of phospho‐ERK1/2 were very low but increased significantly in response to UV irradiation. Activation of ERK1/2 was time‐dependent, with phospho‐ERK1/2 levels peaking at 15 min, decreasing significantly during the next 15 min and returning to baseline by 6 h.
Figure 4.
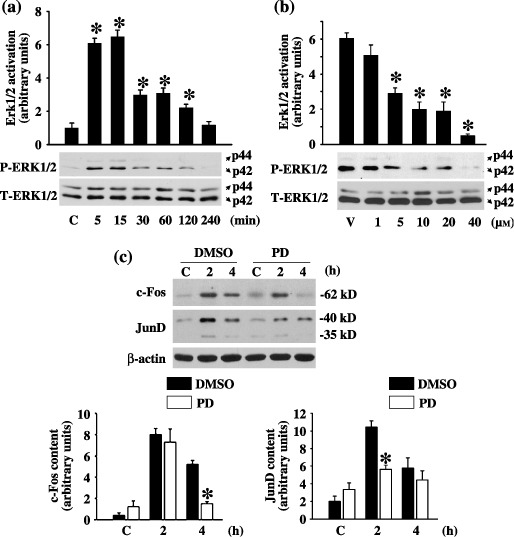
Effect of inhibiting ultraviolet (UV)‐induced ERK on activation of c‐Fos and JunD. (a) UV‐induced ERK1/2 activation. Phosphorylation of ERK1/2 was determined by immunoblot of whole cell lysates collected at the indicated times post‐UV irradiation with an antibody that specifically recognizes the phosphorylated Thr202 and Tyr204 residues. C represents untreated control samples. Total ERK1/2 from the corresponding cell lysates was determined by immunoblot with an ERK antibody. Quantification of at least three independent experiments as well as the representative Western blotting results is shown. * statistically different (P < 0.05) compared to untreated controls. (b) Effect of MEK1 inhibition on UV‐induced ERK1/2 activation. Mouse embryonic fibroblast (MEF) cells were treated with different doses of PD98059 for 1 h prior to UV irradiation. MEF cells were collected 15 min after UV stimulation. V represents dimethyl sulphoxide (DMSO) vehicle control. Quantification of at least three independent experiments as well as the representative Western blotting results is shown. * statistically different (P < 0.05) compared to vehicle control. (c) Effect of MEK1 inhibition on UV‐induced of c‐Fos and JunD expression. DMSO is the vehicle. C represents without UV irradiation after PD98059 pre‐treatment. Representative Western blotting results are to the top and quantification of at least three independent experiments is at the bottom. * statistically different (P < 0.05) compared to DMSO pre‐treated samples at each time point.
In the MAPK/ERK signalling pathway, ERK1/2 is directly activated by MAPK kinase (MEK1). The effect of suppression of ERK1/2 activation on UV‐induced up‐regulation of c‐Fos and JunD was studied by treating the cells with a selective MEK1 inhibitor PD98059. PD98059 was added to the culture medium 1 h prior to UV irradiation. We tried different doses of PD98059 (0, 1 µm, 5 µm, 10 µm, 20 µm and 40 µm) and we found that 10 µm of PD98059 was sufficient to inhibit UV‐induced ERK1/2 activation (~70% decrease compared with dimethyl sulphoxide vehicle control at 15 min after UV irradiation). Inhibition of ERK1/2 activity decreased both c‐Fos and JunD activation compared to dimethyl sulphoxide pre‐treated samples (Fig. 4c). Induction of c‐Fos was reduced from 20‐ to 6‐fold at 2 h and from 13‐fold to almost equal to control levels at 4 h, and the induction of JunD was reduced from 5‐ to 2‐fold at 2 h and from 3‐fold to almost equal to control levels at 4 h.
Effect of inhibiting MEK1 on UV‐induced cell death
To investigate the involvement of the ERK1/2 signalling pathway in UV‐induced cell death, we pre‐treated MEF cells for 1 h with PD98059. We evaluated any cellular apoptotic response to UV irradiation in the presence or absence of ERK1/2 inhibition, based on caspase 3 activities. Pre‐treatment with PD98059 did not change the amount of total PARP (cleaved plus uncleaved) but significantly enhanced the level of cleaved PARP after UV irradiation (Fig. 5a). In addition, as determined using AO/EB nuclear staining, pre‐treatment with PD98059 did not affect cell death in untreated cells, but increased apoptosis by as early as 1 h, sustaining it until 24 h after UV irradiation (Fig. 5b).
Figure 5.
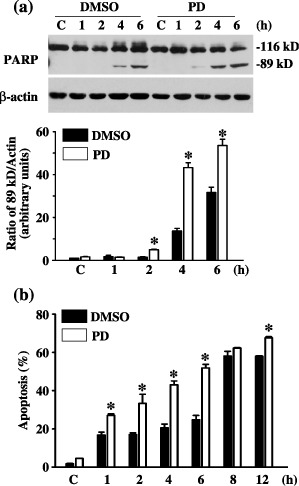
Effect of inhibiting ERK on ultraviolet (UV)‐induced cell death. (a) Effect of MEK1 inhibition on UV‐induced caspase 3 activity. Cleavage of poly(ADP‐ribose)polymerase (PARP) was measured with immunoblot. Dimethyl sulphoxide (DMSO) is the vehicle. C represents without UV stimulation after PD98059 pre‐treatment. Shown here are the results of a typical experiment and the quantification of the densitometric ratio of 89kD PARP fragment and β‐actin. (b) Effect of MEK1 inhibition on UV‐induced cell death. Percentage of cellular apoptosis was measured with acridine orange (AO)/ethidium bromide (EB) nuclear staining. C represents without UV stimulation after PD98059 pre‐treatment. * statistically different (P < 0.05, n = 3) compared to DMSO controls of different time points.
DISCUSSION
In the present study, we have investigated the role of c‐Fos and JunD in stress‐induced cell death. We have demonstrated that exposure of primary MEF cells to UV irradiation or H2O2 significantly increased the activation of c‐Fos/JunD containing AP‐1 transcription complexes and induced caspase 3 activities and cell death. However, overexpression of JunD or JunD together with c‐Fos decreased caspase 3 activity and cell death, suggesting an anti‐apoptotic role of these proteins. We found that UV treatment induced increases of not only the transcriptional activities of c‐Fos and JunD, but also their protein levels, and that these increases preceded initial detection of caspase 3 activity and the eruption of cell death. Because we found that the levels of AP‐1, c‐Fos and JunD complexes increased greatly in response to UV irradiation, and that overexpression of c‐Fos and JunD prevented cell death, we concluded that c‐Fos and JunD play an antagonistic role in UV‐induced cell death.
Our findings are consistent with previous studies showing that UV induces synthesis of c‐fos, c‐jun, junB and junD mRNA and activation of AP‐1 that accompany incidence of cell death (Dosch & Kaina 1996). However, previous studies suggest that c‐fos/c‐jun is up‐regulated in stress‐induced apoptosis, whereas exogenous inhibition of c‐fos and c‐jun mRNA synthesis attenuated cell death (Colotta et al. 1992; Moreno‐Manzano et al. 1999). FosB and JunD containing AP‐1 tanscription complexes are activated and apoptosis is induced in human hepatoma cells treated with TGF‐β1. In addition, ectopic overexpression of FosB and JunD enhances TGF‐β1 induced apoptosis (Yamamura et al. 2000). Based on these findings and ours, it is likely that different combinations of Fos and Jun protein play different roles in stress‐induced cell death. Furthermore, light‐induced retinal apoptosis is correlated to up‐regulation of AP‐1 containing c‐Fos and JunD (Hafezi et al. 1999b). However, whereas c‐Fos is absolutely required for the apoptotic program in these cells (Hafezi et al. 1997), JunD appears to be dispensable (Hafezi et al. 1999a). This finding together with ours suggests that the function of AP‐1 transcription factors in cell death is highly stimulus and cell type‐specific (Shaulian & Karin 2002; Eferl & Wagner 2003). Together, these observations highlight the complexity of AP‐1 functions in cell death and caution against the extrapolation of general principles from single experimental system.
Our studies show that H2O2 treatment has similar effects on cells as UV irradiation. Both the protein expression of c‐Fos and JunD and the activity of c‐Fos/JunD containing AP‐1 transcription factors peaked at around 2 h after UV irradiation or H2O2 treatment. Under both stress conditions, caspase 3 activity, as determined by PARP cleavage, was not detectable until 4 h after treatment. UV‐ or H2O2‐induced cell death could be detected as early as 1 h and reached plateaus between 8 and 24 h. Levels of apoptosis were below 20% in the first 4 h but rose sharply afterwards. Overexpression of JunD alone or c‐Fos plus JunD attenuated UV‐ or H2O2‐induced caspase 3 activity and cell death. Vollgraf et al. showed that H2O2‐induced onset and execution of programmed cell death in mature rat brain oligodendrocytes is accompanied by the increased expression of c‐fos, c‐jun and junD mRNA and the induction of AP‐1 complexes consist of c‐Fos, c‐Jun, JunD and JunB proteins. Furthermore, their findings suggest that the activation of AP‐1 by H2O2 treatment might contribute to signal transduction pathways promoting cell survival (Vollgraf et al. 1999). In agreement with this report, we have directly demonstrated that overexpression of c‐Fos and JunD protected cells from H2O2‐induced cell death.
We have demonstrated that ERK1/2 was activated in response to UV irradiation. Moreover, inhibition of MEK1/ERK decreased expression of c‐Fos and JunD and increased cell death upon UV irradiation. Together, these results suggest that MEK1/ERK mediates c‐Fos and JunD induction. UV irradiation activates cell surface growth factor receptors (Sachsenmaier et al. 1994; Huang et al. 1996; Rosette & Karin 1996) and meanwhile, reactive oxygen intermediates serve as second messengers to activate growth factor receptors in the UV response (Huang et al. 1996). Furthermore, ERK1/2, c‐Fos and JunD can be activated by growth factor binding (Muller et al. 1984; Boulton et al. 1991; Strong et al. 1994). It is possible that UV, mediated by H2O2, activates growth factor receptors and ERK1/2 resulting in expression of c‐Fos/JunD, safeguarding cells from death; further investigation is necessary to testify this hypothesis. Conversely, previous studies have shown that junD expression activated by UV via MEK1/ERK signalling pathway mediates apoptosis in ML‐1 cells (Li et al. 2002). These opposing findings are probably due to different cell types used.
Despite the above unresolved issues, our results indicate that, in primary MEF cells, UV irradiation induces activation of c‐Fos/JunD and this activation is mediated by the MEK1/ERK1/2 signalling pathway. In addition, we show that UV‐ or H2O2‐induced activation of c‐Fos/JunD prevents cell death. These findings extend our understanding of the function of immediate early genes in response to extracelluar stress‐induced cell death.
ACKNOWLEGEMENTS
We would like to thank members in our joint laboratory for helpful discussions. We are also grateful to Dr Basil Rigas and Dr Jennie L. Williams for their invaluable suggestions and critiques. In addition, we thank Xueyu Chen and Taylor B. Guo for their assistance throughout the process of this work. This study was supported in part by core funding from the Chinese Academy of Sciences (CAS), a One Hundred Talent Grant from CAS to M.X., the E‐Institutes of Shanghai Municipal Education Commission Project Number E03003, the Science and Technology Commission of Shanghai Municipality (04DZ14902), a grant from the Shanghai Education Commission (03BZ03) and a National Key Program grant (973) (NO2002CB512805).
REFERENCES
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD (1991) ERKs: a family of protein‐serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65, 663–675. [DOI] [PubMed] [Google Scholar]
- Buttke TM, Sandstrom PA (1994) Oxidative stress as a mediator of apoptosis. Immunol. Today 15, 7–10. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40. [DOI] [PubMed] [Google Scholar]
- Colotta F, Polentarutti N, Sironi M, Mantovani A (1992) Expression and involvement of c‐fos and c‐jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J. Biol. Chem. 267, 18278–18283. [PubMed] [Google Scholar]
- Dhanwada KR, Dickens M, Neades R, Davis R, Pelling JC (1995) Differential effects of UV‐B and UV‐C components of solar radiation on MAP kinase signal transduction pathways in epidermal keratinocytes. Oncogene 11, 1947–1953. [PubMed] [Google Scholar]
- Dosch J, Kaina B (1996) Induction of c‐fos, c‐jun, junB and junD mRNA and AP‐1 by alkylating mutagens in cells deficient and proficient for the DNA repair protein O6‐methylguanine‐DNA methyltransferase (MGMT) and its relationship to cell death, mutation induction and chromosomal instability. Oncogene 13, 1927–1935. [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003) AP‐1: a double‐edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Grimm C, Wenzel A, Abegg M, Yaniv M, Reme CE (1999a) Retinal photoreceptors are apoptosis‐competent in the absence of JunD/AP‐1. Cell Death Differ. 6, 934–936. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Marti A, Grimm C, Wenzel A, Reme CE (1999b) Differential DNA binding activities of the transcription factors AP‐1 and Oct – 1 during light‐induced apoptosis of photoreceptors. Vision Res. 39, 2511–2518. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Steinbach JP, Marti A, Munz K, Wang ZQ, Wagner EF, Aguzzi A, Reme CE (1997) The absence of c‐fos prevents light‐induced apoptotic cell death of photoreceptors in retinal degeneration in vivo . Nat. Med. 3, 346–349. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Meth. Enzymol. 186, 1–85. [DOI] [PubMed] [Google Scholar]
- Huang RP, Wu JX, Fan Y, Adamson ED (1996) UV activates growth factor receptors via reactive oxygen intermediates. J. Cell Biol. 133, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackinger D, Kaina B (2000) Primary mouse fibroblasts deficient for c‐Fos, p53 or for both proteins are hypersensitive to UV light and alkylating agent‐induced chromosomal breakage and apoptosis. Mutat. Res. 457, 113–123. [DOI] [PubMed] [Google Scholar]
- Lakshminarayanan V, Drab‐Weiss EA, Roebuck KA (1998) H2O2 and tumor necrosis factor‐α induce differential binding of the redox‐responsive transcription factors AP‐1 and NF‐κB to the interleukin‐8 promoter in endothelial and epithelial cells. J. Biol. Chem. 273, 32670–32678. [DOI] [PubMed] [Google Scholar]
- Li T, Dai W, Lu L (2002) Ultraviolet‐induced junD activation and apoptosis in myeloblastic leukaemia ML‐1 cells. J. Biol. Chem. 277, 32668–32676. [DOI] [PubMed] [Google Scholar]
- Marti A, Jehn B, Costello E, Keon N, Ke G, Martin F, Jaggi R (1994) Protein kinase A and AP‐1 (c‐Fos/JunD) are induced during apoptosis of mouse mammary epithelial cells. Oncogene 9, 1213–1223. [PubMed] [Google Scholar]
- Mendes AF, Caramona MM, Carvalho AP, Lopes MC (2003) Hydrogen peroxide mediates interleukin‐1β‐induced AP‐1 activation in articular chondrocytes: implications for the regulation of iNOS expression. Cell Biol. Toxicol. 19, 203–214. [DOI] [PubMed] [Google Scholar]
- Moreno‐Manzano V, Ishikawa Y, Lucio‐Cazana J, Kitamura M (1999) Suppression of apoptosis by all‐trans‐retinoic acid. Dual intervention in the c‐Jun n‐terminal kinase‐AP‐1 pathway. J. Biol. Chem. 274, 20251–20258. [DOI] [PubMed] [Google Scholar]
- Muller R, Bravo R, Burckhardt J, Curran T (1984) Induction of c‐fos gene and protein by growth factors precedes activation of c‐myc. Nature 312, 716–720. [DOI] [PubMed] [Google Scholar]
- Price MA, Cruzalegui FH, Treisman R (1996) The p38 and ERK MAP kinase pathways cooperate to activate Ternary Complex Factors and c‐fos transcription in response to UV light. EMBO J. 15, 6552–6563. [PMC free article] [PubMed] [Google Scholar]
- Rosette C, Karin M (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274, 1194–1197. [DOI] [PubMed] [Google Scholar]
- Sachsenmaier C, Radler‐Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ (1994) Involvement of growth factor receptors in the mammalian UVC response. Cell 78, 963–972. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW (2002) Molecular Cloning: A Laboratory Manual, 3rd edn. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Scharffetter‐Kochanek K, Wlaschek M, Brenneisen P, Schauen M, Blaudschun R, Wenk J (1997) UV‐induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem. 378, 1247–1257. [PubMed] [Google Scholar]
- Schreiber M, Baumann B, Cotten M, Angel P, Wagner EF (1995) Fos is an essential component of the mammalian UV response. EMBO J. 14, 5338–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP‐1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136. [DOI] [PubMed] [Google Scholar]
- Strong DD, Merriman HL, Landale EC, Baylink DJ, Mohan S (1994) The effects of the insulin‐like growth factors and transforming growth factor beta on the Jun proto‐oncogene family in MC3T3–E1 cells. Calcif. Tissue Int. 55, 311–315. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog of CED‐3, is a CrmA‐inhibitable protease that cleaves the death substrate poly (ADP‐ribose) polymerase. Cell 81, 801–809. [DOI] [PubMed] [Google Scholar]
- Vollgraf U, Wegner M, Richter‐Landsberg C (1999) Activation of AP‐1 and nuclear factor‐κB transcription factors is involved in hydrogen peroxide‐induced apoptotic cell death of oligodendrocytes. J. Neurochem. 73, 2501–2509. [DOI] [PubMed] [Google Scholar]
- Wang L, Reinach P, Lu L (2005) TNF‐α promotes cell survival through stimulation of K+ channel and NF‐κB activity in corneal epithelial cells. Exp. Cell Res. 311, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman JB, Fiette L, Matsuo K, Yaniv M (2000) JunD protects cells from p53‐dependent senescence and apoptosis. Mol. Cell 6, 1109–1119. [DOI] [PubMed] [Google Scholar]
- Yamamura Y, Hua X, Bergelson S, Lodish HF (2000) Critical role of Smads and AP‐1 complex in transforming growth factor‐β‐dependent apoptosis. J. Biol. Chem. 275, 36295–36302. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M (2002) c‐fos Regulates neuronal excitability and survival. Nat. Genet. 30, 416–420. [DOI] [PubMed] [Google Scholar]