

Abstract
Objectives: This study was performed to explore the strategy of combining Chk1 inhibitors with ionizing radiation (IR) to selectively target p53‐deficient cancer cells.
Materials and methods: Survival and cell cycle progression were measured in response to IR and the Chk1 inhibitors, UCN‐01 and CEP‐3891, in colon carcinoma HCT116 p53+/+ and p53−/− cells, and in osteosarcoma U2OS‐VP16 cells with conditional expression of dominant‐negative p53 (p53DD).
Results: Clonogenic survival was selectively reduced in HCT116 p53−/− compared to p53+/+ cells after treatment with UCN‐01 and IR, and HCT116 p53+/+ cells also displayed strong p53‐dependent G1 arrest in the 1st cell cycle after IR. In contrast, clonogenic survival was affected similarly in U2OS‐VP16 cells with and without expression of p53DD. However, death of U2OS‐VP16 cells was p53 dependent as assessed by cell viability assay at 72 h, and this was associated with p53‐dependent G1 arrest in the 2nd cell cycle after treatment. Notably, HCT116 cells were overall more resistant than U2OS cells to cytotoxic effects of Chk1 inhibitors.
Conclusion: Our results suggest that p53‐dependent G1 arrest in both 1st and 2nd cell cycles may protect human cancer cells from cell death after treatment with IR and Chk1 inhibitors. However, a challenge for future clinical use will be that different cancers display different intrinsic sensitivity to such inhibitors.
Introduction
A common goal for cancer treatment is to achieve selective killing of cancer cells. In recent years, a hypothesis has emerged that abrogation of the G2 checkpoint would selectively sensitize p53‐negative cancer cells to DNA damaging agents (1, 2, 3). The rationale for this hypothesis is that p53‐negative cancer cells would depend more on the G2 checkpoint for repairing DNA damage compared to p53‐positive cells as they lack the p53‐dependent G1 checkpoint (4, 5). As p53 is mutated in about 50% of all human cancers, such a strategy could potentially be of major clinical importance.
The hypothesis that abrogation of the G2 checkpoint would selectively target p53‐negative cells has stimulated development of several small molecule compounds that have efficacy towards human checkpoint kinase 1 (Chk1), a key regulator of the G2 checkpoint (1, 2, 6, 7). Several Chk1 inhibitors have been tested in pre‐clinical studies and some are currently in phase I and phase II clinical trials in combination with chemotherapeutic agents such as cisplatin, irinotecan and gemcitabine (2). Nevertheless, the original hypothesis has not yet been vigorously proven. Only few studies have included isogenic cell pairs that differ only in p53 status, and results of some studies have not supported the hypothesis (8, 9, 10). Moreover, while most pre‐clinical studies, and all phase I/II clinical trials have been performed with Chk1 inhibitors in combination with chemotherapeutic agents, fewer studies have addressed the effects of Chk1 inhibitors combined with ionizing radiation (IR). It is therefore not clear whether Chk1 inhibitors combined with radiation therapy would be a useful strategy to selectively target p53‐defective cancer cells.
To explore this issue, we have studied effects of two distinct Chk1 inhibitors, UCN‐01 and Cep‐3891, combined with IR on two different isogenic human cancer cell systems: HCT116 p53+/+ and p53−/− colon carcinoma cells, and U2OS‐VP16 osteosarcoma cells with conditional expression of p53DD, to suppress p53 transactivation capacity.
Materials and methods
Cell lines, drugs, siRNA treatment and irradiation
Human osteosarcoma U2OS‐VP16 cells expressing p53DD in a tetracycline‐dependent manner (11) were grown in DMEM with 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 2 μg/ml tetracycline (Calbiochem, San Diego, CA, USA). p53DD is a small fragment containing codons 1–13 and 302–390 of wt p53, that binds to endogenous wt p53 and suppresses p53 transactivation capacity (12). Expression of p53DD was induced by culturing the cells in absence of tetracycline for 24 h. HCT116p53+/+ and HCT116 p53−/− cells (13) were grown in DMEM medium. CEP‐3891 Chk1‐inhibitor was provided by Cephalon Inc. (Frazer, PA, USA). UCN‐01 was a gift from R.J. Schultz (National Cancer Institute). Nocodazole (Calbiochem) was used at 0.04 μg/ml. p21 small interfering RNA (siRNA) oligonucleotide sequences were purchased from Dharmacon (Smartpool reagent); Oligofectamine reagent (Invitrogen) was used for transfection. IR was delivered by X‐ray generators (Pantak, Berkshire, UK; HF160; 150 kV; 15 mA; dose rate 2.18 Gy/min, and Siemens, Munich, Germany; 200 kV; 20 mA; dose rate 1.6 Gy/min).
Antibodies and immunohistochemistry
Immunoblotting was performed as described previously (14). Antibodies to p21 and p53 were from Santa Cruz Inc, Santa Cruz, CA, USA, and phospho‐histone H3 from Millipore. The antibody to Mcm7 (DCS‐141) has been described previously (14).
Flow cytometry
To assay cell cycle distribution by DNA content, trypsinized cells were fixed in 70% ethanol, stained with 0.1 mg/ml propidium iodide, and analysed using a FACSCalibur flow cytometer (BD Biosciences, Stockholm, Sweden) with CellQuest software. For two‐parameter flow cytometric analysis to determine number of mitotic cells, fixed cells were incubated for 5 min in 50 μl detergent buffer (0.1% Igepal CA‐630, 6.5 mm Na2HPO4, 1.5 mm KH2PO4, 2.7 mm KCl, 137 mm NaCl, 0.5 mm EDTA, pH 7.5) with 4% (w/v) non‐fat milk, followed by 1 h incubation at room temperature with anti‐phospho H3 (diluted 1:500 in detergent buffer) then 30 min with fluorescein‐conjugated anti‐rabbit immunoglobin (Alexa Fluor 488 anti IgG from Molecular Probes [Invitrogen, Oslo, Norway], diluted 1:500 in detergent buffer). Cells were then stained with Hoechst 33258 (1.5 μg/ml) and analysed using an LSR II Flow cytometer (Becton Dickinson, San Jose, CA, USA) using CellQuest Pro software. To assay cell viability, live cells were stained with Sytox green (0.5 μm Sytox green nucleic acid stain, Molecular probes) and incubated for 10 min, then analysed using the FACSCalibur flow cytometer.
Clonogenic survival assays
Briefly, between 150 and 2000 cells (depending on treatment, to yield 50–100 colonies per dish) were seeded into 6 cm dishes, incubated for around 24 h, and treated with Cep‐3891 (0–500 nm) or UCN‐01 (0–300 nm) and IR (0–6 Gy). The Chk1 inhibitors were added to the culture medium before IR. After 24 or 72 h, medium was removed, cells were washed once with PBS and regular DMEM was added. Cells were cultured for another 12–13 days and stained with crystal violet or methylene blue. Colonies containing more than 50 cells were counted as survivors. Survival fractions were calculated in each experiment as average cloning efficiency (from three parallel dishes) after treatment with IR and Chk1 inhibitors divided by average cloning efficiency for non‐irradiated cells.
Results
To compare effects of G2 checkpoint abrogation in cancer cells that only differ with respect to p53 status, we employed two isogenic human cancer cell systems: HCT116 p53+/+ and p53−/− colon carcinoma cells (13) and U2OS‐VP16 osteosarcoma cells with conditional expression of p53DD, to suppress p53 transactivating capacity (11). Consistent with lack of p53 function, HCT116 p53−/− cells and U2OS‐VP16 cells expressing p53DD lacked induction of p21 in response to IR (Fig. 1a,b). The G2 checkpoint was induced in both systems as cells with G2/M DNA content accumulated after IR (Fig. 1c,d). However, HCT116 p53+/+ cells showed strong p53‐dependent G1 arrest in the first cycle after IR (Fig. 1c), while U2OS‐VP16 cells failed to induce such G1 arrest (Fig. 1d). Lack of normal G1 arrest in the first cycle after IR in U2OS‐VP16 cells, despite wild‐type p53 status, is consistent with previous findings for many other cancer cell lines and may be due to other cancer‐associated defects in the p53 pathway (15).
Figure 1.
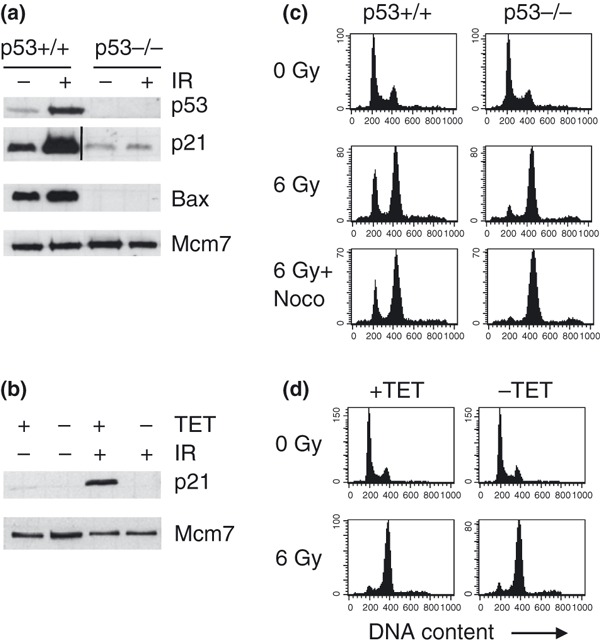
HCT116 and U2OS‐VP16 cell systems. (a) p53, p21 and Bax protein levels in extracts prepared from HCT116 p53+/+ and HCT116 p53−/− cells at 20 h after IR (0 or 6 Gy). Mcm7 demonstrates equal protein input. (The line in p21 blot marks that an excessive lane was cut from this image.).(b) p21 protein levels in extracts prepared from U2OS‐VP16‐p53DD cells at 20 h after IR (0 or 6 Gy). The U2OS‐VP16‐p53DD cells were cultured in the absence (−TET) or presence (+TET) of tetracycline, for 24 h to induce or prevent expression of p53DD respectively. (c) Measurement of 1st‐cycle G1 arrest in HCT116 cell system. HCT116 p53+/+ and p53−/− cells were treated with IR (6 Gy) or IR plus nocodazole (6 Gy + Noco), and harvested at 20 h after treatment. Flow cytometric analysis after staining with propidium iodide was used to obtain DNA histograms. (d) Measurement of 1st‐cycle G1 arrest in U2OS‐VP16‐p53DD cell system. U2OS‐VP16‐p53DD cells were cultured in presence (+TET) or absence (−TET) of tetracycline and DNA histograms, were obtained as in C.
To address whether Chk1 inhibitors combined with IR, in principle, would be a useful strategy to selectively target p53‐deficient cancer cells, we chose to use Chk1 inhibitors, UCN‐01 and CEP‐3891, which are distinct inhibitors of Chk1 known to abrogate the G2 checkpoint (5, 16, 17). Both inhibitors readily abrogated the IR‐induced G2 checkpoint in HCT116 and U2OS cell systems as measured by cell cycle analysis at 20 h after a dose of 6 Gy (Fig. 2a,b and data not shown). To detect smaller differences in rate of G2 checkpoint abrogation, we also assayed number of cells that reached mitosis within 8 h after treatment with UCN‐01 and IR. In these experiments, the microtubule inhibitor, nocodazole, was added at 2 h to allow quantification of accumulating cells that escaped the G2 checkpoint between 2 and 8 h after IR. In HCT116 cells, G2 checkpoint abrogation by UCN‐01 appeared partly dependent on p53 as there were significantly more p53−/− cells compared to p53+/+ cells that had reached mitosis at 8 h (Fig. 2c; two‐tailed P‐values 0.004, 0.03 and 0.0003 for 50, 100 and 300 nm UCN‐01 respectively). On the other hand, no significant dependency on p53 was found in U2OS‐VP16 cells (Fig. 2d; two‐tailed P‐values 0.18, 0.11 and 0.41 for 50, 100 and 300 nm UCN‐01 respectively).
Figure 2.
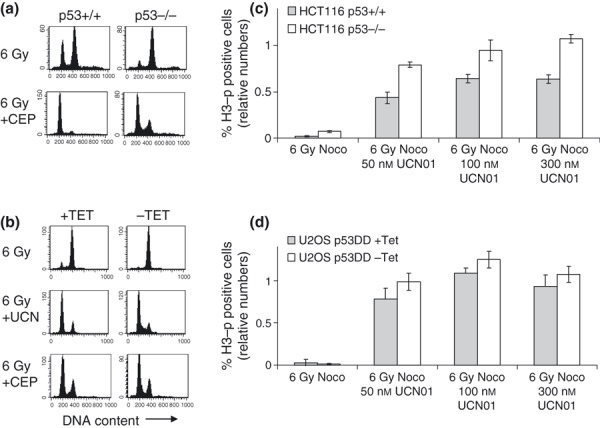
Abrogation of IR‐induced G 2 checkpoint by UCN01 and CEP‐3891. (a) DNA histograms of HCT116 p53+/+ and p53−/− cells shown at 20 h after treatment with IR (6 Gy) or IR plus 500 nm CEP‐3891 (6 Gy + CEP). (b) DNA histograms of U2OS‐VP16‐p53DD cells cultured in presence (+TET) or absence (−TET) of tetracycline shown at 20 h after IR (6 Gy), IR plus 300 nm UCN‐01 (6 Gy + UCN), or IR plus 500 nm CEP‐3891 (6 Gy + CEP). (c) Abrogation of G2 checkpoint in HCT116 p53+/+ and p53−/− cells measured at 8 h after indicated treatments. Nocodazole (Noco) was added to culture medium at 2 h. Cells were stained with Hoechst and antibody to phospho‐histone H3 and analysed by flow cytometry. Numbers of phospho‐H3‐positive cells relative to number of phospho‐H3‐positive cells after treatment with nocodazole in absence of IR are shown. Error bars represent standard errors from three independent experiments. (d) Similar results as in C shown for U2OS‐VP16‐p53DD cells cultured in presence (+TET) or absence (−TET) of tetracycline.
These results (Fig. 2c) were not an artefact due to differences in G1 or S‐phase arrest between HCT116 p53+/+ and p53−/− cells, as dividing number of mitotic cells by number of cells with G2/M DNA content in each sample did not alter the results (data not shown). Thus, rate of G2 checkpoint abrogation appeared partly dependent on p53 in the HCT116 cell system, in agreement with a previous report (13).
Cell survival of IR‐treated cancer cell lines is commonly measured by clonogenic survival assays, which are known to correlate well with in vivo responses after IR (18, 19). In the absence of IR, clonogenic survival of HCT116 p53−/− cells was more reduced than of HCT116 p53+/+ cells after treatment with UCN‐01 (300 nm) (Fig. 3a), although this difference was not quite statistically significant (two‐tailed P‐value 0.07). In irradiation experiments, HCT116 p53−/− cells were selectively radiosensitized after combined treatment with UCN‐01 (300 nm) and IR, when UCN‐01 was added immediately before IR and present in the culture medium for 3 days (Fig. 3b,c). Thus, survival parameter D10, which is radiation dose yielding 10% survival, was 4.6 and 3.8 Gy for ‘p53−/− mock’ and ‘p53−/− UCN‐01’ cells respectively (Fig. 3c). In contrast to p53‐deficient HCT116 cells (Fig. 3c), virtually no effect of Chk1 inhibition was found when p53 was functional in the same cellular background, with D10 values of 3.9 and 3.8 Gy for the ‘p53+/+ mock’ and ‘p53+/+ UCN‐01’ respectively (Fig. 3b).
Figure 3.
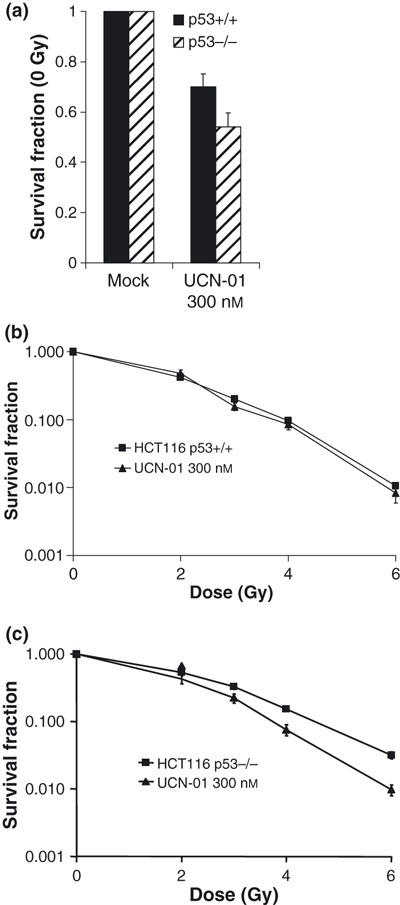
Clonogenic survival assays of IR‐treated HCT116 p53+/+ and p53−/− cells with UCN‐01 (300 nm) present in culture medium for 72 h after irradiation. (a) Survival fractions at 0 Gy for cells treated with 300 nm UCN‐01 for 72 h (UCN‐01 300nm) relative to non‐treated cells (mock). Results shown are average of six independent experiments (each experiment was performed with three parallel dishes for each data point). (b) Survival fractions of HCT116 p53+/+ cells after IR (2, 3, 4 and 6 Gy) relative to non‐irradiated cells (0 Gy). Results shown are average of three to six experiments. (c) Similar to b with HCT116 p53−/− cells.
Unlike in HCT116 isogenic cell system, U2OS‐VP16 cells were sensitized to radiation by Chk1 inhibitors, to a similar degree with or without expression of p53DD, that is, regardless of p53 status (Fig. 4). Specifically, survival parameter D10 was 5.2, 4.0 and 3.5 Gy for ‘−TET mock’, ‘−TET Cep‐3891’ and ‘−TET UCN‐01’, respectively, and 5.1, 3.8 and 3.4 Gy for ‘+TET mock’, ‘+TET Cep‐3891’ and ‘+TET UCN‐01’ respectively (Fig. 4b,c). In these experiments, Chk1 inhibitors (100 nm UCN‐01 or 500 nm CEP‐3891) were present in the medium for 24 h after IR. Analogous conclusions were reached when Chk1 inhibitors were kept in the medium of U2OS‐VP16 cells for 3 days, although the Chk1 inhibitors then became more toxic in absence of IR (data not shown).
Figure 4.
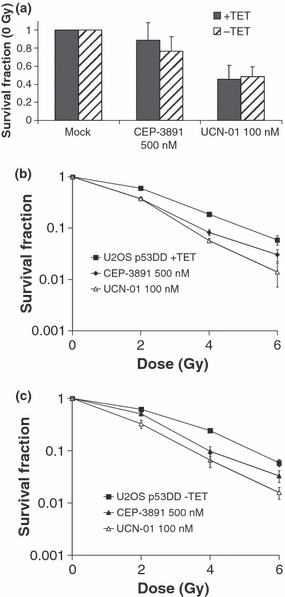
Clonogenic survival assays of IR‐treated U2OS‐VP16‐p53dd cells cultured in presence (+TET) or absence (−TET) of tetracycline. CEP‐3891 (500 nm) or UCN‐01 (100 nm) was present in culture medium for 24 h. (a) Survival fractions at 0 Gy for cells treated for 24 h with 500 nm CEP‐3891 (CEP‐3891 500 nm) or 100 nm UCN‐01 (UCN‐01 100 nm) relative to non‐treated cells (mock). Results shown are average of at least three independent experiments. (b) Survival fractions after IR (2, 4 and 6 Gy) relative to non‐irradiated cells (0 Gy) for U2OS‐p53DD cells cultured in the presence of tetracycline. Results shown are average of at least three independent experiments. (c) Similar to B with U2OS‐p53DD cells cultured in absence of tetracycline.
In addition to clonogenic assays, cell survival may also be assessed by short‐term death assays during the first days after treatment (20, 21). To address whether cell death of U2OS‐VP16 cells was p53‐dependent at 72 h after treatment, we assayed cell viability by intracellular uptake of non‐permeable dye sytox green (Fig. 5a). Cell viability was significantly lower in U2OS‐VP16 cells expressing p53DD (p53‐deficient) compared to parent U2OS‐VP16 cells (Fig. 5a). Thus, p53‐deficient U2OS‐VP16 cells appeared to be selectively targeted when cell viability at 3 days after treatment was measured. To explore whether this might relate to p53‐dependent differences in cell cycle progression, we also assayed DNA profiles by flow cytometry at 48 h after combined treatment with IR and CEP‐3891 or UCN‐01 (Fig. 5b,c). DNA profiles at 48 h indicated that cells expressing p53DD contained a higher fraction of cells in S and G2/M phases compared to parent cells (Fig. 5b,c, upper panels). Furthermore, when nocodazole was added 24 h after treatment and cells were harvested at 48 h, a proportion of parent U2OS‐VP16 cells appeared arrested in G1 (Fig. 5b,c, lower panels). Despite lack of G1 arrest in the 1st cell cycle after treatment (Fig. 1d), these cells thus, showed G1 arrest in the 2nd cell cycle. 2nd‐cycle G1 arrest was p53‐dependent because U2OS‐VP16 cells with expression of p53DD accumulated in G2/M after addition of nocodazole (Fig. 5b,c). Finally, to address whether 2nd‐cycle G1 arrest was also dependent on p21, we transfected U2OS cells with p21 siRNA. Depletion of p21 abrogated 2nd‐cycle G1 arrest (Fig. 5d), confirming that the 2nd‐cycle G1 arrest was p21 dependent. U2OS cells may therefore be protected from cell death at 72 h by p53/p21‐dependent G1 arrest in the 2nd cycle after the combined treatment.
Figure 5.
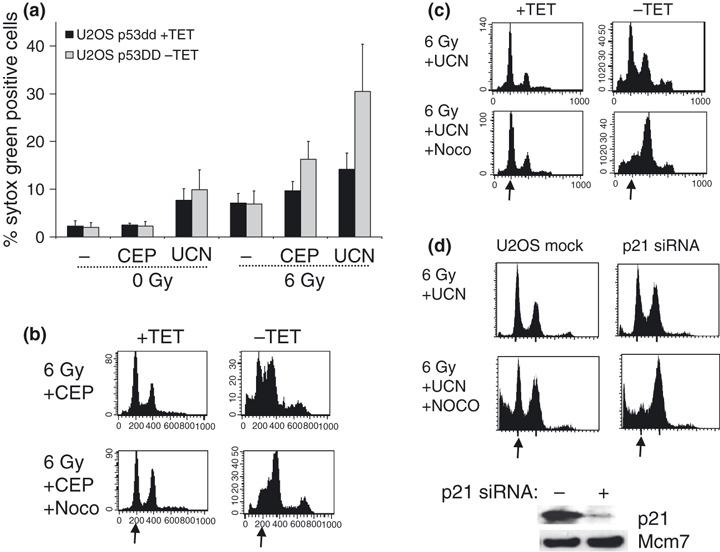
(a) Cell viability of U2OS‐VP16‐p53DD cells at 72 h after treatment with IR and Chk1 inhibitors. Cells cultured in presence (+TET) or absence (−TET) of tetracycline were treated with mock (−), 500 nm CEP‐3891 (CEP), or 300 nm UCN‐01 (UCN) together with IR (0 or 6 Gy) and analysed by flow cytometry. Results shown are average of three to four experiments. (b) Measurement of 2nd‐cycle G1 arrest in U2OS‐VP16‐p53DD cell system. U2OS‐VP16‐p53DD cells cultured in presence (+TET) or absence (−TET) of tetracycline, and DNA histograms shown at 48 h after treatment with IR (6 Gy) and 500 nm CEP‐3891 (CEP) with or without nocodazole (Noco) added to culture medium at 24 h. Representative results are shown. Arrows indicate position of G1‐phase cells. (c) Similar to B with 300 nm UCN‐01 (UCN) instead of CEP‐3891. (d) The 2nd‐cycle G1 arrest is abrogated by p21 siRNA. DNA histograms as in C are shown for parent U2OS cells (U2OS mock) and after transfection with p21 siRNA (p21 siRNA). p21 protein levels are shown in the lower picture.
Discussion
In this study, we employed two different isogenic human cancer cell systems to explore how effects of Chk1 inhibitors combined with IR would be influenced by p53 status of the cells. For the first time, our results show that cancer cells with wild‐type p53, that lack a normal G1 arrest in the 1st cell cycle after IR, may display 2nd‐cycle G1 arrest. We propose that such 2nd‐cycle G1 arrest could contribute to protect against cell death after treatment with IR and Chk1 inhibitors.
2nd‐cycle G1 arrest in U2OS‐VP16 cells seemed to protect against loss of cell viability at 72 h rather than loss of clonogenic survival (4, 5). Similarly, it has previously been reported that UCN‐01 combined with topoisomerase 1 inhibitors caused p53‐dependent mitotic catastrophe measured at 24 h after treatment, while cell death measured by clonogenic survival assays was independent of p53 status (3). Although clonogenic survival assay serves as the most common method to measure survival after IR, cell death assays performed during first days after treatment also correlate with in vivo responses in some cases (21, 22). It has therefore been questioned whether clonogenic survival assay is always superior to short‐term cell death assays for pre‐clinical testing of new treatments (20, 21). Differences in loss of cell viability at 72 h could therefore potentially be important in vivo, although no differences are found in cell survival measured by the clonogenic survival assay.
Notably, HCT116 cells appeared more resistant to cytotoxic effects of Chk1 inhibitors compared to U2OS‐VP16 cells, even though the G2 checkpoint was abrogated in both the cell systems. When UCN‐01 (300 nm) or CEP‐3891 (500 nm) was added to HCT116 cells before IR, and kept in the medium for only 24 h, clonogenic survival of HCT116 cells was almost unaffected (data not shown), while clonogenic survival of U2OS‐VP16 cells was significantly reduced (Fig. 4). These results suggest that cytotoxic effects of UCN‐01 or CEP‐3891 are not solely due to G2 checkpoint abrogation. We have previously found that in addition to abrogation of the G2 checkpoint, Chk1 inhibitors such as UCN‐01 and CEP‐3891 also inhibit homologous recombination repair (23), accelerate entry into unperturbed mitosis because of premature activation of the mitotic kinase CDK1 on centrosomes (24), and cause spontaneous induction of DNA damage in S‐phase cells, due to effects on DNA replication (14). It is likely that one or more of these additional functions of Chk1 kinase targeted by Chk1 inhibitors may also contribute to reducing clonogenic survival on blocking Chk1. Similarly, synergistic effects of UCN‐01 in combination with various chemotherapeutic agents have appeared to be unrelated to G2 checkpoint abrogation (25), and radiosensitizing effect of caffeine, another drug that abrogates the G2 checkpoint, has been attributed to inhibition of homologous recombination repair (26).
Finally, our analysis of two cancer cell systems isogenic for p53 status suggests that loss of p53 can make individual cancers more sensitive to Chk1 inhibitors combined with IR. In response to combined treatment, HCT116 p53−/− cells showed reduced clonogenic survival compared to HCT116 p53+/+ cells, and U2OS cells expressing p53DD showed reduced cell viability at 72 h compared to parent cells. However, when comparing cancers from different patients, p53 status itself is not likely to provide a key predictor. As different cancers display different intrinsic sensitivity to Chk1 inhibitors, as exemplified in this study by enhanced resistance of HCT116 cells to cytotoxic effects of Chk1 inhibitors in the absence of IR, compared to U20S cells, some wt p53 cancers may be even more sensitive to combined treatment than p53 mutant cancers (27). A major challenge for future clinical use of Chk1 inhibitors will therefore be to discriminate between cancers that are intrinsically sensitive and cancers that are resistant to such inhibitors.
Acknowledgements
This study was supported by funds from The Norwegian Research Council, The Norwegian Cancer Society, Harald Andersens Legat, Radiumhospitalets Legater, The Danish Medical Research Council, The Danish Cancer Society, The Danish National Research Foundation, grant MSMT619895921, and the European Commission projects ‘Active p53’, ‘GENICA’ and ‘Mutant p53′.
References
- 1. Ashwell S, Zabludoff S (2008) DNA damage detection and repair pathways – recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 14, 4032–4037. [DOI] [PubMed] [Google Scholar]
- 2. Bucher N, Britten CD (2008) G2 checkpoint abrogation and checkpoint kinase‐1 targeting in the treatment of cancer. Br. J. Cancer 98, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dixon H, Norbury CJ (2002) Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle 1, 362–368. [DOI] [PubMed] [Google Scholar]
- 4. Russell KJ, Wiens LW, Demers GW, Galloway DA, Plon SE, Groudine M (1995) Abrogation of the G2 checkpoint results in differential radiosensitization of G1 checkpoint‐deficient and G1 checkpoint‐competent cells. Cancer Res. 55, 1639–1642. [PubMed] [Google Scholar]
- 5. Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O’Connor PM (1996) UCN‐01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl Cancer Inst. 88, 956–965. [DOI] [PubMed] [Google Scholar]
- 6. Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429. [DOI] [PubMed] [Google Scholar]
- 7. Arrington KL, Dudkin VY (2007) Novel inhibitors of checkpoint kinase 1. ChemMedChem 2, 1571–1585. [DOI] [PubMed] [Google Scholar]
- 8. Hirose Y, Berger MS, Pieper RO (2001) Abrogation of the Chk1‐mediated G(2) checkpoint pathway potentiates temozolomide‐induced toxicity in a p53‐independent manner in human glioblastoma cells. Cancer Res. 61, 5843–5849. [PubMed] [Google Scholar]
- 9. Yu Q, La Rose J, Zhang H, Takemura H, Kohn KW, Pommier Y (2002) UCN‐01 inhibits p53 up‐regulation and abrogates gamma‐radiation‐induced G(2)‐M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res. 62, 5743–5748. [PubMed] [Google Scholar]
- 10. Tse AN, Carvajal R, Schwartz GK (2007) Targeting checkpoint kinase 1 in cancer therapeutics. Clin. Cancer Res. 13, 1955–1960. [DOI] [PubMed] [Google Scholar]
- 11. Falck J, Lukas C, Protopopova M, Lukas J, Selivanova G, Bartek J (2001) Functional impact of concomitant versus alternative defects in the Chk2‐p53 tumour suppressor pathway. Oncogene 20, 5503–5510. [DOI] [PubMed] [Google Scholar]
- 12. Bowman T, Symonds H, Gu L, Yin C, Oren M, Van Dyke T (1996) Tissue‐specific inactivation of p53 tumor suppression in the mouse. Genes Dev. 10, 826–835. [DOI] [PubMed] [Google Scholar]
- 13. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 14. Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F et al. (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 25, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagasawa H, Keng P, Maki C, Yu Y, Little JB (1998) Absence of a radiation‐induced first‐cycle G1‐S arrest in p53+ human tumor cells synchronized by mitotic selection. Cancer Res. 58, 2036–2041. [PubMed] [Google Scholar]
- 16. Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK et al. (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation‐induced accelerated proteolysis of Cdc25A. Cancer Cell 3, 247–258. [DOI] [PubMed] [Google Scholar]
- 17. Syljuasen RG, Sorensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J (2004) Inhibition of Chk1 by CEP‐3891 accelerates mitotic nuclear fragmentation in response to ionizing Radiation. Cancer Res. 64, 9035–9040. [DOI] [PubMed] [Google Scholar]
- 18. Brown JM, Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 5, 231–237. [DOI] [PubMed] [Google Scholar]
- 19. Fertil B, Malaise EP (1985) Intrinsic radiosensitivity of human cell lines is correlated with radioresponsiveness of human tumors: analysis of 101 published survival curves. Int. J. Radiat. Oncol. Biol. Phys. 11, 1699–1707. [DOI] [PubMed] [Google Scholar]
- 20. Tse AN, Schwartz GK (2004) Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN‐01 involves disparate mechanisms resulting in either p53‐independent clonogenic suppression or p53‐dependent mitotic catastrophe. Cancer Res. 64, 6635–6644. [DOI] [PubMed] [Google Scholar]
- 21. Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B et al. (1997) Cell‐cycle arrest versus cell death in cancer therapy. Nat. Med. 3, 1034–1036. [DOI] [PubMed] [Google Scholar]
- 22. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM et al. (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346. [DOI] [PubMed] [Google Scholar]
- 23. Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J et al. (2005) The cell‐cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201. [DOI] [PubMed] [Google Scholar]
- 24. Krämer A, Mailand N, Lukas C, Syljuåsen RG, Wilkinson CJ, Nigg EA et al. (2004) Centrosome‐associated Chk1 prevents premature activation of cyclin‐D‐Cdk1 kinase. Nat. Cell Biol. 6, 884–891. [DOI] [PubMed] [Google Scholar]
- 25. Monks A, Harris ED, Vaigro‐Wolff A, Hose CD, Connelly JW, Sausville EA (2000) UCN‐01 enhances the in vitro toxicity of clinical agents in human tumor cell lines. Invest. New Drugs 18, 95–107. [DOI] [PubMed] [Google Scholar]
- 26. Asaad NA, Zeng ZC, Guan J, Thacker J, Iliakis G (2000) Homologous recombination as a potential target for caffeine radiosensitization in mammalian cells: reduced caffeine radiosensitization in XRCC2 and XRCC3 mutants. Oncogene 19, 5788–5800. [DOI] [PubMed] [Google Scholar]
- 27. Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H et al. (2009) The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 23, 1895–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]