

Abstract
Objectives
Sox9 has recently been reported to be a key mediator during cartilage degradation in osteoarthritis (OA). Our aim was to clarify the role of microRNA‐30a (miR‐30a) and its target gene Sox9 in regulation of extracellular matrix (ECM) degradation in OA.
Materials and methods
Expression of miR‐30a in cartilage tissues and in primary chondrocytes from healthy and OA donors, was determined by real‐time PCR, and levels of Sox9 mRNA and protein were analyzed by real‐time PCR and western blotting, respectively. Subsequently, the target of miR‐30a was predicted by bioinformatics and confirmed using a luciferase assay. Expression of ECM‐related genes was determined by tissue‐specific staining, immunofluorescence, real‐time PCR, and western blotting. The role of miR‐30a in OA was examined in vivo using a collagenase‐induced OA rat model.
Results
miR‐30a was significantly upregulated and Sox9 was downregulated in primary chondrocytes from cartilage taken from OA donors compared to healthy controls. We showed that miR‐30a specifically bound to the 3′ UTR of Sox9, and overexpression of miR‐30a downregulated expression levels of Sox9, proteoglycan aggrecan, and Col II compared to those induced by small interfering RNA transfection to knockdown Sox9. miR‐30a inhibition reversed the effects of ECM degradation in vitro and in vivo.
Conclusions
miR‐30a acts as a virulence MRA in OA, promoting ECM degradation by targeting Sox9 and by modulating activity of its downstream effectors Col II and proteoglycan aggrecan.
Introduction
Osteoarthritis (OA) is the most common chronic joint disease and is characterized by degradation or damage of articular cartilage in joints, including hyaline articular cartilage and subchondral bone. The current clinical treatment of OA is limited to pain management and joint replacement surgery in the late phase of the disease. OA involvesmechanical abnormalities including catabolic and abnormal differentiation of chondrocytes that leads to continuous loss of extracellular matrix (ECM) 1, 2, 3, 4. Thus, molecular mechanisms associated with chondrocyte differentiation during development and maintenance have therapeutic potential as disease‐modifying agents for OA. The matrix proteoglycan aggrecan and type II collagen (Col II) are the main components of the ECM. Degradation of aggrecan is an early event that is crucial for the development of OA lesions 5. Cartilage master regulator transcription factor, Sox9, is important for cartilage development, because it can directly promote the expression of aggrecan and Col II in human osteoarthritic cartilage 6. It was reported that decreased Sox9 expression induces the loss of differentiated phenotype of human articular chondrocytes during subculture in vitro 7, 8.
MicroRNAs (miRNAs) are a class of single‐stranded noncoding RNA molecules of 18–24 nucleotides in length. They play a key role in many biological processes through negative regulation of the expression of target genes post‐transcriptionally by sequence‐specific binding to the 3′ untranslated regions (UTRs) of specific mRNA targets 9. miRNAs also have an important role in diseases affecting articular cartilage and dysregulated expression of miRNAs is found in many pathological conditions, including OA 10, 11. We used microarray analysis technology to screen altered miRNAs in primary chondrocytes isolated from OA and identified miR‐30a. In the present study, we used the Targetscan Program (http//:www.targetscan.org) and predicted Sox9 as a novel target of miR‐30a in OA.
Secreted inflammatory molecules, such as pro‐inflammatory cytokines, are among the critical mediators in OA pathophysiology 12. It was reported that interleukin (IL)‐1β controls the degeneration of articular cartilage matrix, in particular, by reducing the production of cartilage‐specific macromolecules, including Col II and aggrecan 13, 14. In addition, it has been shown that IL‐1β inhibits the expression of cartilage‐specific genes through suppression of Sox9 15. Therefore, this led to our hypothesis that during IL‐1β‐induced OA, overexpression of miR‐30a would markedly suppress the expression of Sox9, resulting in the reduction of ECM‐specific components and ECM degradation.
The aim of this study was to show that miR‐30a participates in the regulation of ECM degradation in articular cartilage cells and that it functions by targeting Sox9 during IL‐1β stimulation. In addition, we investigated whether the effect of miR‐30a inhibition provides a potential approach for preventing IL‐1β‐induced ECM degradation in OA.
Materials and methods
Ethics statement
Experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All patients agreed to participate in the study. Both the study and consents were approved by Ethical Board of Central South University, Changsha, China and complied with the Declaration of Helsinki.
Harvest of human cartilage, cell isolation and culture
Human cartilage samples were obtained from seven healthy donors (three males and four females, aged 39.0 ± 6.3 years) and eleven OA patients (four males and seven females, aged 75.4 ± 8.6 years) who underwent total knee arthroplasty. All samples were obtained from the Department of Orthopaedics at Xiangya Hospital from January 2013 to June 2014. This study was approved by the Ethic Committee of Clinical Investigation of the local investigational review board, and informed consent was obtained from all donors.
Cartilage was removed from each donor and the sliced sections were washed in Dulbecco's modified Eagle's medium (DMEM) (Gibco; Life Technologies, Grand Island, NY, USA) supplemented with penicillin‐streptomycin (Gibco, Grand Island, USA; Invitrogen, Calif., USA). Briefly, the sliced samples were chopped and digested with buffer containing 1% trypsin (Sigma, St. Louis, USA) for 15 min at 37 °C. The supernatant was discarded, a second digestion buffer containing 2 mg/l of type II collagenase (Sigma, St. Louis, USA) was added for 12–16 h at 37 °C overnight. After digestion, the chondrocytes were washed with DMEM and subjected to 200 × g centrifugation for 10 min, and then cultured (5% CO2, 37 °C) in DMEM supplemented with 10% fetal bovine serum (FBS), 1% glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin (Gibco, Grand Island, USA).
Identification of chondrocytes
Adherent cells were digested by trypsin‐EDTA, and then subjected to 200 × g centrifugation for 10 min. The aggregated cells were cultured in DMEM with 10% FBS to develop micropellets. After 1 week, the chondrocyte micropellets from donors were embedded in paraffin or rapidly frozen and cut into 4 μm‐thick sections. These sections were subsequently used for RNA isolation or identification by histological and immunohistochemical staining.
The identification of cartilage chondrocytes was performed using hematoxylin‐eosin (HE), Alcian blue (AB) staining, and Col II immunohistochemistry. For general histological analyses, paraffin sections of the micropellets were stained with HE and AB as described previously 16, 17. HE staining allowed general assessment of the structure of tissues with respect to cytoplasm, synthesized ECM, and the nucleus of the cells. AB showed the presence of aggrecan. Col II is characteristic of hyaline cartilage. Immunodetection was performed to detect Col II. Frozen sections were used to test for the presence of Col II by incubation with anti‐Col II antibody (Abcam Trading Company Ltd., Shanghai, China).
Luciferase reporter assay
The miRNA database (www.microRNA.org) was used to identify the putative miR‐30a binding site in the 3′UTR of Sox9. The forward and reverse oligonucleotides of Sox9‐wild‐type (WT) were annealed to form a segment containing one putative miR‐30a targeting site (5′‐UUUUGUAUAUUAUUGUUUAC‐3′) on the Sox9 3′ UTR, and cloned into the Xhol and Notl sites of psiCHECK‐2‐REPORT vector (Promega Corporation, Madison, WI, USA). Site‐directed mutagenesis of the miR‐30a target‐site in the Sox9 3′ UTR was carried out using a site‐directed mutagenesis kit (Takara Shuzo, Kyoto, Japan), with pmiR‐Sox9‐WT as a template. The psiCHECK‐2‐Sox9‐3′UTR‐WT or psiCHECK‐2‐Sox9‐3′UTR‐mut vector was co‐transfected with control, miR‐NC, or miR‐30a into chondrocytes using Lipofectamine 2000 (Invitrogen), respectively. Reporter gene assays were performed 36 h post‐transfection using the Dual Luciferase Reporter assay system (Promega) according to the manufacturer's protocol. The normalized firefly luciferase activity was obtained by measuring firefly luciferase activity/Renilla luciferase activity. All experiments were performed at least three times.
MiRNA transfection and IL‐1β stimulation in cell culture
Passaged human chondrocytes isolated from healthy donors were seeded at 2.4 × 105 cells per 3.5 cm dish and were subjected to transfection 24 h after seeding. Transfection with 100 nm of miR‐NC, miR‐30a mimic, miR‐Scr (scrambled 25 nucleotides), or 150 nm of miR‐30a inhibitor or miR‐Scr inhibitor (all of these miR mimics and inhibitors were synthesized by Biotend, Shanghai, China) was performed using Lipofectamine 2000 (Invitrogen) for 4 h according to the manufacturer's protocol. Following transfection, the medium was changed to DMEM containing 10% FBS.
Twelve hour after transfection, 10 ng/ml IL‐1β (PeproTech, Rocky Hill, NJ, USA) or phosphate‐buffered saline (PBS) was added to each dish and incubated for 0, 2, 4 or 6 h.
Overexpression or knockdown of Sox9 by Sox plasmid or small interfering RNA transfection
The full‐length or only protein‐coding DNA sequence (CDS) vector of Sox9 was purchased from SinoGeneMax (Beijing, China). Small interfering RNA (siRNA) against Sox9 (siSOX9) and the scrambled siRNA (siScr) were designed and synthesized by RiboBio (Guangzhou, China). The transfection of Sox9 full‐length, Sox9 CDS vector, siScr, and siSox9 in chondrocytes was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Real‐time PCR analysis
Total RNA and small RNA were extracted from clinical OA or normal specimens and from primary chondrocytes isolated from OA or normal human articular chondrocytes using TRIzol (Invitrogen) according to the manufacturer's instructions. The expression of miR‐30a and U6 was examined by the TaqMan miRNA assay system (Life Technologies Corporation). For mRNA detection, RNA was reverse transcribed using the high capacity cDNA reverse transcription kit (Applied Biosystems) including random primers. Reverse transcription was followed by real time PCR using a SYBR Green PCR master mix (Applied Biosystems). Specific primers were used for the different mRNAs are shown in Table 1.
Table 1.
The specific primers used for the different mRNAs
| Primer | Sequence (5′–3′) |
|---|---|
| Sox9‐F | AGGTGCTCAAAGGCTACGACTG |
| Sox9‐R | CCTAATGTTCATGGTCGGCGC |
| Collagen type II‐F | TCCTCTGCGACGACATAAT |
| Collagen type II‐R | TCTACCGACCTCCTAAACT |
| Aggrecan‐F | GAGAAGGAGGTAGTGCTGCTGG |
| Aggrecan‐R | GATGCACAAGGTAATGTCTCGGTA |
| U6‐F | GCTTCGGCAGCACATATACTAAAAT |
| U6‐R | CGCTTCACGAATTTGCGTGTCAT |
| β‐actin‐F | CGTGGACATCCGCAAAG |
| β‐actin‐R | CGGAGCGACAGGTGGAA |
Western blotting
Chondrocytes were cultured as monolayers and Sox9 protein was detected by western blotting using a polyclonal anti‐Sox9 antibody (Abcam, Cambridge, UK) and ECL reagent (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's instructions.
Sulfated‐glycosaminoglycan quantification
A cell suspension was used to analyze soluble sulfated‐glycosaminoglycan (sGAG) secretion/formation with the dimethylmethylene blue (DMMB) assay. sGAG is the main form of aggrecan secreted by chondrocytes in cartilage. A 20 μl of cell suspension was added to 200 μl of DMMB reagent, and the absorbance of the samples was measured at 525 nm using a FlexStation 3 Multi‐Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) and compared with a standard curve based on shark chondroitin 6‐sulfate (Sigma). Total protein contents in the cell lysate from each group were measured by the BCA protein assay kit (Pierce, Rockford, IL, USA). Total sGAG was normalized to total protein content in each group.
Immunofluorescence analysis
Chondrocytes were washed in PBS and fixed with 3.7% formaldehyde in PBS for 10 mins at room temperature. Bovine serum was used to block nonspecific binding sites. Anti‐Col II (1:100) (Abcam) was used as the primary antibody, and goat anti‐rabbit IgG conjugated to fluorescent cy5 dye: (1:100) (Abcam) was used as the secondary antibody. Cells were incubated with these two antibodies in sequence and then developed with Hoechst 33342 (Life Technologies) and observed using a confocal microscope (Nikon A1R MP+multiphoton; Nikon Instruments Inc., Melville, NY, USA).
Establishment of the collagenase‐induced osteoarthritis animal model and treatment
Male Wistar rats aged 10 weeks, weighing 280–300 g, were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and were used to establish a model of collagenase‐induced osteoarthritis (CIOA). To induce CIOA, 1–2 U/10 μl of collagenaseII (Sigma‐Aldrich) was injected into the intra‐articular space of the right and left knee in rats at day 0 and 2, respectively. The control rats received a similar injection of 10 μl endotoxin‐free PBS. PBS, 200 nM of miR‐Scr inhibitor, or miR‐30a inhibitor were subsequently injected into the right and left knee intra‐articular space of CIOA rats, once a day. At day 30, the animals were anesthetized and articular cartilage was obtained for HE assay, immunohistochemical staining or RNA isolation.
Statistical analysis
Data shown are representative of at least 3 independent experiments. The results are expressed as mean ± SD. The significance of the data was estimated by analysis of variance followed by Student Newmann–Keuls multiple comparison tests. All statistical analyses were performed using SPSS software (version 17.0, SPSS Inc., Chicago, IL, USA) and Graphpad Prism 5.01(GraphPad Software, Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
Results
OA chondrocytes show normal morphology, but lack strong staining of aggrecan and Col II
Chondrocyte micropellets were analyzed using different staining methods in order to identify the specific structure and main components of the cartilage ECM. As shown in Fig. 1a, chondrocyte micropellets from both healthy and OA donors showed typical chondrocyte structures, low ECM at the peripheral zone and greater ECM synthesized by the cells in the central area. Chondrocyte micropellets from healthy donors showed positive AB and Col II staining. However, chondrocyte micropellets from OA patients did not show aggrecan and Col II.
Figure 1.
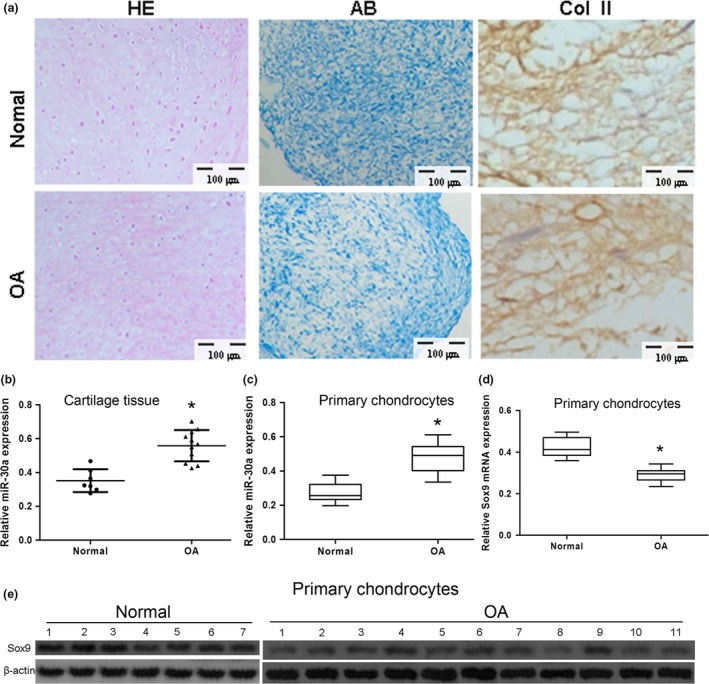
The expression of miR‐30a in clinical specimens and primary chondrocytes. (a) Chondrocyte micropellets from both healthy (normal) donors and donors with osteoarthritis (OA) were identified by hematoxylin‐eosin (HE), Alcian blue (AB) staining, and Col II immunohistochemistry. Scale bars: 100 μm. (b–c) The expression of miR‐30a in cartilage tissue (b) or primary chondrocytes (b) from healthy and OA donors was analyzed by real‐time PCR. Data were normalized to U6. (d–e) The expression of Sox9 mRNA (d) and protein (e) was analyzed by real‐time PCR and western blot, respectively. Data were normalized to β‐actin. All data were expressed as mean ± SD of three independent experiments. Normal, n = 7. OA, n = 11. *P < 0.05 versus normal.
Expression of miR‐30a in OA chondrocytes and targeting of Sox9 by miR‐30a
To assess the putative role of miR‐30a in the pathogenesis of OA, miR‐30a expression was assessed in primary chondrocytes from OA donors. Compared with specimens from healthy donors, the expression of miR‐30a was upregulated in cartilage tissue and in primary chondrocytes from OA specimens (Fig. 1b,c). The expression level of both Sox9 mRNA and protein was downregulated in primary chondrocytes from OA specimens compared with the chondrocytes from normal specimens (Fig. 1d,e).
Sox9, which possesses a putative miR‐30a targeting site, was shown to be a target gene of miR‐30a (Fig. 2a). Luciferase activity of the Sox9 3′ UTR reporter was analyzed to verify Sox9 as a target of miR‐30a in primary chondrocytes. Firefly luciferase activity was measured and normalized to Renilla luciferase activity. The results indicated that miR‐30a repressed luciferase activity in the Sox9‐WT group only, whereas luciferase activity was unaffected in the miR‐control group. Interestingly, with mutated nucleotides in the miR‐30a seed‐binding site, the Sox9‐mut luciferase activity was no longer affected by miR‐30a (Fig. 2b).
Figure 2.
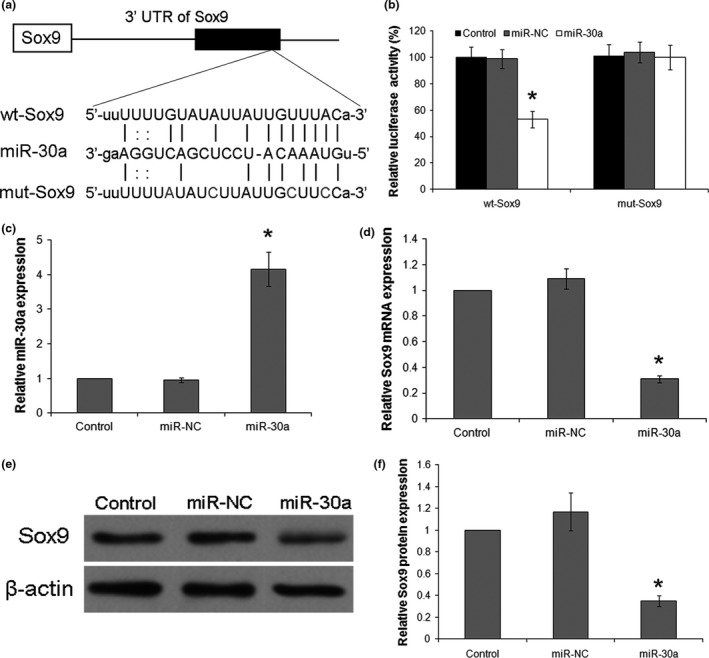
Sox9 is a direct target of miR‐30a. (a) The paired miR‐30a seed sequence and the seed‐recognizing site in the wild‐type (WT) and mutant (mut) 3′ UTR of Sox9 are shown as indicated. (b) Chondrocytes were co‐transfected with firefly luciferase reporter plasmids containing either wt‐Sox9 or mut‐Sox9 binding sites and Renilla plasmids (control), negative miRNA control (miR‐NC) or miR‐30a mimics (miR‐30a). The ratio of firefly/Renilla activity was calculated in the cells and normalized to those of the control. The results were expressed as mean ± SD of three independent experiments. *P < 0.05 versus control of wt‐Sox9. (c–f) Chondrocytes were transfected with miR‐30a mimics (miR‐30a) or a negative miRNA control (miR‐NC), and non‐transfected cells were used as a normal control (control). The levels of miR‐30a (c) and Sox9 mRNA (d) were detected by real‐time PCR. Data were normalized to U6 or β‐actin. (e) Sox9 protein levels were detected by western blotting with β‐actin as the loading control. (f) Quantification of Sox9 protein levels was normalized by β‐actin. Data were expressed as mean ± SD of three independent experiments. *P < 0.05 versus Control.
To further support our hypothesis, the expression level of miR‐30a was assessed in primary chondrocytes transfected with miR‐30a or miR‐control. The average miR‐30a expression level post miR‐30a mimic transfection was4‐fold of the level observed in the miR‐NC group (Fig. 2c). These results demonstrated that miR‐30a was successfully overexpressed. Twenty‐four hours after miRNA transfection, real‐time PCR and western blot showed significant downregulation of Sox9 expression with overexpression of miR‐30a (Fig. 2d–f). These results further confirmed that Sox9 is a direct target of miR‐30a.
Silencing of miR‐30a can reverse the effect of IL‐1β induction on ECM degradation
Studies have shown that IL‐1β can induce ECM degradation by upregulating miR‐30a. In the present study, the expression of miR‐30a was upregulated in normal chondrocytes with IL‐1β treatment compared no treatment group (Fig. 3a). Sox9 expression in the treated chondrocytes was also assessed, and both mRNA and protein levels of Sox9 were reduced in the IL‐1β treated group, especially in the miR‐30a mimic transfected group, but the effects were reversed by transfection with the miR‐30a inhibitor regardless of IL‐1β treatment (Fig. 3b,c). These results indicated that Sox9 had a direct function during IL‐1β induction mediated by miR‐30a. In this study, it was confirmed that Col II and aggrecan genes were downregulated in cells from healthy donors treated with IL‐1β, and transfected with miR‐Scr or with miR‐30a mimic (Fig. 3d,e). However, the gene expression levels of Col II and aggrecan increased with transfection of the miR‐30a inhibitor. This indicated that silencing of miR‐30a in chondrocytes reversed the effect of IL‐1β downregulation on the expression levels of Col II and aggrecan (Fig. 3d,e).In addition, inhibition of miR‐30a increased the level of Col II and aggrecan in the cells without IL‐1β stimulation, suggesting that miR‐30a were also expressed in normal chondrocytes. These results showed that miR‐30a is important in inducing ECM degradation through IL‐1β stimulation, and inhibition of miR‐30a significantly prevents IL‐1β‐induced ECM degradation.
Figure 3.
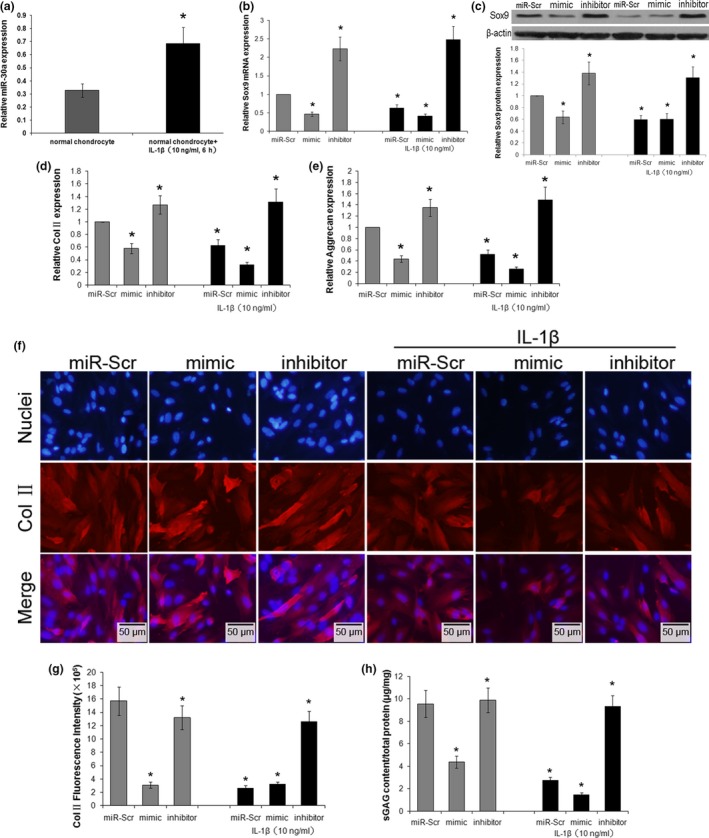
Effects of miR‐30a on IL‐1β‐induced ECM degradation. Chondrocytes were transfected with miR‐Scr, miR‐30a mimic (mimic), or miR‐30a inhibitor (inhibitor), and then treated with or without 10 ng/ml IL‐1β for 6 h. (a) The expression of miR‐30a in normal chondrocytes with or without IL‐1β treatment was analyzed by real‐time PCR. Data were normalized to U6. The expression of Sox9 mRNA (b) and protein (c) were analyzed by real‐time PCR and western blotting, The analysis was performed using images of three independent experiments, respectively. Data were normalized to β‐actin. The mRNA levels of Col II (d) and proteoglycan aggrecan (e) were evaluated by real‐time PCR. Data were normalized to β‐actin. (f) The Col II expression in transfected chondrocytes was assessed by immunofluorescence staining. Scale bars: 50 μm. (g) The fluorescence intensity of Col II expression was analyzed using Image‐Pro Plus 6.0 software. Data were expressed as the average of at least five images. (h) sGAG content in the cell suspension was assessed by DMMB method. Data were normalized by total protein levels in the cell lysate in each group. Data were expressed as mean ± SD of three independent experiments. *P < 0.05 versus miR‐Scr.
In addition to the changes in mRNA, the protein levels of Col II and aggrecan were also assessed by immunofluorescence staining and DMMB assay, respectively. The levels of Col II and sGAG decreased with IL‐1β simulation (Fig. 3f–h). Overexpression of miR‐30a decreased the concentrations of Col II and sGAG, but silencing of miR‐30a restored the levels of Col II and sGAG, regardless of IL‐1β treatment (Fig. 3f–h). In addition, IL‐1β treatment significantly decreased the Col II and sGAG levels compared with untreated healthy chondrocytes, indicating that IL‐1β‐induced upregulation of miR‐30a can induce of ECM‐degradation. Based on these results, IL‐1β stimulates upregulation of miR‐30a, while miR‐30a mediates IL‐1β induced downregulation of Col II and aggrecan, thus affecting the levels of Col II and sGAG.
MiR‐30a mediates IL‐1β‐induced ECM degradation by targeting Sox9
According to the above results, Sox9 is a direct target of miR‐30a. However, it is unknown whether Sox9 participates in miR‐30a function during IL‐1β‐induced chondrocyte ECM degradation. Therefore, we investigated the association of these effects by miR‐30a via Sox9 regulation. And it directly promoted the expression of aggrecan and Col II in chondrocytes.
In order to prove whether the ECM‐degradation effects of miR‐30a were achieved by targeting the 3′ UTR of the Sox9 sequence, a full‐length Sox9 vector (with 3′ UTR) or a Sox9 CDS vector (without 3′ UTR) were co‐transfected with miR‐Scr or miR‐30a mimic into the chondrocytes. It was demonstrated that Sox9 was reduced at both the mRNA and protein level with the miR‐30a mimic together with the full‐length Sox9 vector. However, in the miR‐30a mimic together with the Sox9 CDS vector group, Sox9 expression was only slightly reduced (Fig. 4a,b). These results confirmed that miR‐30a functions mainly by targeting the Sox9 3′ UTR.
Figure 4.
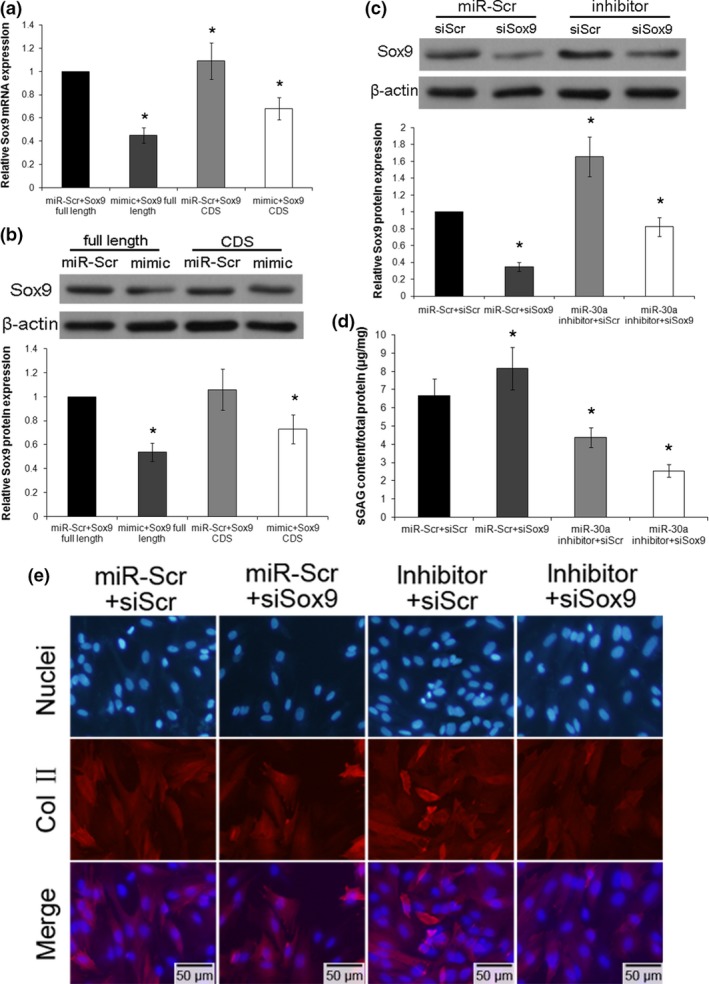
miR‐30a functions during chondrocyte extracellular matrix (ECM) degradation occured through regulation of Sox9. Chondrocytes were cotransfected with miR‐Scr or miR‐30a mimic together with Sox9 full‐length vector (Sox9 expression vector containing both UTR and CDS regions) or Sox9 CDS vector (Sox9 expression vector containing only CDS region, no UTR). The expression of Sox9 mRNA (a) and protein (b) was analyzed by real‐time PCR and western blotting, and the analysis was performed using images of three independent experiments, respectively. Data were normalized to β‐actin. Data were expressed as mean ± SD of three independent experiments. *P < 0.05 versus miR‐Scr + Sox9 full length. Chondrocytes were first transfected with miR‐Scr or miR‐30a inhibitor, and then transfected with either siRNA against Sox9 (siSox9) or scrambled siRNA (siScr). (c) The protein level of Sox9 was analyzed by western blotting. Data were normalized to β‐actin. (d) The sGAG content in the cell suspension was assessed by the dimethylmethylene blue assay. Data were normalized by total protein content in the cell lysate in each group. (e) Col II expression of transfected chondrocytes was assessed by immunofluorescence staining. Scale bars: 50 μm. Data were expressed as mean ± SD of three independent experiments. *P < 0.05 versus miR‐Scr + siScr.
In order to verify the important function of Sox9 during miR‐30a mediated ECM‐degradation, chondrocytes were transfected with the miR‐30a inhibitor or miR‐Scr as the control. Other cell groups were transfected with Sox9 siRNA (siSox9) and Scr siRNA (siScr). In the control group, the expression of Sox9 was significantly reduced by siSox9. In the miR‐30a inhibitor group, the level of Sox9 was increased through the inhibition of miR‐30a. This effect was reduced to the normal level by co‐transfection with siSox9 (Fig. 4c). The protein concentrations of Col II and aggrecan were then assessed in these different treated groups. In the siSox9 alone group, there was a markad decrease in Col II fluorescence as well as a significant reduction in sGAG levels. However, in the co‐transfection with miR‐30a inhibitor and siSox9 group, both the Col II and sGAG levels were reversed to normal levels due to the effect of Sox9 restroed by the miR‐30a inhibitor (Fig. 4d,e). Therefore, Sox9 may be a functional mediator of miR‐30a‐mediated ECM degradation during IL‐1β induction.
The development of OA is via miR‐30a and can be restored by the miR‐30a inhibitor
The severity and progression of CIOAwere evaluated by histology of the joint cartilage. In the control group, histological analyses by HE staining showed intact cartilage with no abnormalities and a homogeneous matrix. However, in the OA group (CIOA was induced in rats by repeated injection of collagenase), the surface of the joint cartilage showed matrix cracks and fissures extending into the deeper cartilage, contributing to cartilage fractures. After 7 days, another group of CIOA rats were injected with PBS. Histological analyses of HE stained sections of CIOA joints showed stronger staining in the cartilage matrix, as well as erosion and matrix loss. In the group of COIA rats treated with miR‐Scr inhibitor, there were small infiltration foci of loose connective tissue and strong HE staining in areas of horizontal sections of articular cartilage. In the CIOA group treated with miR‐30a inhibitor, the articular cartilage surface did not show significantdegradation, suggesting that the CIOA‐induced fissures and fractures in cartilage and bone matrix were induced by miR‐30a (Fig. 5a). The relative expression of miR‐30a in articular cartilage of CIOA rats were analyzed by real‐time PCR, treatment with miR‐30a inhibitor miR‐30a levels were reduced compared with the COIA group treated with PBS or miR‐Scr inhibitor, the results demonstrate reversely that miR‐30a was functional in OA development. Moreover, the relative expression of Col II (Fig. 5b,e) and aggrecan (Fig. 5c,f) were also analyzed by immunofluorescence and real‐time PCR in the bone matrices of different treated groups, respectively. Compared with the control group, there was a marked decrease in Col II and aggrecan in the COIA group. Treatment with the miR‐30a inhibitor restored these ECM component levels to almost normal levels. However, in the COIA group treated with PBS or miR‐Scr inhibitor as controls, the expressions remained low. These results demonstrate that silencing miR‐30a may contribute to the treatment of COIA.
Figure 5.
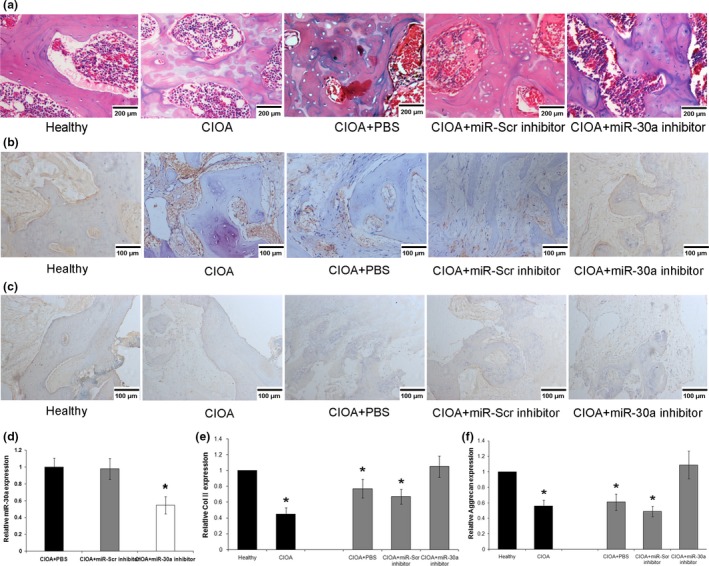
Effect of miR‐30a on the development of collagenase‐induced osteoarthritis (CIOA). (a) Representative joint sections of cartilage and bone matrix in CIOA rats were stained with HE. Scale bars: 200 μm. (b–c) Representative joint sections of cartilage and bone matrix in CIOA rats were immunofluorescence stained with aggrecan and col2. Scale bars: 100 μm. (d) The relative expression of miR‐30a in articular cartilage of CIOA rats. Data were normalized to U6. The mRNA levels of Col II (e) and aggrecan (f) were evaluated by real‐time PCR. Data were normalized to β‐actin. *P < 0.05 versus healthy.
Discussion
OA is the most common joint disease, and affect an increasing number of the aging population. OA is characterized by degeneration of cartilage components inside the joints leading to pain, stiffness, and tenderness 18. The capacity of articular cartilage for repair is very limited. The currentmedical treatment of OA is the reduction of joint pain and stiffness. However, there are no effective pharmacological treatments for OA patients. Many factors contribute to the overall degradation of cartilage observed in OA via a known molecular mechanism 19, including Sox9. Sox9 is essential for cartilage homeostasis and function due to its ability to directly regulate chondrocyte phenotype 20. Therefore, Sox9 may be a new therapeutic target for cartilage repair and identification of its upstream regulator may contribute to the development of new therapeutic option for cartilage repair.
Some miRNA‐related studies have postulated that differential expression of miRNAs is a possible approach for investigating the regulated function of their targets in human diseases. In OA related miRNA research, it has been reported that several miRNAs are involved in the regulation of OA pathogenesis 21, 22, 23. For example, miR‐602 and miR‐608 can regulate sonic hedgehog signaling which is associated with cartilage degradation in OA 24. MiR‐210 can inhibit GPD1L expression and prolyl hydroxylase 2 activity to promote HIF‐1α‐dependent VEGF expression and angiogenesis in human synovial fibroblasts 25. In addition, miR‐146a appears to protect against IL‐1 induced IVD degeneration and inflammation, and thus miR‐146a expression in articular chondrocytes is associated with OA 26.
In the present study, we investigated a possible regulation of miRNA and related genes involved in the pathogenesis of OA. We searched the miRNA database (http//:www.targetscan.org) and found that miR‐30a is cartilage specific as it contains the putative binding site 3′ UTR of Sox9. The literature shows that the function of miR‐30a is important in primary myogenesis and muscle development 27. The miR‐30 family members are downregulated in a variety of malignant and metastatic cancers. miR‐30a plays a putative role in inhibiting epithelial‐mesenchymal transition in prostate cancer 28 and lung cancer 29.
In this study, we hypothesized that upregulated miR‐30a suppressed Sox9 expression and led to ECM degradation in IL‐1β‐induced OA. To verify our hypothesis, the luciferase activity assay was performed and demonstrated that miR‐30a specifically targets Sox9. Furthermore, miR‐30a expression was upregulated in primary chondrocytes isolated from OA specimens, while levels of Sox9 mRNA and protein expression were downregulated. These results confirmed that (i) Sox9 is a direct target of miR‐30a; (ii) upregulation of miR‐30a is involved in OA pathogenesis; (iii) increased miR‐30a may promote OA development by targeting Sox9.
It is known that Sox9 expression may be advantageous in ameliorating the course of OA as it can promote Col II and aggrecan gene expression. However, Col II and aggrecan are chondrocyte ECM genes and are downregulated during chondrocyte ECM degradation 30. IL‐1β, a chondrocyte apoptosis‐inducing agent can trigger cartilage degradation and joint inflammation 31, and thus chondrocytes treated with IL‐1β can be used to mimic OA chondrocytes 32. In the current study, we proposed, for the first time, that miR‐30a is involved in IL‐1β‐induced chondrocyte ECM degradation by targeting Sox9. By overexpressing miR‐30a in chondrocytes, decreased expression of Sox9 was observed by targeting its 3′ UTR regardless of IL‐1β treatment. We demonstrated that silencing miR‐30a reversed the IL‐1β‐induced downregulation of Sox9 as well as its downstream genes Col II and aggrecan. To confirm that miR‐30a mediates ECM degradation through regulation of Sox9, siSox9 was used to demonstrate that knockdown of Sox9 decreased ECM gene expression synthesis. Peck et al.33 demonstrated that there was a conserved target site for miR‐30 in the SOX9 3′ UTR, and miR‐30 and Sox9 expression are positively correlated in the mouse intestine, antisense inhibition of miR‐30 in a human intestinal epithelial cell line (HIECs) led to significantly elevated levels of SOX9, these are similar to our results.
Interestingly, the miR‐30a inhibitor co‐transfected with siSox9 restoredSox9 levels and downstream gene expression. The inhibitor did not induce stress in chondrocytes morphologically. Considering its effect on miR‐30a, the miR‐30a inhibitor could be a potential drug to target miR‐30a in association with ECM degradation together with the targeting of downstream Sox9. Further investigation of this inhibitor will be conducted in our OA animal models and its toxicity and targeting effects determined. Further studies are also planned to elucidate the mechanism of this inhibitor in the prevention of ECM degradation in OA.
It is hoped that, miR‐30a may serve as a new target for preventing IL‐1β‐induced chondrocyte ECM degradation.
Conflict of interest
There is no competing financial interests exist among any authors in relation to the submission.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 81371934).
References
- 1. Goldring MB (2000) The role of the chondrocyte in osteoarthritis. Arthritis Rheumatol. 43, 1916–1926. [DOI] [PubMed] [Google Scholar]
- 2. Kuhn K, D'Lima DD, Hashimoto S, Lotz M (2004) Cell death in cartilage. Osteoarthritis Cartilage 12, 1–16. [DOI] [PubMed] [Google Scholar]
- 3. Lotz M (2001) Cytokines in cartilage injury and repair. Clin. Orthop. Relat. Res. (Suppl 391), S108–S115. [DOI] [PubMed] [Google Scholar]
- 4. Goldring MB (2006) Update on the biology of the chondrocyte and new approaches to treating cartilage diseases. Best Pract. Res. Clin. Rheumatol. 20, 1003–1025. [DOI] [PubMed] [Google Scholar]
- 5. Troeberg L, Fushimi K, Khokha R, Emonard H, Ghosh P, Nagase H (2008) Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 22, 3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haag J, Gebhard PM, Aigner T (2008) SOX gene expression in human osteoarthritic cartilage. Pathobiology 75, 195–199. [DOI] [PubMed] [Google Scholar]
- 7. Lafont JE, Talma S, Murphy CL (2007) Hypoxia‐inducible factor 2alpha is essential for hypoxic induction of the human articular chondrocyte phenotype. Arthritis Rheumatol. 56, 3297–3306. [DOI] [PubMed] [Google Scholar]
- 8. Murphy CL, Polak JM (2004) Control of human articular chondrocyte differentiation by reduced oxygen tension. J. Cell. Physiol. 199, 451–459. [DOI] [PubMed] [Google Scholar]
- 9. Senye M, Mir CF, Morton S, Thie NM (2012) Topical nonsteroidal anti‐inflammatory medications for treatment of temporomandibular joint degenerative pain: a systematic review. J. Orofac. Pain 26, 26–32. [PubMed] [Google Scholar]
- 10. Alvarez‐Garcia I, Miska EA (2005) MicroRNA functions in animal development and human disease. Development 132, 4653–4662. [DOI] [PubMed] [Google Scholar]
- 11. Jones SW, Watkins G, Le Good N, Roberts S, Murphy CL, Brockbank SM et al (2009) The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF‐alpha and MMP13. Osteoarthritis Cartilage 17, 464–472. [DOI] [PubMed] [Google Scholar]
- 12. Lee H, Choi HS, Park Y, Ahn CW, Jung SU, Park SH et al (2014) Effects of deer bone extract on the expression of pro‐inflammatory cytokine and cartilage‐related genes in monosodium iodoacetate‐induced osteoarthritic rats. Biosci. Biotechnol. Biochem. 78, 1703–1709. [DOI] [PubMed] [Google Scholar]
- 13. Gao Y, Wu F, Zhou J, Yan L, Jurczak MJ, Lee HY et al (2014) The H19/let‐7 double‐negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res. 42, 13799–13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kunisch E, Kinne RW, Alsalameh RJ, Alsalameh S (2014) Pro‐inflammatory IL‐1beta and/or TNF‐alpha up‐regulate matrix metalloproteases‐1 and ‐3 mRNA in chondrocyte subpopulations potentially pathogenic in osteoarthritis: in situ hybridization studies on a single cell level. Int. J. Rheum. Dis. doi: 10.1111/1756-185X.12431. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15. Dai L, Zhang X, Hu X, Zhou C, Ao Y (2012) Silencing of microRNA‐101 prevents IL‐1beta‐induced extracellular matrix degradation in chondrocytes. Arthritis. Res. Ther. 14, R268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz‐Prado S, Cicione C, Muinos‐Lopez E, Hermida‐Gomez T, Oreiro N, Fernandez‐Lopez C et al (2012) Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Disord. 13, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stoppoloni D, Politi L, Leopizzi M, Gaetani S, Guazzo R, Basciani S et al (2015) Effect of glucosamine and its peptidyl‐derivative on the production of extracellular matrix components by human primary chondrocytes. Osteoarthritis Cartilage 23, 103–113. [DOI] [PubMed] [Google Scholar]
- 18. Uth K, Trifonov D (2014) Stem cell application for osteoarthritis in the knee joint: a minireview. World J. Stem Cells 6, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tardif G, Hum D, Pelletier JP, Duval N, Martel‐Pelletier J (2009) Regulation of the IGFBP‐5 and MMP‐13 genes by the microRNAs miR‐140 and miR‐27a in human osteoarthritic chondrocytes. BMC Musculoskelet. Disord. 10, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao H, Zhang T, Xia C, Shi L, Wang S, Zheng X et al (2014) Berberine ameliorates cartilage degeneration in interleukin‐1beta‐stimulated rat chondrocytes and in a rat model of osteoarthritis via Akt signalling. J. Cell Mol. Med. 18, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A (2008) Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE 3, e3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martinez‐Sanchez A, Dudek KA, Murphy CL (2012) Regulation of human chondrocyte function through direct inhibition of cartilage master regulator SOX9 by microRNA‐145 (miRNA‐145). J. Biol. Chem. 287, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sekiya I, Tsuji K, Koopman P, Watanabe H, Yamada Y, Shinomiya K et al (2000) SOX9 enhances aggrecan gene promoter/enhancer activity and is up‐regulated by retinoic acid in a cartilage‐derived cell line, TC6. J. Biol. Chem. 275, 10738–10744. [DOI] [PubMed] [Google Scholar]
- 24. Akhtar N, Makki MS, Haqqi TM (2014) MicroRNA‐602 and microRNA‐608 regulate sonic hedgehog expression via target sites in the coding region in human chondrocytes. Arthritis Rheumatol. 67, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu SC, Chuang SM, Hsu CJ, Tsai CH, Wang SW, Tang CH (2014) CTGF increases vascular endothelial growth factor‐dependent angiogenesis in human synovial fibroblasts by increasing miR‐210 expression. Cell Death Dis. 5, e1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK (2008) Upregulated miR‐146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis. Res. Ther. 10, R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Brien JH, Hernandez‐Lagunas L, Artinger KB, Ford HL (2014) MicroRNA‐30a regulates zebrafish myogenesis through targeting the transcription factor Six1. J. Cell Sci. 127, 2291–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J, Shen C, Wang L, Ma Q, Xia P, Qi M et al (2014) Metformin inhibits epithelial‐mesenchymal transition in prostate cancer cells: involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem. Biophys. Res. Commun. 452, 746–752. [DOI] [PubMed] [Google Scholar]
- 29. Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J et al (2011) MicroRNA‐30a inhibits epithelial‐to‐mesenchymal transition by targeting Snai1 and is downregulated in non‐small cell lung cancer. Int. J. Cancer 130, 2044–2053. [DOI] [PubMed] [Google Scholar]
- 30. Legendre F, Dudhia J, Pujol JP, Bogdanowicz P (2003) JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)‐6/soluble IL‐6R down‐regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down‐regulation of SOX9 expression. J. Biol. Chem. 278, 2903–2912. [DOI] [PubMed] [Google Scholar]
- 31. Lin EA, Kong L, Bai XH, Luan Y, Liu CJ (2009) miR‐199a, a bone morphogenic protein 2‐responsive MicroRNA, regulates chondrogenesis via direct targeting to Smad1. J. Biol. Chem. 284, 11326–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyaki S, Nakasa T, Otsuki S, Grogan SP, Higashiyama R, Inoue A et al (2009) MicroRNA‐140 is expressed in differentiated human articular chondrocytes and modulates interleukin‐1 responses. Arthritis Rheumatol. 60, 2723–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peck B, Mah A, Simmons J, Magness S, Kay Lund P, Sethupathy P (2014) MicroRNA‐30 and SOX9 participate in a negative feedback loop to regulate intestinal proliferation. FASEB J. 28, no. 1S. [Google Scholar]