

Abstract
Objectives
To analyse proliferation, differentiation and apoptosis in THP‐1 cells after stimulation with phorbol 12‐myristate 13‐acetate (PMA) and retinoic acid (RA).
Materials and methods
PMA and RA were used in a three‐step‐procedure: (i) treatment with 6, 30, 60 nm PMA, that induced initial, intermediate and advanced levels of monocyte‐macrophage transition, respectively; (ii) recovery in PMA‐free medium; (iii) incubation with 4 μm RA. Cultures were characterized cytokinetically (flow cytometry/bromodeoxyuridine uptake) and immunocytochemically (static cytometry) for expression of CD14, CD11b (monocyte‐macrophage) and DC‐SIGN (dendritic cell: DCs) markers.
Results
Some treatments determined appearance of monocyte/macrophage, dendritic and apoptotic phenotypes, percentages of which were related to PMA dose used in step 1, and dependent on presence/absence of PMA and RA. PMA withdrawal induced dedifferentiation and partial restoration of proliferative activity, specially in 6 and 30 nm PMA‐derived cells. Recovery in the presence of serum (fundamental to DC appearance) indicated that depending on differentiation level, cell proliferation and apoptosis were inversely correlated. Treatment with 30 nm PMA induced intermediate levels of monocytic‐macrophagic differentiation, with expression of alternative means of differentiation and acquisition of DCs without using cytokines, after PMA withdrawal and RA stimulation.
Conclusions
Our experimental conditions favoured differentiation, dedifferentiation and transdifferentiational pathways, in monocytic THP‐1 cells, the balance of which could be related to both cell proliferation and cell death.
Introduction
In many cell systems, proliferation, differentiation and apoptosis are key events in dynamics of cell populations. In some leukaemic cell lines that are able to differentiate into neutrophils or monocytes the process of differentiation can be accompanied by apoptosis 1. Normally, monocytes compose a heterogeneous population characterized by the variety of functions that different subpopulations can display. In vitro comparative analysis of human blood monocytes, and macrophages derived from them, have demonstrated modulation of phenotypes 2, 3, 4. Monocytes isolated from peripheral blood, alternative to bone marrow‐derived stem cells, can also differentiate, after appropriate stimulation, into dendritic cells (DCs) in vitro 5, 6, DCs being the most potent type of antigen‐presenting cells (APC) in vivo and in vitro. Moreover, DCs are considered to be prime targets for modulating anti‐cancer immune responses 7, 8, 9.
Although laboratories worldwide are currently exploring therapeutic applications of DCs obtained by an array of methods, many aspects of DC biology are still poorly understood. This is mainly because studying DC biology in vitro is problematic for a number of reasons. In vitro they can be obtained by differentiation of bone marrow stem cells 10, peripheral blood mononuclear cells 11 or monocytes 12. Isolation of these DC‐precursors is laborious and their subsequent differentiation requires 6–9 days, as well as stimulation with expensive cytokines 13. On the other hand, their limited life span, involvement of apoptosis and difficulties in manipulating them (they are terminally differentiated primary cells) in vitro have seriously hampered studies aimed at exploring their biology. For these reasons, defining in vitro systems for DCs is of paramount importance, as is exploring the possibility of obtaining them from normal and/or leukaemic monocyte cell lines. The latter can be effective models for development of tools to study DC biology. Some workers have indicated that in vitro maturation of monocytes into antigen‐presenting cells (macrophages and dendritic cells) could serve as a model for this in vivo phenomenon 14. For example, monocyte‐derived macrophages could express surface markers not found in their monocyte precursors, with other markers being expressed preferentially on monocytes 2, 4.
In our laboratory, we have previously demonstrated a consolidated cell model of in vitro macrophage (like) differentiation 15 in the THP‐1 human monocytic cell line, in which treatment for 72 h with a variety of concentrations of phorbol ester PMA (phorbol 12‐myristate 13‐acetate ranging from 6 to 60 nm), was able to induce gradual progression of phenotypes from monocyte to terminally differentiated macrophage, that appeared to be related to progressive increase of adhesive capacity 16. Recently, by using this cell model, we have observed that PMA withdrawal, after 72 h, caused cell detachment and death by apoptosis, with different incidence, related to differentiation level induced by the specific PMA dose. For 30 nm PMA, cells expressed intermediate levels of differentiation, during transition from monocytes to macrophages, and cultures were characterized by marked cell heterogeneity. PMA withdrawal from culture medium caused not only cell detachment and apoptosis, but also appearance of typical DC morphology amongst cells still adherent.
Starting from this observation, the aim of the present work was to study the relationship between cell differentiation status modulation and apoptosis; further, these aspects have been correlated with appearance of specific cell features. In general, ability of the cell line to respond to different maturation stimuli was evaluated, highlighting dynamics of DC appearance in relation to conditions of stimulation. For the studies, some cultures were PMA‐deprived for 72 h (pre‐treated with different concentrations: 6, 30 and 60 nm), then subsequently incubated (after one step of recovery with PMA‐free conventional culture medium) with a single dose (4 μm) of retinoic acid (RA). RA is a potent immunomodulator, nevertheless, little is known of its effect on APCs. Some authors have reported controversial data on ability of retinoid derivatives to modulate maturation and functional activities of DCs 17, 18. Studies performed by further authors suggest that all trans retinoic acid (ATRA), at a dose of 10−12 m, can skew monocyte differentiation into cells with APC and DC features 19.
Using techniques of immunofluorescence and static cytometry, initially we examined changes in expression of two cell surface molecules, CD14 and CD11b, in cells treated with different PMA concentrations, and also after absence of this phorbol ester for 72 h (recovery), in the culture medium. CD14 is a monocyte marker 20 downregulated during differentiation to macrophages 21. This molecule acts as co‐receptor for detection of bacterial lipopolysaccharide (LPS). In addition, we evaluated expression of CD11b, a molecule involved in cell adhesion phenomena 22, 23, 24, increase of which in our cell model, is firmly related to differentiation progression 16. CD11b is an integrin family member that pairs with CD18 to form the CR3 heterodimer; in addition to cell adhesion, CD11b is directly involved in phenomena of motility.
In a further series of experiments, after maintaining the cells for 72 h in the absence of PMA, and after subsequent stimulation with RA for 72 h, we investigated whether RA would affect differentiation and phenotype changes of THP‐1 cells. For this purpose, we analysed not only expression of CD14 and CD11b on cell surfaces, but also presence of DC‐SIGN a specific marker of dendritic cells. This molecule (also identified as CD 209) is protein with a C‐type ectodomain, expressed by monocyte‐derived DCs, and is thought to play an important role in establishing initial contact with resting T cells, by interacting with mannosium‐rich glycoproteins, ICAM‐3 and ICAM‐2 25. To measure CD14, CD11b and DC‐SIGN fluorescence emitted after specific immunoreactions, we used static cytofluorometry (SCM) instead of flow cytometry, to avoid cell detachment that could lead to loss of surface molecules. Furthermore, SCM allowed us to correlate quantitative expression of specific molecules at a cellular level and their morphological distributions.
To characterize cultures from a morphofunctional point of view, we studied cell adhesive and migratory properties, in the different experimental conditions, by using cytochemical techniques for fluorochromization of the actin cytoskeleton. Cytokinetic‐related properties such as proliferation, polyploidization and death, related to differentiation, were analysed by flow cytometry after DNA fluorochromization and bromodeoxyuridine (BrdUrdrd) uptake.
Materials and methods
Cell culture and differentiation
THP‐1 cells, derived from a human monocytic leukaemia, were grown in RPMI 1640 supplemented with 15% foetal calf serum (FCS). Cultures were maintained at 37 °C in a humidified atmosphere (95% air/5% CO2) at 0.8–1 × 105 cells/ml, as a stationary suspension culture. For microscopy, THP‐1 cells were grown on glass coverslips, in multiwell plates (0.8–1.0 × 105 cells/cm2).
Activation of THP‐1 cells was obtained by incubation with different concentrations (6, 30 and 60 nm in complete medium) of the phorbol ester PMA. After treatment for 72 h, cells were induced to adhere to their growth substratum and to differentiate. PMA was changed every day and, in relation to different concentrations, cells progressively acquired morphofunctional features related to their level of differentiation.
PMA withdrawal and treatment with retinoic acid
Adherent THP‐1 cells, derived from 72‐h treatment with the three different PMA concentrations, were submitted to recovery conditions for further 72 h in PMA‐free complete medium (presence of serum). In the following series of experiments, PMA‐free complete medium was serum‐deprived. In further experiments, cultures recovered for 72 h in PMA‐free medium containing serum were incubated for 72 h in the presence of 4 μm RA. To evaluate numbers of detached cells, during the range of experimental conditions, aliquots of culture medium were harvested every 24 h.
Before RA stimulation, recovery medium (including detached cells) was removed.
Flow cytometry
Cell cycle and incidence of apoptosis analyses were performed using PARTEC PAS II flow cytometer (PARTEC, Münster, Germany). THP‐1 cells, maintained as a monocyte line in T‐25 cm2 culture flasks, were collected by centrifugation (100 g for 10 min), while PMA/RA‐treated adherent cells (2.5–3.0 × 106 cells/T‐25 cm2) were collected post‐trypsinization (0.25% trypsin in PBS). After being washed in PBS, cells were fixed in 70% ethanol at 4 °C for 30 min. Measurements of DNA/protein relative content were performed after fluorochromization (45 min at room temperature) with 8 μm DAPI (4′,6‐diamidino‐2‐phenylindole) and 50 μm SR 101 (sulphorhodamine 101) in 0.1 m Tris–HCl buffer pH 7.5, respectively. Cells were then filtered through a nylon filter (mesh = 50 μm) to remove aggregates. Cell concentration was adjusted to approximately 4–6 × 105 cells/ml, to minimize errors during flow measurements. Instrumentation equipped with an Hg lamp was used in optic configuration for UV (λexc. = 350 nm), for the simultaneous DAPI (λem. = 435 nm) and SR 101 (λem. = 630 nm) excitation. Data in frequency histograms of DNA content and DNA/protein cytograms, were collected. Calculation of percentage of cells in the various cell cycle phases and in apoptosis was performed on frequency cumulative curves of DNA histograms. Estimation of apoptotic cells was performed by considering the hypo‐2c values and comparison between different experimental conditions was carried out using Student's t‐test; differences were considered to be statistically significant at P < 0.05.
Fluorescence cytochemistry
BrdUrd uptake was used for determination of DNA‐replicating cells (LI). Cultures were submitted to pulse labelling with 10 μm bromodeoxyuridine (BrdUrd) for 30 min at 37 °C. After incubation and after being washed in PBS, cells were fixed in cold 70% ethanol for 30 min and treated with 2 n HCl for 30 min to denature the DNA. Samples were neutralized with Na2B4O7 (0.1 m, pH 8.5) for 2 min and permeabilized with Tween 20 (0.5%) in PBS for 10 min. Then they incubated in anti‐BrdUrd primary antibody (dilution 1:100) for 45 min in a humidity chamber. They were then washed in PBS, incubated with TRITC‐conjugated secondary antibody (dilution 1:60) for 45 min in the humidity chamber. For each condition, three representative slides were examined and by evaluating at least 250 cells per slide. BrdUrd positive immunoreaction was expressed as percentage of labelled nuclei, after DNA nuclear counterstaining with Hoechst 33342.
Actin cytochemical staining was performed after removal of detached cells from samples and washing in PBS. Still adherent THP‐1s on glass coverslips (for the range of experimental conditions, on average in the region of 50% of seeded cells) were fixed in 3% paraformaldehyde (PFA) in PBS containing 1 mm CaCl2 and 1 mm MgCl2 (buffer A), for 30 min at room temperature. After fixation and washing in buffer A, cells were permeabilized with Triton X‐100 (0.2% in buffer A) for 15 min at room temperature. Actin microfilament visualization was performed by incubating cells in the presence of TRITC‐conjugated phalloidin (dilution 1:50) for 45 min in the humidity chamber.
CD11b, CD14, DC‐SIGN immunolabelling was performed to detect expression of these proteins, after removal of detached cells and washing in PBS. Still adherent THP‐1 cells on glass coverslips (for the range of experimental condition, on average, 50% of seeded cells) were fixed in 3% PFA. After being washed in buffer A, cells were immunostained using primary monoclonal antibodies (dilutions: CD11b 1:10, CD14: 1:50; DC‐SIGN: 1:10) for 45 min in a humidity chamber. After being washed in buffer A, cells were incubated in TRITC‐ (CD14) or FITC‐ (CD11b; DC‐SIGN) conjugated secondary antibodies (dilution 1:250), for 45 min in a humidity chamber. Negative reaction controls were obtained by omitting incubation with primary antibodies. For all fluorescence microscopy specimens, nuclear counterstaining was performed by means of Hoechst 33342 (SIGMA, St. Louis, MO, USA) (2 μg/ml in PBS) for 10 min at room temperature. After being washed in appropriate buffer, coverslips were mounted in 1:10 (v/v) mixture of PBS/glycerol containing p‐phenylendiamine as anti‐fading agent. Observation was performed by epifluorescence with a NIKON Eclipse 600 microscope (NIKON, Kanagawa, Japan) equipped with a 100‐W mercury lamp. The following conditions were used: 330–380 nm excitation filter (excf), 400 nm dichroic mirror (dm) and 420 nm barrier filter (bf) for Hoechst 33342; 450–490 nm excf, 510 nm dm and 520 nm bf for FITC; and 540 nm excf, 580 nm dm and 590 nm bf for TRITC.
Static cytofluorometry
After removal of detached cells and washing in PBS, still adherent THP‐1 cells on glass coverslips (for the range of experimental conditions, on average, 50% of seeded cells) were fixed in 3% PFA in buffer A, for 30 min at room temperature. Fluorescence intensity per cell was evaluated after specific immunoreaction for CD11b, CD14 and DC‐SIGN, by measuring fluorescence emitted by each single cell. For this analysis, a NIKON P II (NIKON) cytoflurometer was used. Within the cytometric apparatus, an appropriate system of diaphragms permitted selection of single cells and fluorescence was quantitatively measured after automatic subtraction of background brightness.
The quantitative data, expressed as arbitrary units (a.u.), were collected in frequency histograms of fluorescence intensity. The calculation of the percentage of cells positive to immunoreactions was performed on frequency cumulative curves of histograms, by considering negative the values ranging from 0 to 4 a.u. This range was established by measuring fluorescence emitted by cells in samples in which primary antibodies were omitted (values ranged from 0 to 2 a.u) and by cells in which, after conventional immunoreactions, minimal fluorescence was detected (values ranged from 2 to 4 a.u). For each experimental condition, three representative slides were examined, and at least 250 cells per slide were measured.
Reagents
Chemicals and antibodies, except where specified, were purchased from SIGMA, St. Louis, MO, USA.
Results
Cell cycle, apoptotic index and cell morphology analyses, after PMA withdrawal, in cultures maintained in presence or absence of serum
Comparison between DNA content cytofluorometric measurements (Fig. 1), performed on the THP‐1 monocytic cell line after 6, 30 and 60 nm PMA stimulation for 72 h, illustrated cytokinetic changes during differentiation towards the macrophage phenotype (Fig. 1a–c). Progressive reduction in proliferative activity appeared to be PMA dose‐dependent and was indicated by changes in levels of cells with intermediate ploidy DNA values and by reduction in BrdU cell uptake (see also Table 1). On the whole, treatment of THP‐1 cells with the three different PMA concentrations for 72 h was a representative model of monocyte‐macrophage differentiation, which appeared to be related to increase in cell adhesion to substrates. Acquisition of macrophage‐like morphofunctional features (related to initial, intermediate and high differentiation) was reached after 72 h PMA treatment (6, 30 and 60 nm) and could be maintained for several days in the presence of PMA, changed daily 16. Using this cell model, experiments performed here demonstrate that PMA withdrawal, in cultures maintained in complete medium for 72 h, induced cell detachment in all conditions; this event appeared to be quantitatively related to PMA concentration used in the first step of the procedure (Fig. 2).
Figure 1.
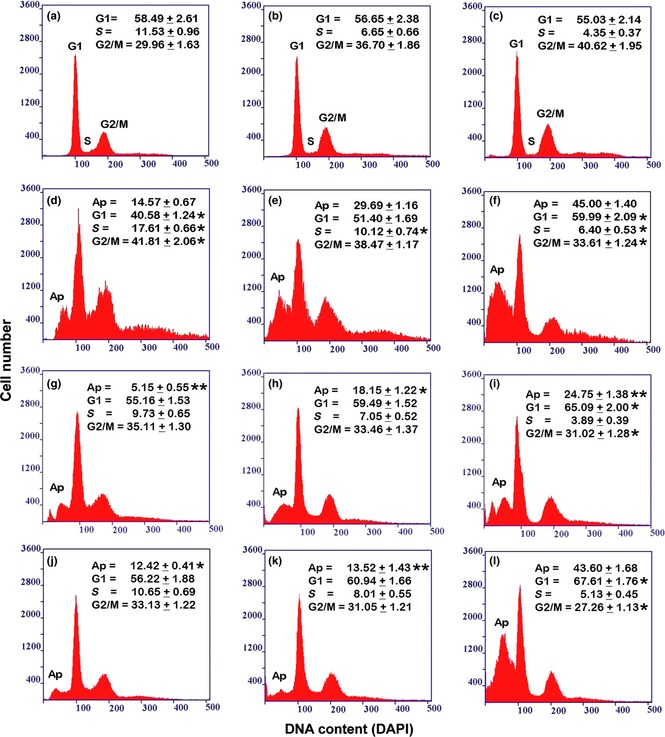
Flow cytometry of DNA cellular content in THP‐1 cell culture in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm). (d, e, f) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment (d: 6 nm; e: 30 nm; f: 60 nm). (g, h, i) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment (g: 6 nm; h: 30 nm; i: 60 nm). (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment (j: 6 nm; k: 30 nm; l: 60 nm). The reported values represent the percentage (mean ± SEM of triplicate experiments) of cells in each phase of the cell cycle (G1, S, G2/M), calculated by excluding, from the cell population, apoptotic (Ap) and over‐4c cells. The asterisks (*P < 0.05; ** P < 0.001 after Student's t‐test) indicate the values that are significantly different from the respective referring condition: left column (d, g, j) versus (a); central column (e, h, k) versus (b) and right column (f, i, l) versus (c) for cell cycle phase percentage; (g, j) versus (d); (h, k) versus (e) and (i, l) versus (f) for apoptotic cell percentage.
Table 1.
Frequency distribution of DNA replicating THP‐1 cells in the different experimental conditions
| Treatment | 2c–4c content | BrdU LI | Apoptosis |
|---|---|---|---|
| 0 nm PMA | 25.38 ± 1.42 | 27.03 ± 1.27 | — |
| 6 nm PMA | 11.53 ± 0.96 | 13.33 ± 0.88 | — |
| 30 nm PMA | 6.65 ± 0.66 | 4.16 ± 0.44 | — |
| 60 nm PMA | 4.35 ± 0.37 | 1.56 ± 0.09 | — |
| 6 nm PMA/Rec‐S | 17.61 ± 0.66* | 20.01 ± 1.18* | 14.57 ± 0.67 |
| 30 nm PMA/Rec‐S | 10.12 ± 0.74* | 11.75 ± 0.81** | 29.69 ± 1.16 |
| 60 nm PMA/Rec‐S | 6.40 ± 0.53* | 7.01 ± 0.73* | 45.00 ± 1.40 |
| 6 nm PMA/Rec‐SF | 9.73 ± 0.65 | 10.85 ± 0.91 | 5.15 ± 0.55** |
| 30 nm PMA/Rec‐SF | 7.05 ± 0.52 | 8.10 ± 0.60* | 18.15 ± 1.22* |
| 60 nm PMA/Rec‐SF | 3.89 ± 0.39 | 4.24 ± 0.40* | 24.75 ± 1.38** |
| 0 nm PMA/RA | 23.95 ± 1.14 | 25.69 ± 1.34 | — |
| 6 nm PMA/Rec‐S/RA | 0.65 ± 0.69 | 9.88 ± 0.98 | 12.42 ± 0.41* |
| 30 nm PMA/Rec‐S/RA | 8.01 ± 0.55 | 7.30 ± 0.37* | 13.52 ± 1.43** |
| 60 nm PMA/Rec‐S/RA | 5.13 ± 0.45 | 3.41 ± 0.28* | 43.60 ± 1.68 |
Comparison between percentage values of cells in the S phase obtained by flow cytometry (2c–4c content) and bromodeoxyuridine immunolabelling (LI: Labelling Index). The percentage of apoptotic cells has been calculated on the basis of the DNA content histograms by considering hypo‐2c signals. The reported values represent the mean ± SEM of triplicate experiments. All values are significantly different (P < 0.05 after Student's t‐test) from that relative to cells in the absence of PMA. The asterisks (*P < 0.05; **P < 0.001) indicate the values that are significantly different from the respective referring condition relative to the only PMA treatment. PMA, treatment with Phorbol 12‐myristate 13‐acetate; PMA/Rec‐S, recovery in PMA‐free medium containing serum, after PMA treatment; PMA/Rec‐SF, recovery in PMA‐free medium in the absence of serum, after PMA treatment; PMA/RA, stimulation with retinoic acid of PMA‐untreated THP‐1 cells; PMA/Rec‐S/RA, stimulation with retinoic acid of cells derived from PMA treatment and recovered in PMA‐free medium containing serum.
Figure 2.
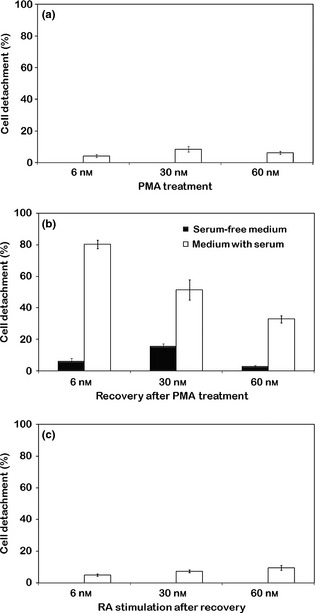
Cell detachment percentage of THP ‐1 cultures. Treatment with PMA at increasing concentrations (a), recovery in PMA‐free medium in presence or absence of serum (b) and stimulation with RA after recovery (c). Reported values represent the mean ± SEM of triplicate experiments.
It must be underlined, that specially for cultures submitted to recovery after 6 and 30 nm PMA treatments, high percentages of suspended cell populations included a fraction of de‐differentiated cells that recovered proliferative activity. In detached cells derived from all PMA‐treatment conditions, high numbers of cells had pre‐apoptotic and apoptotic features (Fig. 3). Total apoptotic events were determined by flow cytofluorometry, as DNA loss (Fig. 1d–f; Table 1). Examination of DNA content histograms revealed progressive increase in signals in sub‐G1 areas in relation to PMA concentrations used in the pre‐treatment step. Moreover, cytofluorometric and immunocytochemical analyses indicated partial restoration of proliferative activity (expressed in terms of DNA content intermediate ploidy values – S phase – and increase in BrdUrd uptake) that appeared to be inversely correlated with increase in PMA concentrations used in the first step of the procedure (Fig. 1d–f). PMA withdrawal also induced appearance of over 4c cells in cultures derived from the different PMA treatment conditions, in particular after recovery in complete medium (Fig. 1d–f).
Figure 3.
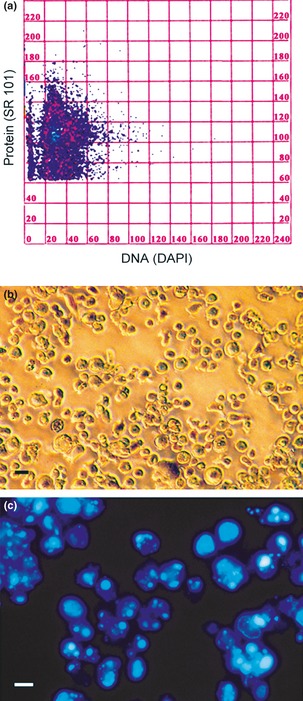
Flow cytometry and morphology of detached cells. DNA/protein cytogram (a) detached cells after 30 nm PMA treatment for 72 h and subsequent recovery for 72 h in complete medium in absence of the phorbol ester. Cytofluorometric data showing two subpopulations with different protien mass: first (SR 101 fluorescence values ranging from 100 to 160 a.u.) indicative of cells in the early phase of apoptosis (or pre‐apoptotic), the second (SR 101 fluorescence values ranging from 60 to 100 a.u.) is representative of cells in advanced stage of apoptosis. (b) Phase contrast microscopy shows the general morphology of detached cells. (c) Fluorescence microscopy (DAPI staining) reveals the high percentage of apoptotic chromatin in floating cells. Bars (b, c) 5 μm.
Regarding cell detachment, in further experiments, performed on cultures after PMA withdrawal and concomitant serum deprivation, a sharp reduction was observed in cell detachment for all PMA concentrations (Fig. 2). This could indicate a direct relationship between levels of suspended cells and partial restoration of proliferative activity that appeared to be dependent on differentiation level acquired by the cells. Moreover, analysis of DNA content histograms and evaluation of BrdUrd uptake related to conditions of recovery in the absence of serum, after PMA treatment (Fig. 1g–i; Table 1), permitted us to reveal a direct relationship between S phase reduction and drop in apoptotic events, with respect to recovery in the presence of serum (Fig. 1d–f; Table 1).
We analysed cultures derived from 30 nm PMA treatment and maintained in complete medium in detail. Figure 3 presents cytofluorometric data expressed as DNA/protein content cytogram, relative only to detached cells (Fig. 3a). Analysis of this cytogram allowed us to identify two subpopulations with different proteic mass: the first (SR 101 fluorescence values ranging from 100 to 160 a.u.) is indicative of cells in early apoptosis (or pre‐apoptotic), the second (SR 101 fluorescence values ranging from 60 to 100 a.u.) is representative of cells in an advanced apoptosis, characterized by a partial protein degradation. Morphological analysis of detached cells (Fig. 3b) demonstrated high incidence of apoptotic nuclei (Fig. 3c). Reasons for interest in 30 nm PMA treatment are that in this condition, cultures were highly heterogeneic and, after PMA withdrawal, among the remaining adherent population, beside macrophages, some cells with features typical of the dendritic phenotype were visible. Lower numbers of stellate cells were also observed in 60 nm PMA derived cultures.
Phase contrast microscopy, performed on all experimental types, is reported in Fig. 4. Progressive changes in shape and adhesion of THP‐1 cells treated with the various PMA concentrations (Fig. 4a–c), and changes induced by PMA withdrawal and recovery in complete medium (Fig. 4d–f), or in absence of serum (Fig. 4g–i), are shown. Phorbol ester withdrawal for 72 h caused general increase in numbers of rounded cells in all cultures derived from previous PMA treatment (6, 30 and 60 nm), and maintained in the presence of serum. In these cultures, derived from 30 nm, and in part from 60 nm PMA treatments, among still adherent populations, some cells with stellate morphology typical of the dendritic phenotype, were visible (Fig. 4e and 4f). Morphological analysis of cultures recovered in the absence of serum, showed a general trend towards reduction in cell detachment, and the absence of cells with dendritic features was evident (Fig. 4g–i). Quantitative data referring to percentages of different cell morphologies observed are reported in Fig. 5.
Figure 4.
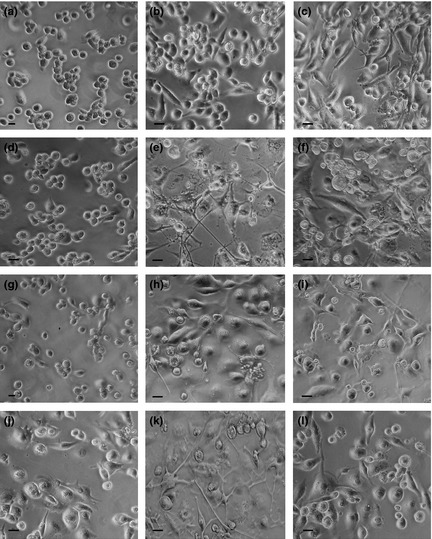
Phase contrast microscopy of THP‐1 cells in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm). (d, e, f) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment (d: 6 nm; e: 30 nm; f: 60 nm). (g, h, i) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment (g: 6 nm; h: 30 nm; i: 60 nm). (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment (j: 6 nm; k: 30 nm; l: 60 nm). In (a, b, c) the significant increase of cell adhesion and spreading to the growth substratum and the trend to morphology change are evident. In (d, e, f), the effects of PMA withdrawal during recovery in complete medium are shown: in e, in particular, the appearance of cytoplasm projections in cells with stellate morphology is appreciable. (g, h, i) In culture submitted to recovery in serum‐free medium after PMA treatment, cells with stellate morphology are not visible. (j, k, l) Effects induced by retinoic acid stimulation are shown: in particular, in (k), increase in numbers of cells with long projections is visible. Bars 5 μm.
Figure 5.
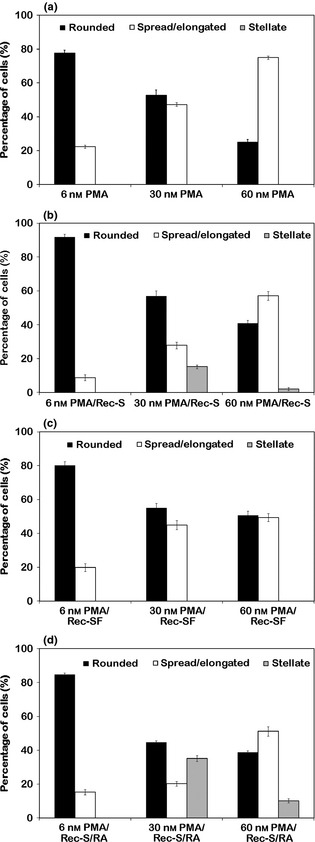
Percentage distribution of main morphological features in still adherent THP ‐1 cells in the different experimental conditions. (a) Incubation for 72 h with PMA at increasing concentrations. (b) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment. (c) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment. (d) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment. The reported values represent the mean ± SEM of triplicate experiments. PMA: treatment with Phorbol 12‐myristate 13‐acetate; PMA/Rec‐S: recovery in PMA‐free medium containing serum, after PMA treatment; PMA/Rec‐SF: recovery in PMA‐free medium in the absence of serum, after PMA treatment; PMA/Rec‐S/RA: stimulation with retinoic acid, after recovery in PMA‐free medium containing serum, of cells derived from PMA treatment.
Cell cycle, apoptotic index and cell morphology analyses, after PMA withdrawal and RA stimulation, in cultures maintained in the presence of serum
To further analyse dynamics of differentiation pathways of the THP‐1 cell line, and in attempt to increase numbers of DCs obtained after PMA treatment and recovery in PMA‐free complete medium, we stimulated cultures with 4 μm RA for 72 h. Choice of this single dose was based on previous experiments in which concentrations from 1 to 10 μm had been used. Over this range, RA stimulation of cultures recovered in the absence of serum, after PMA treatment, did not determine appearance of cells with dendritic morphology (data not shown); however, in the presence of serum, 4 μm RA was seen to be the optimal dose to produce highest numbers of cells with the dendritic phenotype.
With respect to the condition of recovery only in the presence of serum after PMA treatment (Fig. 1d–f; Table 1), flow cytometry measurements of DNA content, after RA stimulation (Fig. 1j–l), showed anti‐apoptotic effects, particularly after 30 nm PMA treatment (Fig. 1k); moreover, in all conditions, proliferative activity was significantly inhibited, as demonstrated by BrdUrd uptake evaluation (Table 1).
Stimulation of PMA‐untreated THP‐1 cells, with 4 μm RA, did not cause cell adhesion and apoptosis with respect to control conditions (PMA‐untreated THP‐1 cells); and there were no significant changes in cell cycle progression, as demonstrated intermediate ploidy DNA content values and BrdUrd uptake evaluation, by flow cytometry (Table 1).
From a morphological point of view (Fig. 4j–l), RA stimulation caused increase in stellate shaped cells, in cultures derived from 30 and 60 nm PMA treatments (Fig. 4k and 4l), with respect to corresponding conditions of recovery only in complete medium after PMA treatment (Fig. 4e and 4f). Cell subset percentage, after RA stimulation, is reported in Fig. 5d and highest increase in stellate cells, observed in 30 nm PMA derived cultures, appeared to be related to reduction in spread/elongated and over 4c (see flow cytometric data in Fig. 1) cells.
Immunolabelling of CD14, CD11b and DC‐SIGN in THP‐1 cells
To study influence of experimental conditions on dynamics of cell subpopulations constituting the cell model used in this work, we evaluated, by fluorescence microscopy and static cytofluorometry, expression of CD14 and CD11b, markers of phenotype modification of the monocyte/macrophage lineage, and expression of DC‐SIGN, a molecule typical of monocyte‐derived dendritic cells.
CD14 immunolabelling at doses of 6 nm PMA, fluorescence microscopy images had intense fluorescence in almost all cells, even in those not of rounded shape (Fig. 6a). At 30 nm PMA, drastic reduction in number of CD14 expressing cells was observed (Fig. 6b), while at 60 nm PMA, CD14 expression was almost completely lost (Fig. 6c). These morphological data are supported by cytofluorometric analyses (Fig. 7a–c) that revealed progressive reduction in percentage of positively immunolabelled cells, in relation to increase in PMA concentrations.
Figure 6.
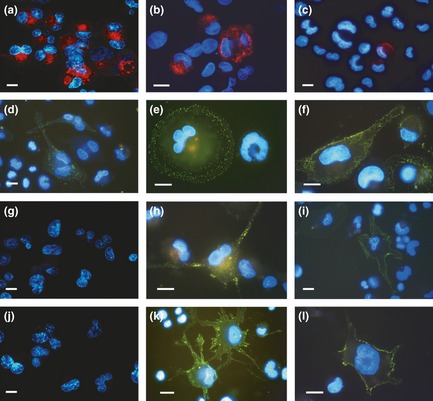
Fluorescence microscopy of THP‐1 cells in the different experimental conditions after immunostaining of cell surface markers. (a, b, c) CD14 (red fluorescence) and (d, e, f) CD11b (green fluorescence) after incubation for 72 h with PMA at increasing concentrations. (g–l) DC‐SIGN immunoreaction (green fluorescence): (g, h, i) recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment. (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment. Nuclei were counterstained with Hoechst 33342. PMA treatments: a, d, g, j: 6 nm; b, e, h, k: 30 nm; c, f, i, l: 60 nm. Bars 5 μm.
Figure 7.
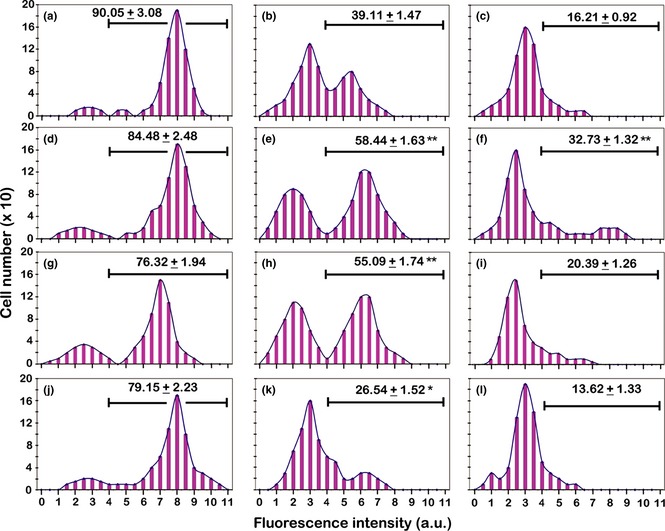
Static cytometric analyses of CD14 immunolabelling in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm). (d, e, f) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment (d: 6 nm; e: 30 nm; f: 60 nm). (g, h, i) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment (g: 6 nm; h: 30 nm; i: 60 nm). (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment (j: 6 nm; k: 30 nm; l: 60 nm). The values relative to each histogram represent the mean ± SEM of the cells that express CD14 and are calculated by considering positive the cells with fluorescence intensity higher than 4 arbitrary units (a.u.) (see Materials and methods). Histograms are representative of three independent experiments. The asterisks (*P < 0.05; **P < 0.001 after Student's t‐test) indicate the values that are significantly different from the respective referring condition: left column (d, g, j) versus (a); central column (e, h, k) versus (b) and right column (f, i, l) versus (c).
As previously described, phorbol ester withdrawal and culture maintenance in complete medium for 72 h caused cell detachment and induction of apoptosis. Moreover, in cultures derived from 30 nm and, to a lesser degree, from 60 nm PMA treatments, some still adherent cells had morphological features typical of dendritic phenotype. In this step of the experimental procedure, CD14 immunolabelling indicated that in cultures derived from 6 nm conditions, PMA withdrawal did not induce evident changes in levels of cells expressing CD14, in both cultures maintained in the presence (Fig. 7d). and in the absence of serum (Fig. 7g). In cultures previously stimulated with 30 nM PMA, withdrawal of phorbol ester caused significant elevation in percentage of cells expressing CD14 in both cultures, in presence (Fig. 7e). and in absence of serum (Fig. 7h). In cultures derived from 60 nm PMA treatment, absence of phorbol ester, in presence of serum, induced elevation in cells expressing CD14 (Fig. 7f), while in absence of serum (Fig. 7i), CD14 expression appeared to be similar to that for PMA treatment only (Fig. 7c).
In a series of parallel experiments, PMA‐treated THP‐1 cells recovered in complete medium were then stimulated with RA, to investigate influence of this substance on appearance and differentiation of dendritic cells in the different experimental conditions. In cultures derived from 6 nm PMA treatment, RA stimulation, after recovery in PMA‐free medium, did not cause evident changes in percentages of cells that expressed CD14 (Fig. 7j), in comparison to recovery only conditions (Fig. 7d), while in cultures derived from 30 and 60 nm PMA treatments recovered in complete medium, RA stimulation (Fig. 7k and 7l) induced a trend towards reduction in CD14 expressing cells.
CD11b integrin immunofluorescence microscopy revealed positivity, after PMA treatment (especially for 30 and 60 nm doses) of adherent cells, with variable immunolabelling intensity and distribution in relation to cell morphology (Fig. 6d–f). After 6 nm PMA treatment, few cells had only low levels of positivity (Fig. 6d). Under 30 nm PMA conditions, higher numbers of cells were positive, and marked fluorescence was observed mainly at the periphery of large, rounded cells (Fig. 6e). Finally, at 60 nm PMA, fibroblastoid cells of elongated shape were strongly positive with integrin expression distributed all over cell surfaces (Fig. 6f).
For quantitative data, analysis of CD11b cytofluorometric histograms (Fig. 8) revealed more positive cells with high fluorescence emissions, depending on PMA concentrations (Fig. 8a–c). Fluorescence values appeared to be distributed with multimodal trends, specially for the 30 nm PMA doses; this confirms heterogeneity of the cells at this point (Fig. 8b). For 60 nm PMA, static cytofluorometry revealed high percentages of CD11b expressing cells with high fluorescence intensity (Fig. 8c).
Figure 8.
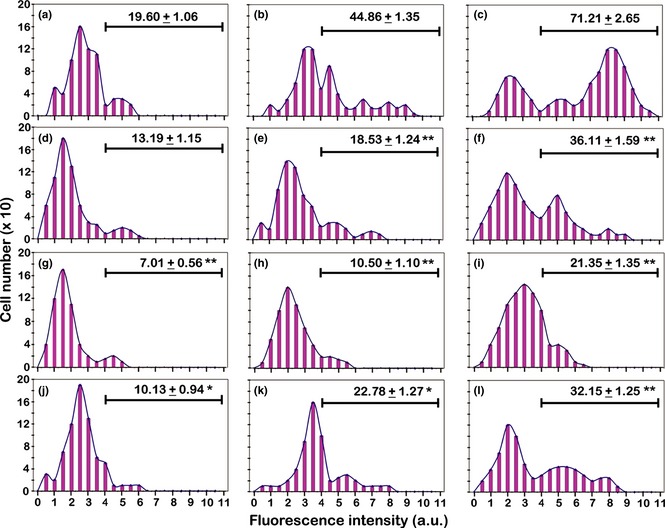
Static cytometric analyses of CD11b immunolabelling in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm). (d, e, f) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment (d: 6 nm; e: 30 nm; f: 60 nm). (g, h, i) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment (g: 6 nm; h: 30 nm; i: 60 nm). (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment (j: 6 nm; k: 30 nm; l: 60 nm). The values relative to each histogram represent the mean ± SEM of the cells that express CD11b and are calculated by considering positive the cells with fluorescence intensity higher than 4 arbitrary units (a.u.) (see Materials and methods). Histograms are representative of three independent experiments. The asterisks (*P < 0.05; **P < 0.001 after Student's t‐test) indicate the values that are significantly different from the respective referring condition: left column (d, g, j) versus (a); central column (e, h, k) versus (b) and right column (f, i, l) versus (c).
PMA withdrawal and recovery in complete medium, in 6 nm‐treated cultures (Fig. 8d), did not induce significant changes in CD11b expression in comparison to 6 nm PMA only treatment (Fig. 8a). For higher doses, withdrawal of phorbol ester (Fig. 8e and 8f) caused reduction in percentages of CD11b positive cells. Cultures recovered in serum‐free medium, after PMA withdrawal, had further reduced CD11b expression, in particular with 30 and 60 nm phorbol ester doses (Fig. 8g–i).
Retinoic acid stimulation recovered cells in complete medium, after PMA withdrawal (Fig. 8j–l), but did not restore CD11b expression to the levels measured for cultures submitted to PMA only treatment (Fig. 8a–c).
DC‐SIGN immunolabelling, analysed by fluorescence microscopy, was negative for cells in 6 nM, and rare cells with very low fluorescence, in 30 and 60 nm PMA conditions (data not shown): this was also demonstrated by cytofluorometric data (Fig. 9a–c). Fluorescence microscopy, performed on cells recovered in complete medium, after PMA stimulation (Fig. 6g–i), allowed us to observe clear DC‐SIGN expression in a fraction of cells derived from 30 and 60 nm treatments (Fig. 6h and 6i). Analysis of cytofluorometric histograms related to these experimental conditions showed expressions to be close to 35% and 15% in cultures derived from 30 and 60 nm PMA treatments, respectively (Fig. 9e and 9f). By correlating cytofluorometric measurements and morphological analysis, it emerged that cells of highly dendritic morphology, with long cytoplasmic projections, had fluorescence intensity higher than 7 a.u, approximately 10% in 30 nm and 5% in 60 nm PMA derived cells. Fluorescence histograms of cells recovered in the absence of serum, after PMA stimulation, did not show significant DC‐SIGN expression (Fig. 9g–i) with respect to cultures derived from 6, 30 and 60 nm PMA only treatment (Fig. 9a–c).
Figure 9.
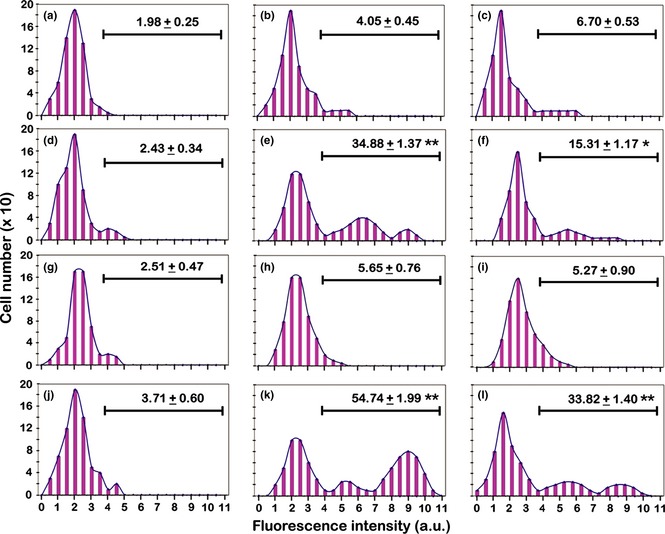
Static cytometric analyses of DC‐SIGN immunolabelling in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm). (d, e, f) Recovery for 72 h, in complete medium, of cultures derived from a previous PMA treatment (d: 6 nm; e: 30 nm; f: 60 nm). (g, h, i) Recovery for 72 h, in serum‐free medium, of cultures derived from a previous PMA treatment (g: 6 nm; h: 30 nm; i: 60 nm). (j, k, l) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from a previous PMA treatment (j: 6 nm; k: 30 nm; l: 60 nm). The values relative to each histogram represent the mean ± SEM of the cells that express DC‐SIGN and are calculated by considering positive the cells with fluorescence intensity higher than 4 arbitrary units (a.u.) (see Materials and methods). Histograms are representative of three independent experiments. The asterisks (*P < 0.05; **P < 0.001 after Student's t‐test) indicate the values that are significantly different from the respective referring condition: left column (d, g, j) versus (a); central column (e, h, k) versus (b) and right column (f, i, l) versus (c).
For RA stimulation after recovery in complete medium, fluorescence microscopy and static cytofluorometry demonstrated that this did not induce DC‐SIGN expression in 6 nm derived cultures, which appeared to be negative across almost in the whole population (Figs 6j,9j). In cultures derived from 30 nm PMA treatment, RA stimulation (Fig. 6k), after recovery in complete medium, induced increase percentages of DC‐SIGN‐positive cells, with high fluorescence values (Fig. 9k), compared to that observed for recovery only types (Fig. 9e). In 60 nm PMA‐treated and recovered cells, after incubation in the presence of RA (Fig. 6l), there was more limited (but significant) elevation in DC‐SIGN expressing cell percentages (Fig. 9l) in comparison to those of recovery only (Fig. 9f).
Actin cytoskeleton and adhesive
Morphological analysis after actin fluorochromization allowed us to observe shape, F‐actin architecture and motility of different cell phenotypes, features of which appeared to be partly related to treatment conditions used in the three‐step protocol described above. After 6 nm PMA treatment, cell populations were constituted of cells generally of small dimension, circular in shape and initially with adhesion to growth substratum, with actin that appeared to be mainly distributed in cortical areas of cytoplasm (Fig 10a). In 30 nm PMA‐treated samples, populations were constituted of larger cells with more evident cell adhesion to substrate. Here, actin accumulated in areas of interaction with the substrate, and elongated cells, indicative of potential motility were observed (Fig. 10b). At 60 nm PMA, cell populations were prevailingly constituted of cells in which actin fluorochromization showed presence of protrusions and ruffles; this is a feature of highly differentiated cells – macrophages (Fig. 10c). In our cell model, adhesive properties are directly related to drop in proliferative activity; moreover, it is to be noted that variability in organization of the actin cytoskeleton and cell motility can be related to cell heterogeneity and changes, over the various steps of differentiation. Panel d in Fig. 10 shows actin reorganization in a macrophage of intermediate level of differentiation, typical of 30 nm PMA treatment – this cytoarchitecture is expression of progressive adhesion to the substratum. With PMA stimulation at 30 nm (partially) and 60 nm (with higher percentage), cells had the ability to migrate, due to cytoplasmic extensions; in several cases, pseudopodia containing clusters of actin expression were visible (Fig. 10e). This distribution pattern, observed specially in differentiated cells prevalent in 60 nm PMA cultures, is indicative of presence of podosomes, dynamic adhesive structures that are a typical means by which macrophages adhere to a substrate. For 6, 30 and 60 nm PMA treatments stellate shaped cells of DC type, were not observed. In all these cultures, PMA withdrawal caused detachment of parts of cells which then detached and floated. In residual adherent cell populations, derived from 30 and 60 nm PMA treatments, appearance of cells that tended to take on a stellate morphology was observed. Mostly these had extended a number of short cell processes around the central cell body outline (Fig. 11a).
Figure 10.
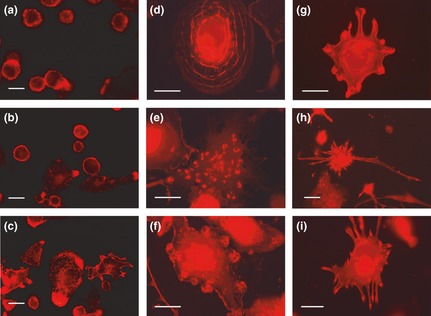
Fluorescence microscopy of actin organization in THP‐1 cells in the different experimental conditions. (a, b, c) Incubation for 72 h with PMA at increasing concentrations (a: 6 nm; b: 30 nm; c: 60 nm): actin distribution in relation to progressive macrophagic differentiation is shown. (d) Typical cell of 30 nm PMA condition, in which actin cytoskeleton organization appears to be related to adhesion to the substratum. (e) A cell representative of the prevalent population in 60 nm PMA culture in which it is possible to observe dot clusters of actin (podosomes). (f) Cell morphology after recovery for 72 h in complete medium of cultures derived from 30 nm PMA treatment: clusters of actin appeared to be accumulated at the cell periphery in areas of possible origin of cellular projections. (g, h, i) Possible dynamic steps of dendritic phenotype acquisition with the progressive extension of cellular processes (micrographs obtained after over‐exposure conditions to show better the thin cytoplasmic projections). Bars 5 μm.
Figure 11.
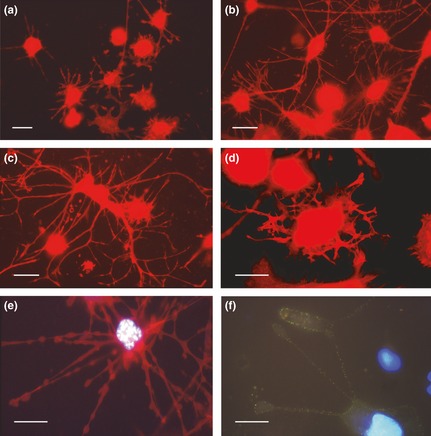
Fluorescence microscopy of actin and CD11b labelling in dendritic cells. (a–e) Actin (TRITC‐phalloidin – red fluorescence) organization and CD11b (f) distribution (FITC – green fluorescence) in dendritic cells. (a) Cell morphology after recovery for 72 h in complete medium of cultures derived from 30 nm PMA treatment: a group of stellate cells with short projections is visible. (b, c) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from 30 nm PMA treatment: cells with a clear dendritic phenotype with long and arborized processes that form an intricate network. (d, e, f) Stimulation with retinoic acid for 72 h, after recovery in complete medium, of cultures derived from 60 nm PMA treatment: (d) dendritic cell that shows retraction of cellular projections. (e, f) Bleb formation in dendritic cellular processes. This aspect was observed in cells with apoptotic features: (e) Hoechst 33342 fluorochromization shows apoptotic chromatin in relation to bleb formation. (f) CD11b immunostaining (low green fluorescence) shows the strong decrease in integrin expression in relation to bleb formation in a cell with hypercondensed chromatin (micrographs are obtained after over‐exposure conditions to show better the thin cytoplasmic projections). Bars 5 μm.
When RA was added to 30 nm PMA‐treated and recovered cultures, there was elevation in numbers of cells with multi‐process, stellate morphology. In this condition, cells with long and matted dendritic projections, tended to aggregate forming intricate networks (Fig. 11b and 11c). When RA was added to 60 nm PMA‐treated and recovered cultures, some cells retracted their dendritic processes (Fig. 11d), assuming typical morphology of migrating cells, or developing shapes compatible with translocation. Furthermore, in this condition in some cases, retraction of cell processes correlated with apoptotic nuclei (Fig. 11e). In these cells, fluorescence microscopy revealed reduction in expression of CD11b integrin (Fig. 11f). Panels f, g, h and i, Fig. 10, present a possible sequence of dynamic events leading to formation of cell projections, typical of the dendritic phenotype. These situations were observed in cultures recovered in complete medium after treatment with 30 or 60 nm PMA. In particular, in panel f, a cell with areas of probable origin of dendritic processes is shown, while in panels g, h and i, successive steps of cytoplasmic projection elongation can be seen.
Discussion
In this study, we analysed cellular aspects related to differentiation and apoptosis in the PMA‐stimulated THP‐1 monocytic cell line; this was subsequently submitted to a recovery step in PMA‐free medium and to a following incubation step with a further differentiating agent, RA. Under some experimental conditions, depending on level of differentiation induced by different PMA concentrations, the sequence of these steps determined not only a variable level of apoptotic events, but also of appearance of cells with the dendritic phenotype. Thus it appeared that differentiation and apoptosis were strictly related phenomena.
In other in vitro cell models, constituted of pluripotent hematopoietic progenitors, PMA treatment after suppression of proliferation, seemed to induce not only appearance of dendritic cells but also of death (in part by apoptosis), in cells that did not differentiate in our way 26. With regard to in vivo conditions, monocytes are circulating precursors of the different types of macrophages and dendritic cells 27, 28. Upregulation of numbers of monocytes may be required during inflammatory responses; in general, the balance of cell proliferation and cell removal is crucial for regulation of cell population numbers 29. As part of normal physiological cell processes, apoptosis regulates the equilibrium between cell proliferation and cell death 30 and in‐depth examination of these biological aspects can also be of fundamental importance to characterization of in vitro experimental models, for analysis of myeloid differentiation applied to studies of neoplastic transformation.
By considering differentiation of monocytes from freshly isolated peripheral blood, it is likely that some of the monocytes would be apoptotic and could not be differentiated into DCs. Moreover, the culture environment 31 may affect level of apoptosis. Thus, when cultured in medium in absence of an appropriate stimulus, monocytes rapidly undergo programmed cell death 32; from a practical point of view, it is important to optimize both isolation procedures and culture conditions of monocytes for DC production. In our experimental model, occurrence of apoptosis and dendritic cell differentiation by the monocyte cell line used, appeared to be dependent on the balance between differentiation and dedifferentiation stimuli, resulting from specific experimental conditions in which cytokines were not used. Monocytes normally differentiate in vitro into DCs following culture with GM‐CSF plus IL‐4 13. In comparison to this seemingly simple procedure however, myeloid differentiation is complex, not fully understood and can be influenced by a number of variables. Yet, combined stimulation of human monoblastic leukaemia cell lines with IL‐4 and protein kinase C activators such as PMA and bryostatin, leads to expression of specific markers of DC phenotype 33. THP‐1 cells used in our work are characterized by monocyte‐like properties and undergo macrophage differentiation after exposure to phorbol ester, PMA 15, 34. According to our previous work, treatment of this cell line for 72 h with 6, 30 and 60 nm PMA is a representative model of gradual differentiation from monocytes to macrophages 16.
Here, THP‐1 cells underwent a three‐step procedure. The first step, treatment with three different PMA concentrations, induced adhesion of the cells and three cultures were obtained that displayed a prevalent phenotype characterized by cells with initial, intermediate and advanced levels of macrophage differentiation. Progressive reduction in proliferative activity correlated with increase in adhesion properties 16. These aspects can be related to variation in expression of differentiation markers indicative of monocyte‐macrophage progression. In particular, analysis of fluorescence microscopy images and cytofluorometric data after anti‐CD14 immunostaining, showed progressive reduction in percentages of CD14 expressing cells correlating with PMA increase. This is indicative of differentiation of the THP‐1 cells that tended to lose, especially at 30 and 60 nm PMA, features of monocytes. With reference to integrin CD11b expression, in general, there was a rise CD11b positive cells in relation to increases in PMA concentration; morphological and cytofluorometric analyses showed variable levels of CD11b expression in both distribution quantity. After 6 nm PMA treatment, few cells had even low levels of CD11b positivity. The intermediate condition (30 nm PMA), revealed cells positive mainly at the periphery, with pattern distribution correlating with actin staining, as seen from the images. Finally, at 60 nm PMA, elongated cells appeared to be strongly positive. Increase in CD11b expression could be related to progressive acquisition of adhesive capacity, specially after 30 or 60 nm PMA treatment.
For the second step, PMA withdrawal and recovery in complete medium caused high frequency of apoptosis demonstrated as floating cells (anoikis). Percentages of detached/apoptotic cells appeared to be related to previous treatment with the PMA concentration; cultures derived from 60 nm PMA treatment had highest apoptotic incidence and as consequence of high level of differentiation, cells demonstrated marked adhesion to the substrate. Removal of phorbol ester induced perturbation of this adherence, also related to molecular modification of the cell surface; this is indicative of high levels of anchorage‐dependent cells. Normally, in 60 nm and partly in 30 nm PMA‐treated cultures, fractions of adherent cells expressing high and intermediate levels of cell adhesion molecules (including of CD11b integrin) were protected from cell death 16. By considering that regulation of cell survival involves distinct mechanisms according to state of differentiation, reduction of expression of integrin after PMA withdrawal can contribute to apoptotic cell death. These molecules can elicit anchorage‐dependent survival signals, and adherent cells undergo anoikis when forced into suspension 35, 36, 37. Moreover, modification of cell adhesion, induced by removal of phorbol ester, also appeared to be related to attempts to recover proliferative activity. From this point of view it must be noted, that in addition to adhesion, spreading and migration, integrins have been found to perform more general functions in modulation of cell physiology, such as proliferation 36. Thus, cell detachment can be correlated to dedifferentiation phenomena, directly linked to a restart of proliferative activity, this is balanced by onset of apoptotic events. Scarcely differentiated cells (deriving from 6 nm PMA treatment and recovered in complete medium) restarted cell division processes more easily and had low levels of apoptosis. Cells at advanced stages of differentiation (derived from 60 nm PMA treatment and recovered in complete medium) had poor capacity for restoring proliferative activity and had high levels of apoptosis. Relationships between these cell properties was supported by parallel experiments in which serum deprivation during recovery steps in PMA‐free medium, drastically inhibited not only cell proliferation, but also cell detachment and death, in all our experimental conditions. As has already noted, morphological analysis performed by means of fluorescence microscopy, demonstrated that large fractions of detached cells (both in presence and in absence of serum) were apoptotic. Comparison of data on cell detachment and apoptosis after recovery, in absence of serum, showed that cells with intermediate levels of differentiation (derived from 30 nm PMA treatment and recovered in PMA‐free medium) were less inhibited from detachment in comparison to those derived from 6 and 60 nm PMA treatments. This finding can be interpreted by considering heterogeneity of culture of this experimental condition, in which subsets of cells with different proliferative and differentiation capacities, probably coexisted. Moreover, considering cultures derived from 30 nm PMA treatment after recovery in complete medium, it was possible that there was a significant fraction of cells with intermediate features, or that a monocyte‐like fraction was bi‐potential being able to develop into either macrophages or dendritic cells. Nevertheless, we must account for how PMA removal lead to dendritic cell generation; it can be hypothesized that these cells might be derived from subpopulations in which PMA withdrawal would induce dedifferentiation (or partial dedifferentiation), followed by switching to a new differentiation pathway, leading to a further cell type, transdifferentiation. This could be a consequence of effects produced by absence of phorbol ester, and by concomitant presence of serum factors in the culture medium. This condition could might provide signals for dendritic cell differentiation. Even though such factors remain unknown, a number of workers have emphasised the importance of presence of FCS, in addition to GM‐CSF and IL‐4, in culture media during dendritic cell differentiation 38, 39. Importance of serum factors in our experimental model is supported by the observation that morphological analysis, and DC‐SIGN immunolabelling performed on serum‐free cultures derived after recovery of 30 and 60 nm PMA‐treated cells, did not reveal presence of any cells with dendritic cell features nor any that were positive after the immunoreaction.
In previous studies on macrophage heterogeneity and differentiation, other authors have reported influence of a variety of factors and serum proteins in promotion of phenotypic modulation of these cells 40. In our experimental model, it must be further underlined that variable differentiation ability of THP‐1 cells into macrophages or DCs could be influenced by their origin from neoplastically transformed cells, that could activate, or not, signal transduction pathways and haematopoietic differentiation programmes in response to different conditions of stimulation.
By considering these biological properties and to analyse effects of RA on dynamics of dendritic cell appearance, THP‐1 cells submitted to recovery in complete medium, were successively treated with a single dose of vitamin A, a derivative RA. In general, cytofluorometric data, relating to DC‐SIGN expression after RA stimulation, had different effects in relation to PMA pre‐treatment conditions. By considering scarce expression of DC‐SIGN in cultures derived from 6 nm pre‐treatment samples, it can be hypothesized that RA was unable to stimulate cells with low differentiative level towards the dendritic phenotype. Conversely, 30 nm PMA pre‐treated cells, dedifferentiating from intermediate levels of monocyte‐macrophage transition, were the most reactive to differentiaton stimuli. After recovery in PMA‐free medium, RA stimulation induced elevation in percentages of cells that expressed high levels of DC‐SIGN, with respect to cultures submitted to recovery only. Under these conditions, cultures (also including cells that did not express the dendritic phenotype), appeared to be highly protected from apoptosis, with respect to the recovery condition. Reduced apoptotic cell death could be also due to presence of these non‐dendritic cells that showed particular levels of differentiation, characterized by low expression of CD14 and CD11b, as a consequence of the de‐stimulation and stimulation conditions.
In comparison to cells derived from 30 nm PMA, RA stimulation after recovery, of 60 nm PMA‐derived cultures, induced a less elevation in DC‐SIGN expression, as indicated by percentages of cells shown to be positive by immunoreactions. This can be interpreted by considering that in cultures derived from 60 nm PMA treatment, cells at advanced stages of differentiation tend to dedifferentiate sparingly and prevailingly choose the apoptotic pathway after PMA withdrawal and recovery in the presence of serum. This could partly explain lower efficiency of RA (in subsequent steps) in both dendritic cell generation and apoptotic inhibition. Thus, it cannot be excluded that combination of subsequent stimuli, such as those exerted by PMA and RA, could be more efficient in cells with an intermediate phenotype (30 nm PMA). Furthermore, these aspects could be related to prevalence, under other experimental conditions, of cells with specific differentiation and maturity levels, in which varied structural features, adhesion and actin distribution could also be expressed.
In PMA‐treated cultures, cell shape, spreading and adhesion appeared to be dose‐dependent and strictly related to actin cytoskeleton organization. After differentiation induced by PMA, in particular at 30 and 60 nm doses, areas of actin predominance were evident, indicative of podosomes – adhesion complexes 41. In several cases, these structures were particularly observed at cell peripheries and in protrusions, pseudopodia and lamellipodia.
In 30 and 60 nm PMA‐treated cultures, after recovery in PMA‐free complete medium, podosomes could only be observed in lower numbers of cells. This could be related to appearance of cells expressing alternative differentiative ways. In this context, as far as DCs are concerned, their shape is directly related to adhesive and migration properties and their regulation is an important aspect of DC‐behaviour. Immature DCs are strongly adherent cells that spread and can form podosomes 42. In our experimental model, cells with similar features, could be observed specially in 30 nm PMA‐treated cells and samples recovered in complete medium cultures – tending to be flattened and prevailingly extended by numbers of short processes around the cell body. In general, these in vivo phenotypes were incompletely differentiated exhibiting behaviour patterns typical of immature DCs 43. After RA stimulation of 30 nm PMA‐treated and recovered cultures, dendritic cells had features similar to those seen of stationary condition cells, with wide branching ramifications (arborization) forming webs, these could be compared to in vivo shapes of DCs, positioned in peripheral tissues to capture foreign antigens. Moreover, we observed DCs able to retract their protrusions, showing some dynamic ability. This latter morpho‐functional observation was expressed prevailingly when RA was added to 60 nm PMA pre‐treated and recovered cultures. There, some cells retracted their dendritic processes, assuming typical morphology of migrating cells or shapes compatible with translocation 43. These could be related to maturation processes in which DCs lose podosomes and change from being slowly migrating cells into cells capable of high‐speed migration 44. Referring to other cell types constituting our cultures, presence of podosomes in only a fraction of cells (especially in 30 nm PMA pre‐treated and recovered cultures) indicated elements maintaining features of differentiated macrophages. The other cell subsets that lose podosomes, could possibly be either cells designed to follow a differentiation pathway towards mature dendritic phenotype or alternatively, could be programmed to undergo cell death. In some cases, particularly when RA was added to cultures derived from 60 nm PMA and recovered in complete medium, blebs were seen to have formed on cell processes and their retraction was correlated with typical apoptotic nuclear characteristics, such as chromatin hyper‐condensation. In cells with these features, molecules with adhesive roles such as CD11b, appeared to be poorly expressed. In general, apoptosis is an important event in biology of dendritic cells; defects in DC apoptosis have been linked to development of general autoimmunity with systemic autoimmune diseases appearing in murine models 45. Furthermore, high levels of spontaneous DC apoptosis have also been observed in breast cancer patients, and the significance of this is still unclear 46. In these biological situations, reciprocal influence that can be exerted by cell subsets constituting different cell systems may play important roles. Some evidence indicates that viable DCs are affected by death of other, neighbouring, cells 47, 48, 49.
These lines of evidence illustrate how obtaining cells of suitable in vitro systems are the basis for development of knowledge that can be also be important clinical fields; models of DCs have been already applied in the field of cancer immunotherapy. DCs have emerged to be good cellular tools, to initiate immune responses, induce immunological memory and break immunological tolerance of a resident tumour 50, 51. DC vaccination is a very promising approach for the immunotherapy for cancer. As tumour cells can express a whole array of tumour associated antigens (TAA), an ideal anti‐cancer vaccine could consist of DCs loaded with TAAs expressed by the tumour of that particular patient 52. In recent years, strategic attempts have been exploited for generation of DCs for use in clinical trials and many kinds of protocols have been designed to load antigens on to DCs. Together, these findings make it possible to initiate clinical studies with antigen‐loaded DCs for cancer patients. To date, DCs of different sources have been exploited for clinical use – circulatory DCs can be directly isolated from peripheral blood, but numbers are small. On the other hand, isolation of CD34+ cells requires pre‐treatment of patients with cytokines such as G‐CSF to increase the percentage of scarce CD34+ cells in the blood, and DC generation from isolated CD34+ cells requires prolonged culture in a complex set of cytokines. Alternatively, monocytes can be enriched by using a range of methods and converted into immature DCs, which can then be differentiated into populations of fully mature DCs, using autologous monocyte‐conditioned medium or its mimic, based on interleukin cocktails 53. Even though these methods are used for studying both basic DC biology and cancer vaccination trials, the workload to generate large numbers of DCs for clinical application, according to standard methods, is huge. In contrast, from a methodological point of view, our work, after further cell model characterization and definition, could represent a new basis for development of simple and cheap procedures for obtaining DCs from monocytes without using cytokines. Moreover, it is useful to consider that our study reveals a cell model in which DC differentiation from monocytes is related to appearance of apoptotic dendritic‐like cells. In our opinion, this is a further interesting consideration, as previously discussed, as DC apoptosis has been observed in patients with cancer, and DC apoptosis has been implicated in development of autoimmune disease. Prospectively also, our cell model could be used to elucidate and modulate relationships between apoptosis and differentiation of DCs during their maturation.
In conclusion, referring to our experimental cell model, variability of cell populations obtained and induction of apoptosis under different conditions, could be the result of a balance of cell stimulation and de‐stimulatory conditions, in which PMA treatment and withdrawal play a central role. Our findings suggest that interruption/modification of intracellular signalling pathway(s) mediated by activation of protein kinase C PMA‐induction can determine onset of apoptotic phenomena or cell reprogramming towards alternative differentiation ways, depending on level of differentiation and in relation to the cytokinetic features of the cells. In this context, interactions through diffusible molecules and/or direct contacts between different cell subsets constituting our cultures, could be of paramount importance.
Conflict of interest
None declared.
Acknowledgements
Our work is supported by grants from Fondazione Banco di Sardegna.
References
- 1. Mollinedo F, López‐Pérez R, Gajate C (2008) Differential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL‐60 cells. Gene 419, 16–26. [DOI] [PubMed] [Google Scholar]
- 2. Andreesen R, Brugger W, Scheibenbogen C, Kreutz M, Leser HG, Rehm A et al (1990) Surface phenotype analysis of human monocyte to macrophage maturation. J. Leukoc. Biol. 47, 490–497. [DOI] [PubMed] [Google Scholar]
- 3. Prieto J, Eklund A, Patarroyo M (1994) Regulated expression of integrins and other adhesion molecules during differentiation of monocytes into macrophages. Cell. Immunol. 156, 191–211. [DOI] [PubMed] [Google Scholar]
- 4. Poli G, Wang JM, Ruco L, Rossini S, Biondi A, Mantovani A et al (1993) Expression and modulation of a mononuclear phagocyte differentiation antigen (PAM‐1) during in vitro maturation of peripheral blood monocytes. Int. J. Clin. Lab. Res. 23, 83–87. [DOI] [PubMed] [Google Scholar]
- 5. Peters JH, Gieseler R, Thiele B, Steinbach F (1996) Dendritic cells: from ontogenetic orphans to myelomonocytic descendants. Immunol. Today 17, 273–278. [DOI] [PubMed] [Google Scholar]
- 6. Romani N, Reider D, Heuer M, Ebner S, Kämpgen E, Eibl B et al (1996) Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J. Immunol. Methods 196, 137–151. [DOI] [PubMed] [Google Scholar]
- 7. Palucka K, Ueno H, Roberts L, Fay J, Banchereau J (2010) Dendritic cells: are they clinically relevant? Cancer J. 16, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stevenson FK, Palucka K (2010) Understanding and activating immunity against human cancer. Curr. Opin. Immunol. 22, 212–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palucka K, Ueno H, Zurawski G, Fay J, Banchereau J (2010) Building on dendritic cell subsets to improve cancer vaccines. Curr. Opin. Immunol. 22, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S et al (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J. Exp. Med. 176, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Romani N, Gruner S, Brang D, Kämpgen E, Lenz A, Trockenbacher B et al (1994) Proliferating dendritic cell progenitors in human blood. J. Exp. Med. 180, 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kiertscher SM, Roth MD (1996) Human CD14 + leukocytes acquire the phenotype and function of antigen‐presenting dendritic cells when cultured in GM‐CSF and IL‐4. J. Leukoc. Biol. 59, 208–218. [DOI] [PubMed] [Google Scholar]
- 13. Sallusto F, Lanzavecchia A (1994) Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony‐stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kreutz M, Andreesen R, Krause SW, Szabo A, Ritz E, Reichel H (1993) 1,25‐dihydroxyvitamin D3 production and vitamin D3 receptor expression are developmentally regulated. Blood 82, 1300–1307. [PubMed] [Google Scholar]
- 15. Kohro T, Tanaka T, Murakami T, Wada Y, Aburatani H, Hamakubo T et al (2004) A comparison of differences in the gene expression profiles of phorbol 12‐myristate 13‐acetate differentiated THP‐1 cells and human monocyte‐derived macrophage. J. Atheroscler. Thromb. 11, 88–97. [DOI] [PubMed] [Google Scholar]
- 16. Spano A, Monaco G, Barni S, Sciola L (2007) Expression of cell kinetics and death during monocyte‐macrophage differentiation: effects of Actinomycin D and Vinblastine treatment. Histochem. Cell Biol. 127, 79–94. [DOI] [PubMed] [Google Scholar]
- 17. Tao Y, Yang Y, Wang WP (2006) Effect of all‐trans‐retinoic acid on the differentiation, maturation and functions of dendritic cells derived from cord blood monocytes. FEMS Immunol. Med. Microbiol. 47, 444–450. [DOI] [PubMed] [Google Scholar]
- 18. Zapata‐Gonzalez F, Rueda F, Petriz J, Domingo P, Villarroya F, de Madariaga A et al (2007) 9‐cis‐Retinoic acid (9cRA), a retinoid X receptor (RXR) ligand, exerts immunosuppressive effects on dendritic cells by RXR‐dependent activation: inhibition of peroxisome proliferator‐activated receptor gamma blocks some of the 9cRA activities, and precludes them to mature phenotype development. J. Immunol. 178, 6130–6139. [DOI] [PubMed] [Google Scholar]
- 19. Mohty M, Morbelli S, Isnardon D, Sainty D, Arnoulet C, Gaugler B et al (2003) All‐trans retinoic acid skews monocyte differentiation into interleukin‐12‐secreting dendritic‐like cells. Br. J. Haematol. 122, 829–836. [DOI] [PubMed] [Google Scholar]
- 20. Goyert SM, Ferrero E, Rettig WJ, Yenamandra AK, Obata F, Le Beau MM (1988) The CD14 monocyte differentiation antigen maps to a region encoding growth factors and receptors. Science 239, 497–500. [DOI] [PubMed] [Google Scholar]
- 21. Steinbach F, Thiele B (1994) Phenotypic investigation of mononuclear phagocytes by flow cytometry. J. Immunol. Methods 174, 109–122. [DOI] [PubMed] [Google Scholar]
- 22. Arnaout MA (1990) Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 75, 1037–1050. [PubMed] [Google Scholar]
- 23. Hickstein DD, Smith A, Fisher W, Beatty PG, Schwartz BR, Harlan JM et al (1987) Expression of leukocyte adherence‐related glycoproteins during differentiation of HL‐60 promyelocytic leukemia cells. J. Immunol. 138, 513–519. [PubMed] [Google Scholar]
- 24. Sokoloski JA, Sartorelli AC, Rosen CA, Narayanan R (1993) Antisense oligonucleotides to the p65 subunit of NF‐kappa B block CD11b expression and alter adhesion properties of differentiated HL‐60 granulocytes. Blood 82, 625–632. [PubMed] [Google Scholar]
- 25. Mummidi S, Catano G, Lam L, Hoefle A, Telles V, Begum K et al (2001) Extensive repertoire of membrane‐bound and soluble dendritic cell‐specific ICAM‐3‐grabbing nonintegrin 1 (DC‐SIGN1) and DC‐SIGN2 isoforms. Inter‐individual variation in expression of DC‐SIGN transcripts. J. Biol. Chem. 276, 33196–33212. [DOI] [PubMed] [Google Scholar]
- 26. Davis TA, Saini AA, Blair PJ, Levine BL, Craighead N, Harlan DM et al (1998) Phorbol esters induce differentiation of human CD34+ hemopoietic progenitors to dendritic cells: evidence for protein kinase C‐mediated signaling. J. Immunol. 160, 3689–3697. [PubMed] [Google Scholar]
- 27. Fahy RJ, Doseff AI, Wewers MD (1999) Spontaneous human monocyte apoptosis utilizes a caspase‐3‐dependent pathway that is blocked by endotoxin and is independent of caspase‐1. J. Immunol. 163, 1755–1762. [PubMed] [Google Scholar]
- 28. Lund PK, Westvik AB, Joo GB, Ovstebo R, Haug KB, Kierulf P (2001) Flow cytometric evaluation of apoptosis, necrosis and recovery when culturing monocytes. J. Immunol. Methods 252, 45–55. [DOI] [PubMed] [Google Scholar]
- 29. Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vermes I, Haanen C, Reutelingsperger C (2000) Flow cytometry of apoptotic cell death. J. Immunol. Methods 243, 167–190. [DOI] [PubMed] [Google Scholar]
- 31. Lund PK, Namork E, Brorson SH, Westvik AB, Joo GB, Ovstebo R et al (2002) The fate of monocytes during 24 h of culture as revealed by flow cytometry and electron microscopy. J. Immunol. Methods 270, 63–76. [DOI] [PubMed] [Google Scholar]
- 32. Mangan DF, Welch GR, Wahl SM (1991) Lipopolysaccharide, tumor necrosis factor‐alpha, and IL‐1 beta prevent programmed cell death (apoptosis) in human peripheral blood monocytes. J. Immunol. 146, 1541–1546. [PubMed] [Google Scholar]
- 33. Li Y, Mohammad RM, al‐Katib A, Varterasian ML, Chen B (1997) Bryostatin 1 (bryo1)‐induced monocytic differentiation in THP‐1 human leukemia cells is associated with enhanced c‐fyn tyrosine kinase and M‐CSF receptors. Leuk. Res. 21, 391–397. [DOI] [PubMed] [Google Scholar]
- 34. Traore K, Trush MA, George M Jr, Spannhake EW, Anderson W, Asseffa A (2005) Signal transduction of phorbol 12‐myristate 13‐acetate (PMA)‐induced growth inhibition of human monocytic leukemia THP‐1 cells is reactive oxygen dependent. Leuk. Res. 29, 863–879. [DOI] [PubMed] [Google Scholar]
- 35. Xia H, Nho RS, Kahm J, Kleidon J, Henke CA (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3‐kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J. Biol. Chem. 279, 33024–33034. [DOI] [PubMed] [Google Scholar]
- 36. Bouchard V, Demers MJ, Thibodeau S, Laquerre V, Fujita N, Tsuruo T et al (2007) Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state‐specific uncoupling with the PI3‐K/Akt‐1 and MEK/Erk pathways. J. Cell. Physiol. 212, 717–728. [DOI] [PubMed] [Google Scholar]
- 37. Chiarugi P, Giannoni E (2008) Anoikis: a necessary death program for anchorage‐dependent cells. Biochem. Pharmacol. 76, 1352–1364. [DOI] [PubMed] [Google Scholar]
- 38. Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N (1996) Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods 196, 121–135. [DOI] [PubMed] [Google Scholar]
- 39. Ramadan G, Schmidt RE, Schubert J (2001) In vitro generation of human CD861 dendritic cells from CD341 haematopoietic progenitors by PMA and in serum‐free medium. Clin. Exp. Immunol. 125, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mollah ZU, Aiba S, Nakagawa S, Hara M, Manome H, Mizuashi M et al (2003) Macrophage colony‐stimulating factor in cooperation with transforming growth factor‐b1 Induces the differentiation of CD34+ hematopoietic progenitor cells into Langerhans cells under serum‐free conditions without granulocyte‐macrophage colony‐stimulating factor. J. Invest. Dermatol. 120, 256–265. [DOI] [PubMed] [Google Scholar]
- 41. Lehto VP, Hovi T, Vartio T, Badley RA, Virtanen I (1982) Reorganization of cytoskeletal and contractile elements during transition of human monocytes into adherent macrophages. Lab. Invest. 47, 391–399. [PubMed] [Google Scholar]
- 42. Linder S, Aepfelbacher M (2003) Podosomes: adhesion hot‐spots of invasive cells. Trends Cell Biol. 13, 376–385. [DOI] [PubMed] [Google Scholar]
- 43. Shutt DC, Daniels KJ, Carolan EJ, Hill AC, Soll DR (2000) Changes in the motility, morphology, and F‐actin architecture of human dendritic cells in an in vitro model of dendritic cell development. Cell Motil. Cytoskeleton 46, 200–221. [DOI] [PubMed] [Google Scholar]
- 44. van Helden SF, Krooshoop DJ, Broers KC, Raymakers RA, Figdor CG, van Leeuwen FN (2006) A critical role for prostaglandin E2 in podosome dissolution and induction of high‐speed migration during dendritic cell maturation. J. Immunol. 177, 1567–1574. [DOI] [PubMed] [Google Scholar]
- 45. Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ et al (2006) Dendritic cell apoptosis in the maintenance of immune tolerance. Science 311, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 46. Pinzon‐Charry A, Ho CS, Maxwell T, McGuckin MA, Schmidt C, Furnival C et al (2007) Numerical and functional defects of blood dendritic cells in early‐ and late‐stage breast cancer. Br. J. Cancer 97, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711. [DOI] [PubMed] [Google Scholar]
- 48. Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD et al (2000) A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N (2000) Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 191, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banchereau J, Palucka AK (2005) Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 5, 296–306. [DOI] [PubMed] [Google Scholar]
- 51. Schuler G, Schuler‐Thurner B, Steinman RM (2003) The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 15, 138–147. [DOI] [PubMed] [Google Scholar]
- 52. Novellino L, Castelli C, Parmiani G (2005) A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 54, 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feuerstein B, Berger TG, Maczek C, Roder C, Schreiner D, Hirsch U et al (2000) A method for the production of cryopreserved aliquots of antigen‐preloaded, mature dendritic cells ready for clinical use. J. Immunol. Methods 245, 15–29. [DOI] [PubMed] [Google Scholar]