

Abstract
Objectives: Forkhead Box M1 (FoxM1) transcription factor regulates expression of cell cycle effective genes and is stabilized by checkpoint kinase 2 (Chk2) to stimulate expression of DNA repair enzymes in response to DNA damage. This study intended to test whether FoxM1 is involved in cell cycle checkpoint pathways.
Materials and methods: Analysis of senescence and cell proliferation in FoxM1 transgenic (TG) mouse embryonic fibroblasts (MEFs) with 2‐fold elevation of FoxM1, and overexpression or knockdown of FoxM1 in an inducible FoxM1 expression cell line, or FoxM1 siRNA. Chromatin immunoprecipitation (ChIP), electrophoretic mobility shift assays (EMSA), and cotransfection to determine FoxM1 transcription targets, as well as RNase protection assays and western blot analysis, were performed.
Results: Two‐fold elevation of FoxM1 in FoxM1‐TG‐MEFs resulted in low levels of cell proliferation and increase in permanent cell cycle arrest at early passages (from passage 6 to 9). These phenotypes correlated with increased phosphorylation of p53 on Ser15, elevated expression of cell cycle inhibitor p21 and Chk1 at passage 3. FoxM1 was stabilized in response to DNA damage in MEFs and FoxM1 overexpression induced p21. Knockdown of FoxM1 resulted in decrease in Chk1. ChIP, EMSA and cotransfection assays confirmed that FoxM1 stimulated promoters of p21 and Chk1.
Conclusions: Chk1 and p21 are direct transcription targets of FoxM1 and FoxM1 participates in transcriptional responses to stress in normal cells.
Introduction
The term ‘cell senescence’ is used to describe that normal cells have a limited ability of proliferation in culture (1, 2). Normal cells, such as mouse embryonic fibroblasts (MEFs), eventually cease proliferation after initial robust cell division in vitro. It is believed that appearance of the senescent phenotype of normal cells is induced by continual stress from exogenous or endogenous sources during cell culture (progressive splitting and serial passaging), and relies on intact cell cycle checkpoint pathways, which prevent DNA replication and arrest the cell cycle in response to cell damage (2). Stimuli for senescence include genotoxic damage such as dysfunctional telomeres (3, 4, 5) or non‐telomeric DNA damage (6, 7) and non‐genotoxic considerations such as oxidative stress (8) or perturbations in chromatin organization (9, 10). Senescence‐inducing signals usually engage the p53 pathway, which plays an important role in cell cycle arrest in both stress‐ and telomere‐initiated senescence (4, 6, 11). For example, DNA damage activates a checkpoint network that consists of damage‐sensing kinase ataxia‐telangiectasia mutated (ATM), ATM and Rad3 related (ATR) (12), and downstream signalling effectors checkpoint kinase 1 (Chk1) and Chk2 (13). Damage signals are enhanced by subsequent phosphorylation of p53, whose transcriptional activity is stimulated by these kinases (14, 15, 16). p53 directly induces expression of cyclin‐dependent kinase inhibitor p21 (also termed CDKN1a, p21Cip1, Waf1 or SDI1), which is one of the cell cycle inhibitors often expressed by senescent cells (17). The p53‐p21 pathway, in concert with other checkpoint pathways, can establish and maintain permanent cell cycle arrest of MEFs by late passages (after passage 10).
The mammalian Forkhead Box (Fox) family of transcription factors consists of more than 50 mammalian proteins (18) that share homology in the winged helix DNA‐binding domain (19). FoxM1 (the splicing FoxM1b isoform) is expressed in all proliferating mammalian cells and tumour‐derived cell lines (20, 21), where it regulates transcription of cell cycle genes critical for progression to DNA replication and mitosis (22, 23, 24, 25). Mouse hepatocytes with conditional deletion of the FoxM1 gene show 80% reduction in DNA replication and complete inhibition of mitosis during liver regeneration (23). During G2/M and mitotic progression, FoxM1 stimulates transcription of genes coding for: cyclin B1, Cdc25B phosphatase, polo‐like kinase 1, aurora B kinase, survivin, centromere protein A (CENPA) and CENPB (22, 23, 24, 26, 27). FoxM1 transcriptional activity requires binding of Cdk‐cyclin complexes and subsequent phosphorylation at Thr596 of the FoxM1 carboxyl‐terminal region, to recruit CREB‐binding protein (CBP) coactivator protein (28). In G1/S phase of the cell cycle, there is evidence to show that FoxM1 stimulates DNA replication by activating transcription of S‐phase kinase‐associated protein 2 (Skp2) and CDK subunit 1 (Cks1) (24), which are subunits of the Skp1‐Cullin1‐F‐box (SCF) ubiquitin ligase complex that targets CDK inhibitor p21 and p27 for ubiquitin‐mediated proteasome degradation (29, 30, 31). Interestingly, FoxM1 is degraded through Cdh1 interaction in early G1 by anaphase‐promoting complex/cyclosome (APC/C) E3 ubiquitin ligase, and proteolysis of FoxM1 is important for entry into S phase (32), implicating that FoxM1 may possess functions other than stimulating DNA replication and mitosis. Recently, we provided evidence to show that FoxM1 is phosphorylated at Ser361 by DNA damage‐induced Chk2 and this modification resulted in stabilization of FoxM1 protein (33). FoxM1 directly stimulates transcription of base excision repair factor X‐ray cross‐complementing group 1 (XRCC1) (34) and breast cancer‐associated gene 2 (BRCA2), the latter being involved in homologous recombination repair of DNA double strand breaks (35, 36). Furthermore, FoxM1‐deficient cells display increased DNA breaks, suggesting that FoxM1 is involved in transcription regulation during DNA damage‐induced checkpoint signalling and DNA repair (33). This finding was supported by further recent work showing that induction of DNA repair gene XRCC1 and Chk 2/1 expression was mediated by FoxM1 in irradiated tumour cells (37).
During DNA replication for cell cycle progression, mechanisms balancing DNA synthesis, DNA damage checkpoints and DNA repair assure stability and integrity of genetic material. Based on published results showing that FoxM1 stimulates DNA synthesis and DNA repair, we have hypothesized that FoxM1 regulates and coordinates all three aspects of DNA replication during proliferation of normal cells. To determine whether FoxM1 participates in regulation of cell cycle checkpoints, it is necessary to establish a normal cell model that has intact cell cycle checkpoint pathways at experimental starting points. Primary culture of mouse embryonic fibroblasts provides such a normal cell model. In the present study, we have generated MEFs containing −800‐bp Rosa26 promoter‐driven 2.7‐kb human FoxM1b cDNA from transgenic (TG) mice, and have demonstrated that 2‐fold elevation of FoxM1 expression in TG MEFs resulted in decrease in cell proliferation and increase in permanent cell cycle arrest (cell senescence) in early passages (from passage 6 to passage 9), compared to wild‐type controls. Such phenotypes correlated with early onset expression of Chk1 and increased phosphorylation of p53 on Ser 15 residue at passage 3. Cell cycle inhibitor p21 expression was also induced in FoxM1 TG cells at passage 3. Chromatin immunoprecipitation (ChIP) assays determined that FoxM1 binds to endogenous promoters of Chk1 and p21. Furthermore, FoxM1 stimulated Chk1 and p21 promoters in cotransfection assays, suggesting that Chk1 and p21 genes were direct transcription targets of FoxM1 protein. These results identify a novel role for FoxM1 in transcription responses during normal cell senescence induced by cell stress.
Materials and methods
Generation and cell population doubling analysis of mouse embryonic fibroblasts
FoxM1 FVB/N TG mice containing −800‐bp Rosa26 promoter‐driven 2.7‐kb human FoxM1b cDNA in a transthyretin (TTR) minigene construct, in which FoxM1b cDNA was inserted into the second exon of the TTR minigene adjacent to SV40 virus transcriptional termination sequence (25), have been described previously (38). FoxM1 TG mice were mated to generate 13.5‐day embryos, for isolation of mouse embryonic fibroblasts. To isolate them from the embryos, liver, heart and head were removed and the remaining embryo was digested with 0.25% trypsin in 2.21 mm EDTA (Cellgro, Manassas, VA, USA) to isolate single cell suspensions of MEFs using standard procedures described in Hogan et al. (39). The heart tissue was used to generate DNA for PCR genotyping of the embryos as described previously (38). MEFs were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal calf serum (FCS), 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mm l‐glutamine, 0.1 mm MEM non‐essential amino acids and 55 μm 2‐mercaptoethanol, in a humidified 9% CO2 incubator under conditions described by Zindy et al. (40). To measure cell population doublings, 3T9 MEF culture protocol was used. Briefly, MEFs were trypsinized and each time 9 × 105 cells were plated on to one new 60 mm tissue culture plate. After 3 days culture, number of cells was counted and number of cell population doublings of each passage was calculated according to the formula log(cell number after 3 days growth/9 × 105)/log2. For each passage, cell counting was repeated three times. 3T9 protocols were repeated with three individually isolated MEF preparations. To measure cell proliferation capability, passage 7 MEFs were trypsinized and 1 × 104 cells were plated into each well of six‐well tissue culture plates and counted every day thereafter.
Procedure for senescence‐associated β‐galactosidase staining and BrdU staining of MEFs
In situ SA‐β‐galactosidase activity was detected as described previously (41, 42) with minor modifications. Passage 7 wild type (WT) or FoxM1 TG MEFs were washed in phosphate‐buffered saline (PBS) and fixed in 2% formaldehyde, 0.2% glutaraldehyde in PBS for 15 min at room temperature, washed twice in PBS, and stained with 1 mg/ml 5‐bromo‐4‐chloro‐3‐indolyl‐β‐galactoside (X‐gal) (pH 6.0) in 40 mm citric acid/sodium phosphate buffer containing 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 2 mm MgCl2 and 150 mm NaCl, for 16 h at 37 °C. Micrographs of β‐galactosidase‐stained MEFs were taken at ×200 magnification using a TE2000 microscope (Nikon, Tokyo, Japan). To measure DNA replication capability, bromodeoxyuridine (BrdU) was added to passage 7 MEFs 1 h before harvesting and cells were stained with BrdU‐specific antibody according to the manufacturer’s protocol (Roche, Basel, Switzerland).
Cell culture and transient transfection
Human osteosarcoma U2OS and mouse hepatoma Hepa1‐6 cells were grown in DMEM (Cellgro) supplemented with 10% FCS. U2OS clone C3 cell line (U2OS C3 cell) (28) that allowed doxycycline (Dox)‐inducible expression of green fluorescent protein (GFP)‐FoxM1b fusion protein was maintained in DMEM supplemented with 10% FCS plus 50 μg/ml of hygromycin B (Invitrogen, Carlsbad, CA, USA). Transient transfections were carried out using Fugene 6 (Roche) as described previously (28) with pCMV‐T7FoxM1b expression vector and one of the following reporter constructs: mouse Chk1 promoter regions or p21 promoter regions were PCR amplified from mouse MEF genomic DNA with the following primers: mChk1 −1818 bpXhoI: 5′‐TCT CTC GAG TAG TTA AGG AAT ATT GAT‐3′ or mChk1 −1618 bpXhoI: 5′‐ TCT CTC GAG GCA TCT TGG TTG GGT ATC TCC‐3′ and mChk1 + 22 bpHindIII: 5′‐TCT AAG CTT ATG ACT CCA AGC ACA GCG‐3′; or mp21 −1837 bpXhoI: 5′‐TCT CTC GAG GAC TCC AGT CTC TGC TTT‐3′ or mp21 −1705 bpXhoI: 5′‐TCT CTC GAG GTG CCG GGA TTA CAG ATG‐3′ and mp21 + 22 bpHindIII: 5′‐TCT AAG CTT CGG CTC ACA CCT CTC GGC‐3′. Resulting PCR mouse promoter fragments were cloned into corresponding XhoI and HindIII sites of pGL3 basic luciferase vector (Promega, Madison, USA). Assays of dual luciferase enzyme activity were performed according to the manufacturer’s protocol (Promega).
For siRNA treatment, hFoxM1 siRNA has been described previously (24). Human p53 siRNA (sc‐29435) and control siRNA (sc‐37007) were purchased from Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA. siRNA transfection was performed according to the manufacturer’s instructions.
RNase protection assay (RPA), RT‐PCR and western blotting
To measure RNA levels, total RNA was prepared using RNA‐STAT‐60 reagent (Tel‐Test “B” Inc., Friendswood, USA). RPAs were performed with [32P]UTP‐labelled antisense RNA synthesized from mouse FoxM1b exon4 genomic clone (43) and TTR transgene (25) as described previously (44). For RT‐PCR, cDNAs were synthesized using RevertAid™ first strand cDNA synthesis kits (Fermentas, Canada) with total RNA as template. PCR amplification was performed with Taq DNA polymerase (Promega) using the following sense(S) and antisense(AS) primers, annealing temperature (T a) and number of PCR cycles (N): mp21‐S, 5′‐AAC AGG ACG GTG ACT CCT ACT TCT G‐3′ and mp21‐AS, 5′‐GAA CGC GCT CCC AGA CGA AGT TGC‐3′ (T a: 60 °C, N: 33); mp53‐S, 5′‐AGT ATT TCA CCC TCA AGA TCC GCG G‐3′ and mp53‐AS, 5′‐AGA CCT GAC AAC TAT CAA CCT ATT CCC‐3′ (T a: 60 °C, N: 33); and mCyclophilin‐S, 5′‐GGC AAA TGC TGG ACC AAA CAC‐3′ and mCyclophilin‐AS, 5′‐TTC CTG GAC CCA AAA CGC TC‐3′ (T a: 57.5 °C, N: 22). mRNA levels of FoxM1 or Chk1 were monitored by real‐time PCR as described previously (24). The following sense (S) and antisense (AS) primer sequences and annealing temperatures (T a) were used for human mRNA: FoxM1‐S: 5′‐GGA GGA AAT GCC ACA CTT AGC G‐3′ and FoxM1‐AS: 5′‐TAG GAC TTC TTG GGT CTT GGG GTG‐3 (T a: 55.7 °C) and Chk1‐S: 5′‐CAC AGG TCT TTC CTT ATG GGA TAC C‐3′ and Chk1‐AS: 5′‐TGG GGT GCC AAG TAA CTG ACT ATT C‐3′ (T a: 55.7 °C). These real‐time RT‐PCR RNA levels were normalized to human cyclophilin mRNA levels: hCyclophilin‐S 5′‐GCA GAC AAG GTC CCA AAG ACA G‐3′ and hCyclophilin‐AS 5′‐CAC CCT GAC ACA TAA ACC CTG G‐3′ (T a: 55.7 °C).
To measure protein levels, cell lysates were prepared as described previously (24), and nuclear/cytosol fractions of cell lysates were prepared using nuclear/cytosol extraction kit according to the manufacturer’s protocol (BioVision Inc., San Francisco, USA). Protein samples were resolved by denaturing gel electrophoresis before electrotransfer to Protran nitrocellulose membranes. These were subjected to western blot analysis with antibodies to the proteins of interest. Signals from the primary antibody were amplified by HRP‐conjugated anti‐mouse IgG (Bio‐Rad, Philadelphia, PA, USA), and detected using Enhanced Chemiluminescence Plus (ECL‐plus; Amersham Pharmacia Biotech, Amersham, UK). The following antibodies and dilutions were used for western blotting: rabbit anti‐FoxM1 (1:5000) (24), mouse anti‐Chk1 (1:200; Santa Cruz, CA, USA, sc‐8408), mouse anti‐β‐Actin (1:20 000; Sigma, St. Louis, MO, USA, AC15), p53 antibody (1:1000; Cell Signaling, No. 9282), phospho‐p53ser15 antibody (1:1000; Cell Signaling, Denvers, MA, USA, No. 9284), rabbit anti‐p21 (1:500; Oncogene, PC55), mouse anti‐lamin A/C (1:200; Santa Cruz, sc‐7293) and mouse anti‐α‐tubulin (1:15 000; Sigma, T 9026).
Quantitative chromatin immunoprecipitation and electrophoretic mobility shift assays
ChIP assays were used to measure FoxM1 binding to endogenous promoter regions of mouse Chk1 and p21, as described previously (24). Primers used to amplify the following mouse gene promoter fragments were annotated with the binding position upstream of transcription start site, annealing temperature (T a) and whether in sense (S) or antisense (AS) orientation: Chk1 −1717S: 5′‐CCA ACC TGA ATC ACT TTC CCT G‐3′ and Chk1 −1602AS: 5′‐ATA CCC AAC CAA GAT GCT CGC‐3′ (T a: 54 °C) and p21 −2178S: 5′‐TGG ATG GAC GAC TTG GAT GC‐3′ and p21 −2096AS: 5′‐CAA ACC AGG ACA GAC ATA ATG GC‐3′ (T a: 54 °C). Normalization was carried out using the ΔΔC T method. Briefly, IP samples and total input threshold cycles (C T) for each treatment were subtracted from C T of the corresponding serum control IP (rabbit serum). The resulting corrected value for total input was then subtracted from corrected experimental IP value (ΔΔC T), and these values were raised to the power of two (2ΔΔC T). These were then expressed as relative promoter binding ± standard deviation (SD).
For electrophoretic mobility shift assays (EMSA), nuclear proteins were extracted from cells as described previously (45). Binding of FoxM1 to DNA oligonucleotides was initiated by mixing the following components: 1 ng [γ‐32P]ATP‐labelled DNA oligonucleotide fragment, binding buffer [20 mm Tris–HCl (pH 7.5), 5% glycerol, 40 mm KCl, 1 mm MgCl2, 0.5 mm DTT, 1 mm EDTA], 3 μg poly(dI‐dC), and 10 μg of nuclear extract. One microlitre of rabbit anti‐FoxA1 antiserum was added where indicated, in total reaction mixture of 30 μl. After 30 min incubation at room temperature, samples were loaded on 5% polyacrylamide gel. Electrophoresis was performed for 2 h at 150 V. Gels were subjected to autoradiography with Kodak XAR film and Fisher Biotech L‐Plus screens. The following DNA oligonucleotides were used: mouse Chk1 promoter −1651 to −1628 bp, 5′‐TCT ACT TTG CTG TTT GTT TTT TCT‐3′.
Statistical analysis
Microsoft Excel Program was used to calculate SD and statistically significant differences between samples, with the help of Student’s t‐test. Asterisks in each graph indicate statistically significant changes, P‐values calculated by Student’s t‐test: *P < 0.05, **P ≤ 0.01 and ***P ≤ 0.001. P‐values <0.05 were considered statistically significant.
Results
FoxM1 TG MEFs exhibit abolished cell proliferation and severe senescent phenotypes appeared in early passages
DNA damage‐induced Chk2 phosphorylated FoxM1 at Ser361 to stabilize the protein, which in turn stimulated expression of DNA repair genes XRCC1 and BRCA2 (33). FoxM1 was degraded through Cdh1 interaction in early G1 by anaphase‐promoting complex/cyclosome (APC/C) E3 ubiquitin ligase; proteolysis of FoxM1 is important for entry into S phase (32), suggesting that FoxM1 possesses a role in DNA damage‐induced checkpoint and DNA repair, in addition to its functions in cell proliferation. As studies of FoxM1 in cell cycle controls were usually performed with tumour cell lines or immortalized cells (such as NIH3T3) (46) whose cell cycle checkpoint machinery is not always intact, it has been difficult, if not biased, to observe any influence of FoxM1 on checkpoint control. To test whether FoxM1 was involved in damage or stress‐induced responses in normal cells, primary MEF cultures were prepared from either wild type (WT) or Rosa26‐FoxM1 TG 13.5 dpc embryos. The −800‐bp Rosa26 promoter region has been reported to drive ubiquitous tissue expression of exogenous genes in TG mice (47). Rosa26‐FoxM1 TG mice do not have any obvious deficiency during their lifetimes and have been successfully used in studies of pulmonary cell proliferation (38) and lung tumour development (48). In FoxM1 TG MEFs, FoxM1 expression was increased a modest 2‐fold compared to WT MEFs (Fig. 1a). It is known that passaging MEFs continually in culture results in cell senescence through oxidative stress‐induced DNA damage, which activates cell cycle checkpoints and DNA repair pathways (49). Here we first measured cell population doubling frequency of MEFs during cell culture, at each passage, according to 3T9 protocols. After initial robust cell proliferation (doubling number >2), TG MEFs exhibited dramatic decrease in cell doubling as early as passage 6 (no cell proliferation when doubling number = 1) (Fig. 1b). On the other hand, WT MEFs showed deficiency of cell proliferation until passage 11 (Fig. 1b), suggesting that elevated FoxM1 in TG MEFs abolished the ability of cell proliferation even at early passages. Early onset of cell proliferation deficiency in TG MEFs correlated with early appearance of senescent cell morphology; for example, passage 7 TG cells seemed enlarged and had flat morphology typical of senescent cells, although WT MEFs at passage 7 exhibited normal fibroblast morphology (Fig. 1c). The senescent phenotype can be characterized using positive staining of β‐galactosidase (β‐gal) activity with 5‐bromo‐4‐chloro‐3‐indolyl‐β‐galactoside (X‐Gal) to detect senescence‐associated β‐gal in situ (50). TG MEFs at passage 7 had increased numbers of β‐gal positive cells compared to WT controls (Fig. 1d). Furthermore, cell growth curves of MEFs from passage 7 illustrated the diminished proliferation capability of TG MEFs (Fig. 1e). Deficiency of TG MEF proliferation at early passages was further confirmed by detection of 5‐bromodeoxyuridine (BrdU) incorporation at passage 7. TG and WT MEFs at passage 7 had been cultured with BrdU for 1 h before staining with BrdU‐specific monoclonal antibody. TG MEFs exhibited dramatic decreases in cell numbers with BrdU‐positive staining which represented active DNA synthesis (Fig. 1f). This indicated that 2‐fold elevation of FoxM1 expression in TG MEFs resulted in earlier appearance of senescent phenotypes than in control WT MEFs.
Figure 1.
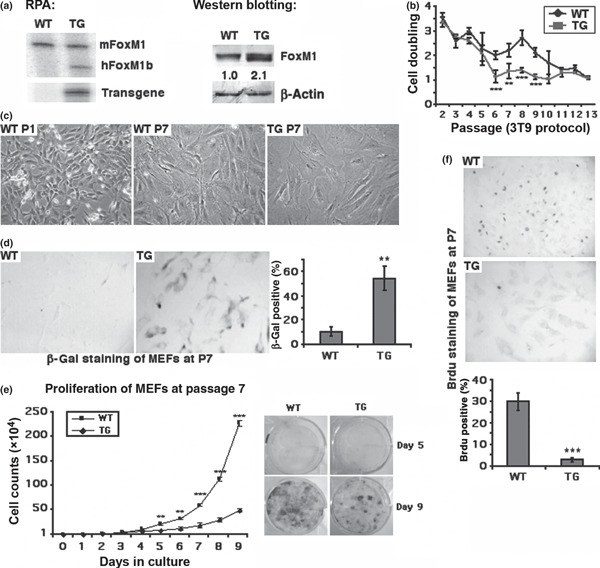
Two‐fold elevation of FoxM1 expression in FoxM1 TG MEFs abolished cell proliferation and resulted in early appearance of the senescent phenotype during MEF culture. MEFs were isolated from WT or Rosa26‐FoxM1 TG embryos at 13.5 dpc. (a) Elevated FoxM1 expression in FoxM1 TG MEFs. Total RNA or protein lysates were isolated from WT and TG MEFs at passage 1. mRNA levels of mouse endogenous FoxM1 and human FoxM1 transgene expression were measured by RNase Protection Assays (RPA) and protein levels of FoxM1 were measured by western blotting. (b) Cell population doubling events were measured from cultured WT and TG MEFs according to 3T9 protocol. Experiments were independently repeated three times with WT or TG MEF cultures isolated from separate embryos. (c) Morphology of WT MEFs at passage 1 (WT P1) or passage 7 (WT P7) and FoxM1 TG MEFs at passage 7 (TG P7) exhibit enlarged cell size and expanded cell shape. All images taken at 200× magnification using a TE2000 microscope (Nikon). (d) TG MEFs at passage 7 exhibit increased levels of β‐galactosidase (β‐gal) staining. WT and TG MEFs at passage 7 stained with 5‐bromo‐4‐chloro‐3‐indolyl‐β‐galactoside (X‐Gal) to detect senescence‐associated β‐gal activity in situ, and β‐gal positive cells were counted. (e) TG MEFs from passage 7 exhibit lower proliferation rates. Growth curves were calculated for WT and TG MEFs from passage 7 of MEF culture. 1 × 104 cells were seeded in each well (in triplicate) of six‐well tissue culture plates and cell numbers were counted every day thereafter. Day 5 and day 9 cultures were chosen to be stained with trypan blue and cell density of WT or TG MEFs are shown (f) TG MEFs at passage 7 exhibit decreased levels of DNA synthesis. WT and TG MEFs at passage 7 were cultured with bromodeoxyuridine (ΒrdU) for 1 h before staining with BrdU‐specific monoclonal antibody to detect DNA synthesis; BrdU positive cells were counted. Asterisks indicate statistically significant differences between WT and TG MEFs: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Two‐fold elevation of FoxM1 in MEFs increased p53 phosphorylation and p21 levels at passage 3 of MEF culture, indicating p21 as being one of FoxM1’s transcriptional targets
Stress‐induced signals, including those that trigger DNA‐damage responses, usually engage the p53 pathway. To test whether early onset of senescent phenotypes in FoxM1 TG MEFs correlated with activation of p53, we measured levels of p53 Ser 15 residue phosphorylation mediated by DNA‐integrity checkpoint pathways (14, 51). We chose WT and FoxM1 TG MEFs at passage 3, when both types of cell had no signs of senescent phenotypes, to prepare cell lysates. We found that FoxM1 TG MEFs at passage 3 already displayed p53 phosphorylation at Ser 15 (Fig. 2a, western blot). Expression of cell cycle inhibitor p21, which is one of the transcriptional targets of p53, was also induced in TG MEFs at passage 3 at both mRNA and protein levels compared to WT samples (Fig. 2a, RT‐PCR and western blot). Our group has already shown that FoxM1 is stabilized in response to DNA damage, by Chk2‐mediated phosphorylation, in tumour cell lines (33). To test whether DNA damage‐induced stabilization of FoxM1 occurred in normal cells, we treated WT MEFs at passage 3 with γ‐radiation (20 Gy). Induction of FoxM1 proteins after DNA damage correlated with increase in phosphorylation of p53 at Ser 15 residue and p21 proteins (Fig. 2b). We also found that FoxM1 protein was stabilized and localized to the nucleus after DNA damage (Fig. 2c). These results suggested that 2‐fold elevation of FoxM1 in MEFs enhanced stress‐induced signals during cell culture processes and might mediate early onset of senescence.
Figure 2.
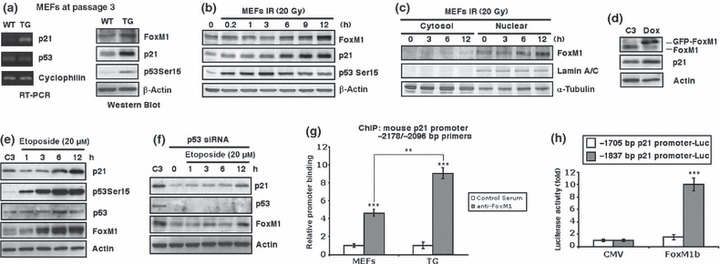
FoxM1 TG MEFs display increased levels of phosphorylation of p53 at Ser15 and p21 and FoxM1 stimulates p21 expression. (a) Passage 3 WT and TG MEFs were used to prepare RNA and cell extracts. Levels of p21, p53 and cyclophilin mRNA were analysed by RT‐PCR; levels of p53 Ser15 phosphorylation, p21, FoxM1 and β‐actin were determined by western blotting. (b) Stabilization of FoxM1 protein after genotoxic stress correlates with increased levels of phosphorylation of p53 at Ser15 and p21 protein. Twenty Gray of γ‐radiation (IR) was used to induce DNA damage in WT MEFs and extracts were prepared at indicated time points after IR exposure. Protein levels of FoxM1, p21, phosphorylation of p53 at Ser15, or β‐actin were measured by western blotting. (c) Stabilized FoxM1 by DNA damage stays in nucleus. Extracts were prepared from the cytosol and nuclear portions of MEFs at indicated time points after exposure to 20 Gy of IR and immunoblotted for FoxM1, Lamin A/C, or α‐tubulin protein levels. (d) Conditional expression of FoxM1 protein in U2OS cells stimulates p21 protein. U2OS C3 cells were induced for GFP‐FoxM1 expression by doxycycline treatment and then cell extracts were prepared 48 h later and analysed for FoxM1, p21 and β‐actin protein levels by western blotting. (e, f) Stabilization of FoxM1 protein after genotoxic stress correlates with increased p21 with or without p53. U2OS C3 cells (e) or cells transfected with p53 siRNA (f) were treated with etoposide (20 μm) to induce DNA damage. Extracts were prepared at the indicated time points and protein levels of p21, phosphorylation of p53 at Ser15, p53, FoxM1, or β‐actin were measured by Western blotting. (g) Chromatin immunoprecipitation (ChIP) assays show direct binding of FoxM1 to the endogenous mouse p21 promoter regions. WT or TG MEF chromatin was cross‐linked, sonicated and then immunoprecipitated (IP) with either FoxM1 antiserum (24) or rabbit serum (control), and the amount of promoter DNA associated with the IP chromatin was quantified using qRT‐PCR with primers specific to the mouse p21 promoter region. (h) Mouse p21 −1837 bp promoter stimulated by FoxM1 in cotransfection assays. Cotransfection assays were performed in Hepa1‐6 cells with the CMV‐FoxM1b expression vector and luciferase plasmid containing −1837 bp or −1705 bp of the mouse p21 promoter region, prepared protein extracts at 24 h after transfection and used them to measure dual luciferase enzyme activity. The asterisks indicate statistically significant changes: **P ≤ 0.01; ***P ≤ 0.001.
To further test whether FoxM1 directly regulated p21 expression, we used constructed human osteosarcoma U2OS C3 cells, in which GFP‐FoxM1b fusion protein could be induced by doxycycline treatment (52). We found that increased FoxM1b levels stimulated expression of endogenous p21 proteins (Fig. 2d). In this cell line, treatment with DNA‐damaging agent etoposide also induced levels of FoxM1, which correlated with increase in phosphorylation of p53 at Ser15 and p21 proteins (Fig. 2e). To confirm that increased FoxM1 levels contributed to enhanced p21 expression after DNA damage, we transfected the cells with p53 siRNA to knockdown expression of this known stimulator of p21 (Fig. 2f, p53 panel). After p53 knock down, levels of p21 decreased as expected, and interestingly FoxM1 also decreased (Fig. 2f, p21 and FoxM1 panels). In p53‐depleted cells, etoposide still caused increase in levels of FoxM1 protein at 12 h after treatment (Fig. 2f, FoxM1 panel). Increased levels of FoxM1 correlated with increased p21 at this time point (Fig. 2f, p21 panel), suggesting that FoxM1 could truly transactivate p21 after DNA damage.
After the −3‐kb promoter region of mouse p21 gene was analysed with FoxM1 DNA binding consensus sequence, we found that four FoxM1 putative binding sites overlapped between −1812 and −1792 bp (ATT TTT GTT TGT GTT TGT TTT). We next used quantitative chromatin immunoprecipitation assays to determine whether FoxM1 is bound to endogenous mouse p21 promoter regions. WT or TG MEF chromatin was cross‐linked, sonicated to DNA fragments of 500–1000 nucleotides in length, and then immunoprecipitated (IP) with antibodies specific to either FoxM1 (24) or rabbit serum (control), and amount of promoter DNA associated with IP chromatin was quantified using qRT‐PCR with mouse p21 promoter region (−2178 to −2096 bp)‐specific primers. These primers were made to DNA sequences situated near the potential FoxM1‐binding sites. Quantitative ChIP assays showed that FoxM1 protein was bound to endogenous mouse p21 promoter region, and in TG MEFs, there were nearly 2‐fold more FoxM1 bindings at the p21 promoter than that in WT MEFs (Fig. 2g). We performed negative control ChIP assays with primers of the TTR promoter region. This control ChIP experiment demonstrated that neither FoxM1 antibody nor IgG serum immunoprecipitated significant levels of this proximal TTR promoter region, from either untransfected or FoxM1‐depleted U2OS cell extracts (Fig. S1a). As positive controls, we performed ChIP assays with primers of promoter regions of Cdc25B, XRCC1 and BRCA2, known FoxM1 transcription target genes (Fig. S1b).
To determine whether FoxM1 regulates transcription of mouse p21 gene, luciferase reporter gene was linked to the −1837 to +22 bp region or −1705 to +22 bp region of the mouse p21 promoter. We performed cotransfection in mouse hepatoma Hepa1‐6 cells with the CMV‐FoxM1b expression vector and promoter luciferase plasmids, and prepared protein extracts 24 h after transfection. Measurement of dual luciferase enzyme activity confirmed that cotransfection of FoxM1b expression vector induced activity of −1837 bp p21 promoter, but not −1705 bp p21 promoter (that did not contain the putative FoxM1‐binding site) (Fig. 2h). These results demonstrated that the p21 gene is a potential transcription target of FoxM1 protein.
FoxM1 stimulates Chk1 expression
Senescence of normal cells is induced by cytotoxic and genotoxic stresses, which always involve DNA‐integrity checkpoint pathways (53). One of the major components of such pathways is the serine/threonine kinase Chk1 (54). Interestingly, protein levels of Chk1 were higher in FoxM1 TG MEFs at very early passages (passage 3) (Fig. 3a). Therefore, we tested whether FoxM1 would participate in transcriptional regulation of Chk1. Human osteosarcoma U2OS cells were transfected with FoxM1 siRNA or control siRNA, then 72 h later protein and RNA were prepared to examine any expression of FoxM1 or Chk1 proteins and mRNA, by western blotting or qRT‐PCR respectively. We determined that FoxM1‐depleted U2OS cells exhibited reduced expression of Chk1 protein and mRNA (Fig. 3b,c). We analysed the −2 kb promoter region of mouse Chk1 gene and found that the FoxM1 DNA binding consensus sequence existed between −1646 and −1631 bp (TTT GCT GTT TGT TTT T) of the promoter. ChIP assays showed direct binding of FoxM1 to endogenous mouse Chk1 promoter regions when the MEF chromatin was immunoprecipitated with FoxM1 antibodies and quantified by real‐time PCR with primers made to sequences situated near the FoxM1 binding site in the promoter region (−1717 to −1602 bp) (Fig. 3d). This result was further confirmed by EMSA. Nuclear extract was prepared from FoxM1 TG MEFs and used for EMSA with a 32P‐labelled DNA probe synthesized from mouse Chk1 promoter sequence from −1651 to −1628 bp. FoxM1 protein bound to the probe and addition of 100‐fold unlabelled probe or FoxM1‐specific antiserum (24) disrupted formation of FoxM1/DNA complex (Fig. 3e). To determine whether FoxM1 regulates transcription of mouse Chk1 gene, luciferase reporter gene was linked to the region from −1818 to +22 bp or the region from −1618 to +22 bp of the mouse Chk1 promoter. We performed cotransfection with the CMV‐FoxM1b expression vector and promoter luciferase plasmids. Only the −1818 bp but not the −1618 Chk1 promoter‐driven luciferase was induced with the CMV‐FoxM1b expression vector, when dual luciferase enzyme activity was measured with protein extracts at 24 h after transfection (Fig. 3f). These results demonstrated that Chk1 gene is a direct transcriptional target of FoxM1 protein.
Figure 3.
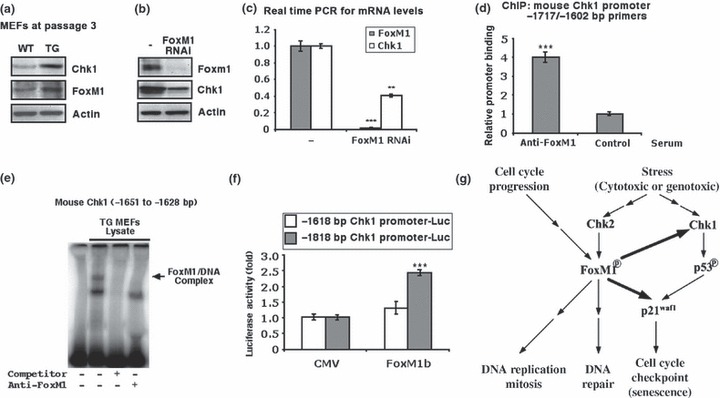
FoxM1 stimulates Chk1 expression and participates in cellular stress responses. (a) FoxM1 TG MEFs display increased protein levels of Chk1. Cell extracts were prepared from WT or TG MEFs at passage 3, and levels of Chk1 (top), FoxM1 (middle) and β‐actin (bottom) were analysed by western blotting. (b and c) FoxM1‐depleted human osteosarcoma U2OS cells exhibit reduced expression of Chk1 mRNA and protein. U2OS cells were transfected with FoxM1 siRNA or control siRNA and 72 h later, protein and RNA were prepared to examine for the expression of Chk1 or FoxM1 protein by western blotting (b) and Chk1 or FoxM1 mRNA using qRT‐PCR (c). (d) ChIP assays show direct binding of FoxM1 to the endogenous mouse Chk1 promoter regions. WT MEF chromatin was cross‐linked, sonicated, and then IP with either FoxM1 antiserum or rabbit serum (control) was performed, and the amount of promoter DNA associated with the IP chromatin was quantified using qRT‐PCR with primers specific to the mouse Chk1 promoter region. (e) FoxM1 bound to Chk1 promoter. Nuclear extract was prepared from FoxM1 TG MEFs and used for EMSA with a 32P‐labelled DNA probe synthesized from the mouse Chk1 promoter sequence position −1651 to −1628 bp. (f) The mouse Chk1 −1818 bp promoter is stimulated by FoxM1. Cotransfection assays were performed in mouse hepatoma Hepa1‐6 cells with the CMV‐FoxM1b expression vector and luciferase plasmid containing −1818 or −1616 bp of the mouse Chk1 promoter region, prepared protein extracts at 24 h after transfection, and used them to measure dual luciferase enzyme activity. In this figure the asterisks indicate statistically significant changes: **P ≤ 0.01 and ***P ≤ 0.001. (g) Model of the function of FoxM1 in response to cellular stress. The FoxM1 transcription factor regulates the expression of cell cycle genes essential for progression into DNA replication and mitosis. In response to genotoxic stress, Chk2 phosphorylates and stabilizes the FoxM1 protein and increased levels of FoxM1 activate the transcription of the DNA repair genes (33). In this study, modestly increased levels of FoxM1 in normal cells (MEFs) stimulate the expression of Chk1 that phosphorylates and activates p53. FoxM1 also stimulates p21 expression in concert with p53 and enhances the senescent phenotype during passaging MEFs. Stress signalling also induces DNA repair enzymes and cell cycle checkpoint independent of the FoxM1 transcription factor (not shown in the figure).
Discussion
In previously published studies, FoxM1 transcription factor has been found to regulate expression of cell cycle genes essential for progression into DNA replication and mitosis, and reduced expression of FoxM1 significantly diminished development of mouse tumours in response to carcinogens or oncogenes (24, 52, 55, 56, 57). In response to genotoxic stress, Chk2 phosphorylates and stabilizes FoxM1 protein and increased levels of FoxM1 activate transcription of DNA repair genes (33, 37). In this study, we have shown that modestly higher levels of FoxM1 in normal cells (MEFs) stimulated expression of Chk1, which phosphorylates and activates p53 (15). FoxM1 also stimulated p21 expression in concert with p53, and enhanced appearance of the senescent phenotype at early passages (passage 6 to 9) of MEFs (summarized in fig. 5c). The present study identified a novel role for FoxM1 in transcriptional response of cell cycle checkpoints. These results suggested that in response to cell stress, FoxM1 might enhance mechanisms of DNA integrity in normal cells whose checkpoint pathways are intact. We believe that in normal cells, FoxM1 participates in cell cycle checkpoints and DNA repair mechanisms, in addition to its functions in stimulating DNA replication and mitosis, to maintain integrity of the genome and aid accomplishment of the cell cycle without DNA damage.
Recently, roles of Chk1 in protecting cells from apoptosis induced by DNA damage or replication stress have been established. Chk1 can suppress caspase‐2‐mediated apoptotic response to DNA damage that bypasses the caspase‐3 pathway, in human and zebrafish cells treated with IR (58). On the other hand, disruption of DNA replication in Chk1‐depleted cells with DNA replication inhibitors results in apoptosis that correlates with activation of caspase 3 (59). In this study, we found that FoxM1 stimulated Chk1 transcription in response to cell stress, suggesting a potential anti‐apoptotic role for FoxM1 through up‐regulation of Chk1. This hypothesis is worthy of being tested and has important implications for anti‐cancer therapy.
This study was performed in a FoxM1 TG MEF model that possesses two significant differences compared to other FoxM1 studies. First, cells used here are primary MEFs. Second, there was only 2‐fold, that is modest, elevation in FoxM1 expression in TG MEFs compared to WT controls. FoxM1 TG MEFs have allowed us to study whether FoxM1 participates in regulation of cell cycle checkpoints of normal cells, which contain intact cell cycle checkpoint pathways at the experimental starting point. Many studies on FoxM1 in cell models have been carried out in either tumour cell lines or immortalized cell models, which always contain mutations or deficiencies in cell cycle checkpoint pathways. With those cells, it has been difficult to observe influences of FoxM1 in controlling cell cycle checkpoints. In addition, levels of FoxM1 overexpression in many published studies are usually much higher than endogenous FoxM1 expression levels. For example, overexpression of FoxM1 in immortalized NIH3T3 fibroblasts suppresses H2O2‐induced cell cycle arrest along with decreased expression of p53 and p21 (46). As NIH3T3 cells are immortalized fibroblasts that have already passed the senescent stage of normal MEFs, we believe that in such cells, FoxM1 functions prominently as a stimulator of cell cycle progression and its strong overexpression enhances cells to bypass cell cycle arrest.
Reduced cell proliferation during aging is associated with progressive decline in FoxM1 expression (43, 60). In previously published studies, −3 kb TTR promoter was used to maintain hepatocyte expression of the FoxM1b transgene in 12‐month‐old (that is, old) TG mice during liver regeneration (43). Maintaining hepatocyte expression of FoxM1b alone in old‐aged TTR‐FoxM1b TG mice is sufficient to restore regenerating hepatocyte DNA synthesis, mitosis and expression of cell cycle genes, to levels found in young regenerating mouse liver cells (43). Based on our current study, reduction in FoxM1 levels may also cause defects in cell cycle checkpoints and lead to decrease in responses to cell stress during aging.
Supporting information
Figure S1. The negative (a) or positive (b) control of ChIP assays. We performed control ChIP assays with cross‐linked extracts from U2OS cells and FoxM1 antibody or control IgG serum. The IP genomic DNA was analysed for the presence of the liver‐specific Transthyretin (TTR) promoter region by real time PCR. This control ChIP experiment demonstrated that neither the FoxM1 antibody nor IgG serum immunoprecipitated significant levels of this proximal TTR promoter region from either untransfected or FoxM1 depleted U2OS cell extracts (a). As positive controls, we performed ChIP assays with primers of promoter regions of Cdc25B, XRCCI and BRCA2, the known FoxM1 transcriptional target genes (b).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
This study was supported by Natural Science Foundation of China [30771096, 30871244 to Y.T.]; Hunan Natural Science Foundation of China [07JJ1006 to Y.T., 2008SK3085 to X.M.]; the Ministry of Education of China [NCET‐06‐0698 to Y.T.]; the Ministry of Science and Technology of China [SKLCBSC‐2007‐05 to Y.T.].
References
- 1. Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636. [DOI] [PubMed] [Google Scholar]
- 2. Campisi J, D’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. [DOI] [PubMed] [Google Scholar]
- 3. Hemann MT, Strong MA, Hao L‐Y, Greider CW (2001) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107, 67–77. [DOI] [PubMed] [Google Scholar]
- 4. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513. [DOI] [PubMed] [Google Scholar]
- 5. Martens UM, Chavez EA, Poon SS, Schmoor C, Lansdorp PM (2000) Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp. Cell Res. 256, 291–299. [DOI] [PubMed] [Google Scholar]
- 6. Di Leonardo A, Linke SP, Clarkin K, Wahl GM (1994) DNA damage triggers a prolonged p53‐dependent G1 arrest and long‐term induction of Cip1 in normal human fibroblasts. Genes Dev. 8, 2540–2551. [DOI] [PubMed] [Google Scholar]
- 7. Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MH (2001) Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide‐treated fibroblasts. Exp. Cell Res. 265, 294–303. [DOI] [PubMed] [Google Scholar]
- 8. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH (1996) Human fibroblast commitment to a senescence‐like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol. Cell. Biol. 16, 5210–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK (2004) Histone deacetylase inhibitors induce a senescence‐like state in human cells by a p16‐dependent mechanism that is independent of a mitotic clock. Exp. Cell Res. 295, 525–538. [DOI] [PubMed] [Google Scholar]
- 11. D’Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T et al. (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- 12. Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- 13. Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429. [DOI] [PubMed] [Google Scholar]
- 14. Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh S‐Y et al. (1999) A role for ATR in the DNA damage‐induced phosphorylation of p53. Genes Dev. 13, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shieh S‐Y, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage‐inducible sites. Genes Dev. 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- 16. Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- 17. Brown JP, Wei W, Sedivy JM (1997) Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277, 831–834. [DOI] [PubMed] [Google Scholar]
- 18. Kaestner KH, Knochel W, Martinez DE (2000) Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 14, 142–146. [PubMed] [Google Scholar]
- 19. Clark KL, Halay ED, Lai E, Burley SK (1993) Co‐crystal structure of the HNF‐3/fork head DNA‐recognition motif resembles histone H5. Nature 364, 412–420. [DOI] [PubMed] [Google Scholar]
- 20. Costa RH, Kalinichenko VV, Holterman AX, Wang X (2003) Transcription factors in liver development, differentiation, and regeneration. Hepatology 38, 1331–1347. [DOI] [PubMed] [Google Scholar]
- 21. Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG et al. (1997) Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 17, 1626–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A et al. (2005) FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 7, 126–136. [DOI] [PubMed] [Google Scholar]
- 23. Wang X, Kiyokawa H, Dennewitz MB, Costa RH (2002) The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc. Natl. Acad. Sci. USA 99, 16881–16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang I‐C, Chen Y‐J, Hughes D, Petrovic V, Major ML, Park HJ et al. (2005) Forkhead Box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2‐Cks1) ubiquitin ligase. Mol. Cell. Biol. 25, 10875–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye H, Holterman A, Yoo KW, Franks RR, Costa RH (1999) Premature expression of the winged helix transcription factor HFH‐11B in regenerating mouse liver accelerates hepatocyte entry into S‐phase. Mol. Cell. Biol. 19, 8570–8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krupczak‐Hollis K, Wang X, Kalinichenko VV, Gusarova GA, Wang I‐C, Dennewitz MB et al. (2004) The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev. Biol. 276, 74–88. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Krupczak‐Hollis K, Tan Y, Dennewitz MB, Adami GR, Costa RH (2002) Increased hepatic Forkhead Box M1B (FoxM1B) levels in old‐aged mice stimulated liver regeneration through diminished p27Kip1 protein levels and increased Cdc25B expression. J. Biol. Chem. 277, 44310–44316. [DOI] [PubMed] [Google Scholar]
- 28. Major ML, Lepe R, Costa RH (2004) Forkhead Box M1B transcriptional activity requires binding of Cdk‐cyclin complexes for phosphorylation‐dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 24, 2649–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin‐mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1, 193–199. [DOI] [PubMed] [Google Scholar]
- 30. Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A et al. (1999) Ubiquitination of p27 is regulated by Cdk‐dependent phosphorylation and trimeric complex formation. Genes Dev. 13, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M et al. (2001) The cell‐cycle regulatory protein Cks1 is required for SCF(Skp2)‐mediated ubiquitinylation of p27. Nat. Cell Biol. 3, 321–324. [DOI] [PubMed] [Google Scholar]
- 32. Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P (2008) Anaphase‐promoting complex/cyclosome‐Cdh1‐mediated proteolysis of the Forkhead Box M1 transcription factor is critical for regulated entry into S phase. Mol. Cell. Biol. 28, 5162–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan Y, Raychaudhuri P, Costa RH (2007) Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 27, 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP (2000) Domain specific interaction in the XRCC1‐DNA polymerase beta complex. Nucleic Acids Res. 28, 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY (1999) BRCA2 is required for ionizing radiation‐induced assembly of Rad51 complex in vivo. Cancer Res. 59, 3547–3551. [PubMed] [Google Scholar]
- 36. Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G et al. (2001) Mutation in Brca2 stimulates error‐prone homology‐directed repair of DNA double‐strand breaks occurring between repeated sequences. EMBO J. 20, 4704–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chetty C, Bhoopathi P, Rao JS, Lakka SS (2009) Inhibition of matrix metalloproteinase‐2 enhances radiosensitivity by abrogating radiation‐induced FoxM1‐mediated G2/M arrest in A549 lung cancer cells. Int. J. Cancer 124, 2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalinichenko VV, Gusarova GA, Tan Y, Wang I‐C, Major ML, Wang X et al. (2003) Ubiquitous expression of the Forkhead Box M1B transgene accelerates proliferation of distinct pulmonary cell types following lung injury. J. Biol. Chem. 278, 37888–37894. [DOI] [PubMed] [Google Scholar]
- 39. Hogan B, Beddington R, Constantini F, Lacy E (1994) Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 40. Zindy F, Quelle DE, Roussel MF, Sherr CJ (1997) Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 15, 203–211. [DOI] [PubMed] [Google Scholar]
- 41. Palmero I, Serrano M (2001) Induction of senescence by oncogenic Ras. Methods Enzymol. 333, 247–256. [DOI] [PubMed] [Google Scholar]
- 42. Wei W, Sedivy JM (1999) Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp. Cell Res. 253, 519–522. [DOI] [PubMed] [Google Scholar]
- 43. Wang X, Quail E, Hung N‐J, Tan Y, Ye H, Costa RH (2001) Increased levels of Forkhead Box M1B transcription factor in transgenic mouse hepatocytes prevents age‐related proliferation defects in regenerating liver. Proc. Natl. Acad. Sci. USA 98, 11468–11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan Y, Yoshida Y, Hughes DE, Costa RH (2006) Increased expression of hepatocyte nuclear factor 6 stimulates hepatocyte proliferation during mouse liver regeneration. Gastroenterology 130, 1283–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan Y, Adami G, Costa RH (2002) Maintaining HNF‐6 expression prevents AdHNF3b mediated decrease in hepatic levels of Glut2 and glycogen. Hepatology 35, 790–798. [DOI] [PubMed] [Google Scholar]
- 46. Li SKM, Smith DK, Leung WY, Cheung AMS, Lam EWF, Dimri GP et al. (2008) FoxM1c counteracts oxidative stress‐induced senescence and stimulates Bmi‐1 expression. J. Biol. Chem. 283, 16545–16553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kisseberth WC, Brettingen NT, Lohse JK, Sandgren EP (1999) Ubiquitous expression of marker transgenes in mice and rats. Dev. Biol. 214, 128–138. [DOI] [PubMed] [Google Scholar]
- 48. Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV (2008) Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene 17, 17. [DOI] [PubMed] [Google Scholar]
- 49. Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J (2003) Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell 2, 287–294. [DOI] [PubMed] [Google Scholar]
- 50. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shieh S‐Y, Ikeda M, Taya Y, Prives C (1997) DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334. [DOI] [PubMed] [Google Scholar]
- 52. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM et al. (2004) Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 18, 830–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pearce AK, Humphrey TC (2001) Integrating stress‐response and cell‐cycle checkpoint pathways. Trends Cell Biol. 11, 426–433. [DOI] [PubMed] [Google Scholar]
- 54. Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica‐Worms H et al. (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 55. Kalin TV, Wang I‐C, Ackerson TJ, Major ML, Detrisac CJ, Kalinichenko VV et al. (2006) Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY Transgenic Mice. Cancer Res. 66, 1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim I‐M, Ackerson T, Ramakrishna S, Tretiakova M, Wang I‐C, Kalin TV et al. (2006) The Forkhead Box m1 Transcription Factor Stimulates the Proliferation of Tumor Cells during Development of Lung Cancer. Cancer Res. 66, 2153–2161. [DOI] [PubMed] [Google Scholar]
- 57. Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D et al. (2009) Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res. 69, 3501–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R et al. (2008) Chk1 suppresses a caspase‐2 apoptotic response to DNA damage that bypasses p53, Bcl‐2, and caspase‐3. Cell 133, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Myers K, Gagou ME, Zuazua‐Villar P, Rodriguez R, Meuth M (2009) ATR and Chk1 suppress a caspase‐3‐dependent apoptotic response following DNA replication stress. PLoS Genet. 5, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ly DH, Lockhart DJ, Lerner RA, Schultz PG (2000) Mitotic misregulation and human aging. Science 287, 2486–2492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The negative (a) or positive (b) control of ChIP assays. We performed control ChIP assays with cross‐linked extracts from U2OS cells and FoxM1 antibody or control IgG serum. The IP genomic DNA was analysed for the presence of the liver‐specific Transthyretin (TTR) promoter region by real time PCR. This control ChIP experiment demonstrated that neither the FoxM1 antibody nor IgG serum immunoprecipitated significant levels of this proximal TTR promoter region from either untransfected or FoxM1 depleted U2OS cell extracts (a). As positive controls, we performed ChIP assays with primers of promoter regions of Cdc25B, XRCCI and BRCA2, the known FoxM1 transcriptional target genes (b).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item