

Abstract
Abstract. Introduction: Epidemiologic studies point towards a significant correlation between the dietary intake of isothiocyanate‐containing foods and the reduced risk for cancer. Methods and results: In the current investigation, we examined the consequence of activating of signalling pathways during the release the cells from the block at G1/S boundary by synthetic isothiocyanate E‐4IB. Using synchronized leukaemic HL60 cells, we show that activation of mitogen‐activated protein kinases ERK1/2, c‐Jun N‐terminal kinase and p38 signalling pathways by E‐4IB are coupled with delayed transition through the cell cycle and rapid cell cycle arrest resulted in diminished mitochondrial membrane potential culminating in apoptosis. These events were accompanied by histone deacetylase inhibition, increase of double strand DNA breaks detected by histone H2AX phosphorylation and up‐regulation of cell cycle regulatory protein p21 and phosphorylation of CDC25C phosphatase. Conclusion: These findings suggest that the activation of mitogen‐activated protein kinases signalling pathways, followed by the induction cell cycle arrest and apoptosis, might be responsible for anticancer activities of E‐4IB.
INTRODUCTION
Several isothiocyanates (ITC) have been found to inhibit rat lung, liver, small intestine, colon and bladder tumorigenesis (Hecht 2000; Kelloff et al. 2000; Zhao et al. 2001; Zhang 2004). ITCs are also known to be associated with oxidative stress as they may cause oxidative DNA damage by depleting glutathione and/or an alternation of the redox potential (Liu et al. 1998; Payen et al. 2001). The complex pattern of ITCs effects may depend on both the genetic background of cells and/or the concentration ranges used. In this regard, preferential cytotoxic effects of ITCs in tumour cells in comparison to normal cells, induction of acute phase II enzymes and ATP‐binding cassette transporters, disruption of microtubule network formation, G2/M arrest and mitotic catastrophe, as well as involvement of mitogen‐activated protein kinase (MAPK) pathways were found (Hu et al. 2003; Jackson & Singletary 2004a; Smith et al. 2004; Svehlikova et al. 2004; Fimognari et al. 2005; Jakubikova et al. 2005b). It has been also shown that ITCs induce apoptosis in various tumour cells (Fimognari et al. 2003; Xiao et al. 2004; Jakubikova et al. 2005a).
The wide differences in the cytotoxicity of synthetic ITCs were observed. To mention several, esters of isothiocyanatocarboxylic acids, such as 4‐methylisothiocyanatobutanoate, 5‐ethyl isothiocyanato‐pentanoate and ethyl‐4‐isothiocyanatobutanoate (E‐4IB) belong to the most active compounds (1971, 1989). We have recently shown that E‐4IB exerted cytotoxicity and exhibited potential synergy in combination with cisplatin in human ovarian carcinoma cells (Bodo et al. 2005, 2006a).
The aim of the current study was to examine the activation of MAPKs ERK, c‐Jun N‐terminal kinase (JNK) and p38 and regulation of cell cycle progression after releasing the cells from the block at G1/S boundary. Our findings demonstrate that E‐4IB treatment led to the delay of cell cycle progression into mitosis and was associated with augmented p21 expression and CDC25C phosphorylation.
MATERIALS AND METHODS
Reagents
Ethyl‐4‐isothiocyanatobutanoate (E‐4IB) was synthesized as described (Floch et al. 1997). 2′‐deoxythymidine was received from Applichem (Darmstadt, Germany). Rabbit polyclonal antibodies against, actin, FasL, poly(ADP‐ribose) polymerase (PARP), p21, p53, ERK1/2 (Ser 216), phospho‐ERK1/2, JNK1/2, p38 MAPK, goat polyclonal antibodies against CDC25C, phospho‐CDC25C, horseradish peroxidase‐conjugated antirabbit, antigoat and antimouse antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal antibody 14‐3‐3ɛ was received from Becton Dickinson Transduction laboratories (San Diego, CA, USA). Rabbit polyclonal antibodies Anti‐ACTIVE® JNK (pTPpY) and Anti‐ACTIVE® p38 (pTGpY) were purchased from Promega (Madison, WI, USA). Anti‐phosphohistone H2AX polyclonal antibody was from Upstate (Lake Placid, NY, USA). Fluorescein isothiocyanate‐conjugated antirabbit and antimouse F(ab′)2 fragments were obtained from Beckman Coulter (Fullerton, CA, USA). Other chemicals used in this study were obtained from Sigma Chemical Co. (St. Louis, MO, USA).
Cell culture, synchronization, cytotoxic assay and treatments
HL60 cells were routinely cultured in RPMI 1640 medium supplemented with 10% foetal calf serum, 100 µg/ml penicillin and 50 µg/ml streptomycin in humidified 5% CO2 atmosphere, at 37 °C. Cells were plated at 0.5 × 106 cells/ml density and cultured in 96‐, 24‐ or 6‐well plates (Greiner, Germany).
Cells were synchronized at the G1/S‐phase boundary using single thymidine block as previously described (Dangi et al. 2003). Briefly, cells were treated with 2 mm thymidine in complete medium for 18 h. The cells were released back into the cell cycle by washing with phosphate‐buffered saline (PBS) and then incubated for an additional 3 h in complete medium in the absence of thymidine. Then, synchronized cells were treated with 5 or 10 µm E‐4IB for 3, 6, 9 and 24 h. Stock solution of E‐4IB (40 mm) was originally dissolved in dimethyl sulfoxide (DMSO) and an equal volume of DMSO (final concentration <0.05%) was added to the control cultures. All experiments here were carried out with synchronized cells.
To detect cytotoxicity (Mosmann 1983), cells were incubated with E‐4IB for 24 h in triplicates. The absorbance was measured at 540 and 690 nm by Multisoft plate reader (Labsystems Oy, Helsinki, Finland). The concentration of drug that inhibited cell survival to 50% (IC50) was determined by Calcusyn software (Biosoft, Ferguson, MO, USA).
Cell cycle analysis and apoptosis
To measure cell cycle, cells (5 × 105/ml) were washed twice with PBS and 0.05% Triton X‐100 and 15 µl RNA‐se A (10 mg/ml) were added to each sample for 20 min at 37 °C and then supplemented with propidium iodide (PI) (50 µg/ml) and incubated on ice for 15 min. Finally, the stained cells were analyzed using photomultipliers log FL2 for sub‐G1, lin FL3 for DNA cell cycle histogram and FL4 peak versus FL4 integral for doublets discrimination.
Apoptotic cells were quantified using fluorescein diacetate (FDA)/PI staining according to Bartkowiak et al. (1999). Briefly, 5 × 105 cells were twice washed with PBS and re‐suspended in 400 µl of PBS/0.2% bovine serum albumin (BSA) containing 10 nm of FDA (Sigma Chemical Co., St. Louis, MO, USA) for 30 min at room temperature. Then, cells were cooled and 4 µl of PI (1 mg/ml) was added. Finally, cells were analyzed using a Coulter Epics ALTRA flow cytometer with FL1 and FL2 photomultipliers for fluorescein and PI, respectively.
Cytofluorometric analysis of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) Ψm was assessed by JC‐1 dye measurement of red/green fluorescence ratio as described (Jakubikova et al. 2005a). Briefly, cells (5 × 105) were washed twice with PBS and incubated with 400 µl of PBS/0.2% BSA containing 4 µm of JC‐1 (Molecular Probes, Eugene, OR, USA) at 37 °C for 30 min. Fluorescence was measured by Coulter Epics ALTRA flow cytometer. Data (FL1, FL2, and ratio FL2/FL1) were analyzed by the WinMDI version 2.8 software (J. Trotter, Scripps Research Institute, La Jolla, CA, USA).
Histone H2AX phosphorylation
Cells (1 × 106/ml) were fixed in 1% methanol‐free paraformaldehyde in PBS on ice for 3 min, washed and kept in 70% ethanol for 2 h at –20 °C. Then, the cells were diluted in PBS/BSA containing antiphosphohistone H2AX polyclonal antibody. Extensively washed cells were diluted in 100 µl PBS/BSA with Fluorescein isothiocyanate‐conjugated antirabbit immunoglobulin (diluted 1 : 60). Finally, cells were counterstained with 5 µg/ml of PI in PBS, for 15 min at 4 °C and cellular fluorescence was measured using Coulter Epics ALTRA flow cytometer with photomultipliers FL1 and FL2 for analyzing γ‐H2AX and PI.
Flow cytometry measurements and data analysis
Coulter Epics ALTRA flow cytometer was equipped with 488 nm excitation laser and fluorescence emission was measured using bandpass filter set 525, 575, 610, 675 nm (FL1‐4). Forwards/side light scatter characteristic was used to exclude the cell debris from the analysis. For each analysis, 1 × 104 cells were required. Data were analyzed with WinMDI version 2.8 software (J. Trotter, Scripps Research Institute). The cell cycle calculations were performed with MULTI‐CYCLE Software (Phoenix Flow System, San Diego, CA, USA).
Fluorometric detection of histone deacetylase activity
Histone deacetylase (HDAC) activity was evaluated using the HDAC Assay kit (Upstate, Lake Placid, NY, USA) according to the manufacturer's instructions. The assay is a two‐step procedure performed in a microtitre plate. Briefly, nuclear extracts (5 µg) were incubated with HDAC substrate (67 µm) for 30 min at 37 °C allowing deacetylation of the fluorescent substrate. Then, activation solution containing 4 µm HDAC inhibitor trichostatin A was added to release a fluorophore from the deacetylated substrate and the fluorescence was evaluated using plate reader POLARstar OPTIMA (BMG Labtech, Offenburg, Germany), using excitation 355 nm and emission 460 nm.
Western blot analysis
To examine effects of E‐4IB on membrane protein expression, HL60 cells were harvested at indicated times, lysed in a buffer as described (Bodo et al. 2006b). For each lane 50 µg of protein were loaded. Blots were incubated with the indicated antibodies (all primary antibodies were used at final concentration 1 µg/ml) and horseradish peroxidase‐conjugated antirabbit‐, antigoat‐ or antimouse‐secondary antibody (1 µg/ml). Enhanced chemiluminescence (ECL, Amersham Arlington Heights, IL, USA) was used for detection. Actin was used as a control for equal gel loading.
RESULTS
Cell viability and E‐4IB induced apoptosis
To assess cytotoxic effect of E‐4IB, HL60 cells were treated with 0.5–20 µm concentrations of E‐4IB. The IC50 value was determined from the cell survival plot after 24 h of treatment of the drug and was evaluated 4.2 ± 0.3 µm (data not shown). Although the concentration of DMSO in cell cultures treated with 10 µm E‐4IB is close to the DMSO level able to induce differentiation of HL60 cells no induction of CD14 antigen expression was observed at 24 h of treatment (data not shown).
To gain an insight into cytotoxic effect of E‐4IB, we determined cell portions undergoing apoptosis by flow cytometric technique. As demonstrated in Fig. 1(a), E‐4IB induced significant increase of FDA−/PI− apoptotic cells (up to 18% at the highest concentration tested) and apoptotic/necrotic FDA−/PI+ cells (up to 52%) associated with proteolytic cleavage of PARP and a moderate increase of FasL, observed after 24 h of the treatment (Fig. 1b).
Figure 1.
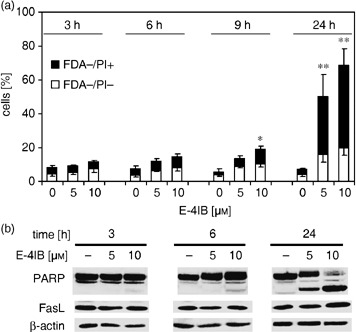
E‐4IB induces apoptosis. (a) Thymidine‐synchronized HL60 cells were exposed to 5 or 10 µm E‐4IB for 3, 6, 9 and 24 h and FDA−/PI− apoptotic cells and apoptotic/necrotic FDA−/PI+ cell populations were calculated from flow cytometry measurements. Means of at least three independent experiments ± SD are shown. Statistical significance from the controls was evaluated, *P < 0.05, **P < 0.01. (b) Western blot analysis of PARP and FasL (50 µg protein lysate) is demonstrated. β‐actin was used as a loading control.
E‐4IB modulation of mitochondrial membrane potential
The lipophilic cationic dye JC‐1 was utilized to determine E‐4IB‐induced alterations in MMP. Cytofluorometric analysis showed that proportion of cells with reduced MMP coincided with the appearance of apoptotic cells. As demonstrated in Fig. 2, E‐4IB caused a concentration‐ and time‐dependent decrease of MMP after 24 h of the treatment by more than 40% in comparison to control cells.
Figure 2.
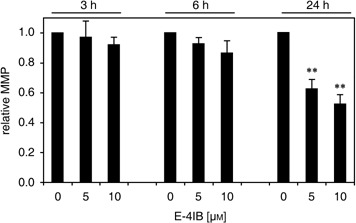
E‐4IB induced depolarization of mitochondrial membranes. Synchronized cells were stained with the mitochondrial‐selective JC‐1 dye and measured by flow cytometry. Experimental conditions were applied as shown in Fig. 1. The ordinate represents relative mitochondrial potential (MMP) levels compared with control DMSO‐treated cells. Error bars are ± SD of at least three independent measurements. Statistical significance was inferred at *P < 0.01.
E‐4IB effects on cell cycle transition
Effects of E‐4IB on cell proliferation were assessed by evaluation the distribution of cells in the phases of the cell cycle. Figure 3(a) shows histograms of HL60 cells exposed to E‐4IB (5 or 10 µm) for 3, 6, 9 and 24 h. Delayed transition through the cell cycle without appearance of significant apoptotic sub‐G1 fraction was found in E‐4IB treated cells after 3, 6 and 9 h in comparison to control DMSO treated cells. Despite the delayed transition the majority of cells treated with 5 µm E‐4IB progress through the cell cycle to G2/M phase. However, higher concentration of E‐4IB caused the S‐phase arrest and appearance of sub‐G1 apoptotic cell population after 9 h of treatment. The appearance of G1 cell population in histogram of control cells after 24 h suggests that significant proportion of cells completed mitosis and entered the next cell cycle. Contrariwize, progressive decrease of post‐mitotic cell population in E‐4IB treated cells was determined. Higher proportion of affected cells, at the expense of S and G2/M phases, was present in cell cultures treated with 10 µm E‐4IB for 24 h. Despite the apoptosis induction with 5 µm E‐4IB, there were still cells that entered the new cell cycle. These findings indicate that an onset of E‐4IB‐induced apoptosis might be linked with different cell cycle checkpoints in the dose‐dependent manner.
Figure 3.
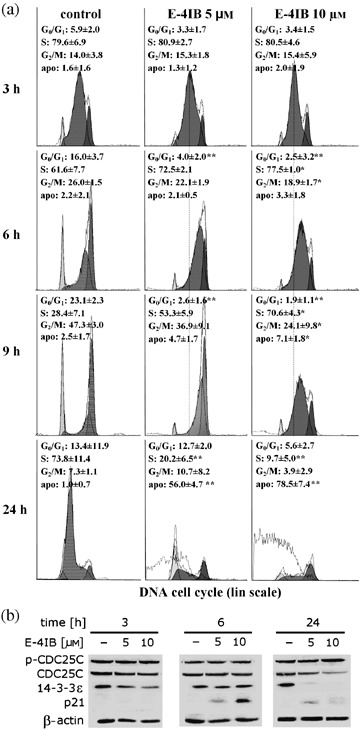
E4IB effects on cell cycle transition. (a) Flow cytometry‐based cell cycle analysis of E‐4IB‐treated synchronized cells. The distribution of cells in G0/G1, S and G2/M phase was analyzed with Multi‐cycle software and the results are shown in corresponding histograms. The histograms of one representative experiment out of three are shown. Means of three independent experiments ± SD are shown and statistical significance from the controls was evaluated, *P < 0.05, **P < 0.01. Apo‐fragmented DNA of apoptotic cells. (b) Western blot analyses of CDC25C‐ and p‐CDC25C phosphatase, p21 and 14‐3‐3ɛ (50 µg protein lysate) at indicated times are shown. Shown here are representative Western blots from one of two independent experiments.
To corroborate the alterations in cell proliferation measured by cell cycle analysis, CDC25C, phospho‐CDC25C and Cdk inhibitor p21 protein levels were evaluated by Western blotting (Fig. 3b). Exposure of cells to E‐4IB resulted in accumulation of phospho‐CDC25C, in a dose‐dependent manner, whereas, interestingly, total CDC25C levels were decreased. Furthermore, p21 expression reached its maximum at 6 h, however, at 24 h this was not detected in control and treated cells, respectively. 14‐3‐3ɛ protein has been shown to regulate the cell progression through mitosis. In our experimental conditions, it diminished in a dose‐ and time‐dependent manner during the cell cycle.
Effect of E‐4IB on histone deacetylase activity and histone H2AX phosphorylation
To determine the effect of E‐4IB on HDAC activity of cells and their nuclear extracts, cell samples were exposed to 5 and 10 µm E‐4IB for 3, 6 and 24 h. E‐4IB effect was compared to trichostatin A‐induced inhibition of HDAC activity. Significant concentration‐dependent decreases of HDAC activity in E‐4IB (~13%) treated cells for 3 h were found (Fig. 4a). On the contrary, no statistically significant inhibitory effect of E‐4IB upon HDAC activity was noticed in nuclear extracts and cell samples treated longer than 3 h (data not shown).
Figure 4.
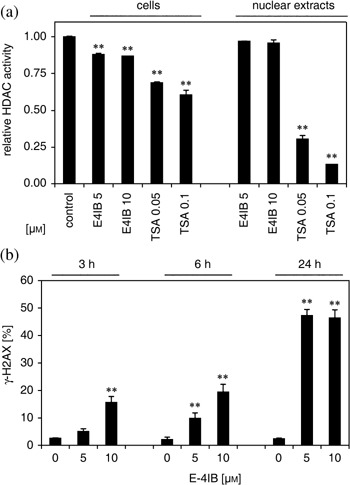
E‐4IB effect on histone deacetylase activity and histone H2AX phosphorylation. (a) E‐4IB inhibition of HDAC activity in synchronized HL60 cells calculated by fluorimetry. The ordinate shows relative HDAC activity in cells and nuclear extracts exposed to E‐4IB (5 or 10 µm) or TSA (50 or 100 nm) for 3 h and compared to corresponding controls. Results of the two independent experiments are shown. The data are expressed as mean ± SD. Statistical significance was inferred at *P < 0.01. (b) γ‐H2AX levels were indicated. The ordinate represents means of percentage of γ‐H2AX positive cells and error bars are ± SD of at least three independent measurements. Statistical significance was inferred at *P < 0.01.
Double‐strand breaks in the DNA backbone were measured by histone H2AX phosphorylation, when Ser 139 that lies within an ataxia telangiectasia mutated (ATM) consensus sequence was utilized as the major H2AX residue phosphorylated in response to DNA damage. After E‐4IB treatment, a significant time‐ and dose‐dependent increase in phosphorylated histone H2AX levels were indicated compared to untreated controls (Fig. 4b).
E‐4IB activation of MAPKs
Because MAPKs have been implicated in regulation of proliferation and/or cell death in response to various stimuli a potential participation of p38, JNK and ERK in cells treated with E‐4IB was evaluated (Fig. 5). Immunoblot analysis revealed a significant concentration‐dependent increase of p38 MAPK, JNK and ERK1/2 phosphorylation even after 3 h of the treatment. An augmentation of ERK1/2 and JNK phosphorylation sustained for the next 3 h. The constitutive cellular levels of all three MAPK remained relatively constant throughout the time‐course used, although a slight decrease of p38 MAPK and JNK1/2 protein levels was determined after 24 h.
Figure 5.
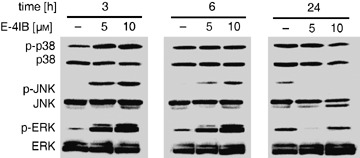
E‐4IB activation of MAPKs. Western blot analyses of MAP kinases (p38 MAPK, JNK1/2 and ERK1/2 and their phosphorylated forms) in synchronized HL60 cells treated with indicated E‐4IB concentrations and times (50 µg protein lysate) are demonstrated. Shown here are representative Western blots from one of two independent experiments.
DISCUSSION
To date, the most important biological property of ITCs is their ability to inhibit chemical carcinogenesis. ITCs are also known to induce both cell cycle arrest and apoptosis in several types of cultured human cancer cells. Early studies suggested that chemopreventive activity of ITCs is based on the modulation of enzymes required for the activation or detoxification of carcinogens (Fimognari et al. 2003; Jackson & Singletary 2004b; Singh et al. 2004; Xiao et al. 2004). In this study, we examined effects of synthetic isothiocyanate E‐4IB on human promyelocytic leukaemia HL60 cells. Using synchronized cells, this study demonstrates that E‐4IB initiated rapid delay in cell cycle progression and induced concentration‐dependent appearance of apoptotic cells. Considering that higher concentration of E‐4IB induced a more profound delay with subsequent S‐phase arrest, it is intriguing to speculate that the observed effects might be compatible with ataxia telangiectasia and Rad3 related (ATR)‐dependent replication checkpoint during perturbed S‐phase (Petermann & Caldecott 2006).
The cell cycle modulation and induction of apoptosis within relatively narrow concentration range is often observed in ITC‐treated cells (Kuang & Chen 2004; Miyoshi et al. 2004b; Jakubikova et al. 2006). Fragmentation of DNA, cleavage of PARP and FasL induction was determined after prolonged time of E‐4IB treatment. Thus, these results suggest that apoptosis was induced by E‐4IB in different cell cycle checkpoints, which depend on the drug dosage.
Perturbations in the mitochondrial function are manifested by dissipation of the MMP and are generally associated with cell‐programmed death. The exposure of asynchronous cells to ITCs resulted in dose‐dependent damage of mitochondria accompanied by disruption of MMP (Nakamura et al. 2002; Hu et al. 2003; Zhang et al. 2003). Measurements of MMP after E‐4IB treatment in synchronized HL60 cells showed that E‐4IB‐induced MMP dissipation is associated with cell cycle‐dependent apoptosis.
The ITCs‐induced cell cycle arrest is associated with significant reduction in the amount of several proteins that regulate G2/M progression, including CDC25 proteins (Singh et al. 2004; Srivastava & Singh 2004; Xiao et al. 2004). ITCs treatments resulted in a rapid and sustained phosphorylation of CDC25C at S‐216 leading to the increased binding with 14‐3‐3ɛ followed by its translocation from the nucleus to the cytoplasm and proteasomal degradation (Singh et al. 2004). In accordance with these observations, we determined an increase of p‐CDC25C (S‐216) and diminution of total CDC25C and 14‐3‐3ɛ protein levels. In this context, some studies report that p38 MAPK may play an important role in early G2/M arrest due to phosphorylation of CDC25B and leading to the formation of CDC25B/14‐3‐3ɛ complex (Bulavin et al. 2001). In the line of these observations, we assume that activation of p38 MAPK may also have an important role in E‐4IB‐induced cell cycle arrest.
Increased S‐216 phosphorylation of CDC25C upon treatment with sulforaphane resulted in Chk2 activation that was associated with ATM‐ and S‐139 phosphorylation of histone H2AX, a sensitive marker for the presence of DNA double‐strand breaks (Singh et al. 2004). Recently, H2AX phosphorylation was observed in cells synchronized by the inhibitors of DNA replication (Kurose et al. 2006). In our studies, we determined an increased phosphorylation of H2AX (S‐139), and thus we suppose that increased H2AX phosphorylation may represents DNA breaks induced by E‐4IB. In this context, in the line with some authors (Celeste et al. 2002), we could hypothesize that E‐4IB‐induced phosphorylation of H2AX would lead to the recruitment of BRCA1 to the site(s) of DNA damage. Furthermore, phosphorylated ATM/ATR activated ERK through BRCA1 resulting in p21 elevation in a p53‐independent manner (Gartel & Tyner 1999). In fact, we present here an increased p21 levels paralleled with ERK activation. It agrees with observations where activated ERK pathway was associated with increased p21, presumably through its stabilization by phosphorylation (Kim et al. 2002), or with ERK‐mediated delay in M‐phase entry that was dependent on de novo synthesis of p21 during G2‐phase (Dangi et al. 2006).
The concept of the use of low affinity HDAC inhibitors in cancer management was recently proposed (Dashwood et al. 2006; Myzak & Dashwood 2006). We here showed that E‐4IB‐inhibited HDAC activity in the whole cells but no inhibitory effect was detected in nuclear extracts. From these findings, we assume that for effective inhibition of HDAC activity either metabolic conversion of E‐4IB and/or processing of intracellular signals are required. E‐4IB‐induced inhibition of HDAC activity might be another plausible explanation for significant increase of p21 levels referred by others in HDAC inhibitor‐induced apoptosis (Komata et al. 2005).
Several lines of evidence were accumulated to indicate that ITCs can induce cellular stress followed by stimulation of variety of signal transduction pathways (Miyoshi et al. 2004a; Jakubikova et al. 2005b). In certain cell types, a balance between activation of JNKs, ERKs and p38 MAPKs appeared to be a critical for apoptosis (Xia et al. 1995). In many tumour cell types, MAPKs activation was observed shortly after ITCs treatment (Kong et al. 2001; Hu et al. 2003). We presented here that in synchronized cells E‐4IB induced phosphorylation of JNK1, ERK1/2 and p38 MAPK that culminated shortly after the treatment. Our results are in agreement with findings when increased activation of JNK in synchronized HL60 cells after benzyl isothiocyanate treatment was found (Miyoshi et al. 2004b).
In summary, our results demonstrate E‐4IB‐induced concentration‐dependent delay of cell cycle transition and apoptosis induction in HL60 cells. Furthermore, we showed MAPKs activation, the delay of cell cycle progression that was associated with augmented p21 synthesis and HDAC inhibition. Further research on the molecular mechanisms and cellular effects of E‐4IB might help in the understanding of anticancer mechanism(s) of this compound.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge expert technical assistance by Mrs M. Sulikova. This work was supported in part by Slovak Governmental Research and Development Sub‐Programme Food – quality and safety no. 2003SP270280E010280E01, National Programme ‘Use of Cancer Genomics to Improve the Human Population Health’ project 2003SP510280800/0280801, Slovak Academy of Sciences Centre of Excellence ‘Molecular Medicine’, European Social Fund (project code 13120200038) and VEGA project 2/4069.
REFERENCES
- Bartkowiak D, Hogner S, Baust H, Nothdurft W, Rottinger EM (1999) Comparative analysis of apoptosis in HL60 detected by annexin‐V and fluorescein‐diacetate. Cytometry 37, 191–196. [DOI] [PubMed] [Google Scholar]
- Bodo J, Chovancova J, Hunakova L, Sedlak J (2005) Enhanced sensitivity of human ovarian carcinoma cell lines A2780 and A2780/CP to the combination of cisplatin and synthetic isothiocyanate ethyl 4‐isothiocyanatobutanoate. Neoplasma 52, 510–516. [PubMed] [Google Scholar]
- Bodo J, Hunakova L, Kvasnicka P, Jakubikova J, Duraj J, Kasparkova J, Sedlak J (2006a) Sensitisation for cisplatin‐induced apoptosis by isothiocyanate E‐4IB leads to signalling pathways alterations. Br. J. Cancer 95, 1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodo J, Jakubikova J, Chalupa I, Bartosova Z, Horakova K, Floch L, Sedlak J (2006b) Apoptotic effect of ethyl‐4‐isothiocyanatobutanoate is associated with DNA damage, proteasomal activity and induction of p53 and p21 (cip1/waf1). Apoptosis 8, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ Jr (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez‐Capetillo O, Chen HT, Sedelnikova OA, Reina‐San‐Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini‐Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A (2002) Genomic instability in mice lacking histone H2AX. Science 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangi S, Cha H, Shapiro P (2003) Requirement for phosphatidylinositol‐3 kinase activity during progression through S‐phase and entry into mitosis. Cell Signal. 7, 667–675. [DOI] [PubMed] [Google Scholar]
- Dangi S, Chen FM, Shapiro P (2006) Activation of extracellular signal‐regulated kinase (ERK) in G phase delays mitotic entry through p21. Cell Prolif. 39, 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood RH, Myzak MC, Ho E (2006) Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis 27, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimognari C, Berti F, Iori R, Cantelli‐Forti G, Hrelia P (2005) Micronucleus formation and induction of apoptosis by different isothiocyanates and a mixture of isothiocyanates in human lymphocyte cultures. Mutat. Res. 582, 1–10. [DOI] [PubMed] [Google Scholar]
- Fimognari C, Nusse M, Berti F, Iori R, Cantelli‐Forti G, Hrelia P (2003) Sulforaphane modulates cell cycle and apoptosis in transformed and non‐transformed human T lymphocytes. Ann. N. Y. Acad. Sci. 1010, 393–398. [DOI] [PubMed] [Google Scholar]
- Floch L, Kuban J, Gogova A, Jakubik T, Pronayova N (1997) Synthesis of ethyl 4‐isothiocyanatobutanoate derivatives. Chem. Pap.-Chem. Zvesti 51, 416–420. [Google Scholar]
- Gartel AL, Tyner AL (1999) Transcriptional regulation of the p21 (WAF1/CIP1) gene. Exp. Cell Res. 246, 280–289. [DOI] [PubMed] [Google Scholar]
- Hecht SS (2000) Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 32, 395–411. [DOI] [PubMed] [Google Scholar]
- Horakova K, Drobnica L, Nemec P (1971) Cytotoxic and cancerostatic effect of isothiocyanates and related compounds. VII. Effect of isothiocyanatechalkones on HeLa cells. Neoplasma 18, 355–359. [PubMed] [Google Scholar]
- Horakova K, Strakova Z, Jantova S, Muckova M, Floch L (1989) Biological effects of 2‐isothiocyanate carboxylic‐acid esters.1. Cytotoxic, antimicrobial and genetoxic screening amino‐acid isothiocyanate derivatives. Biologia 44, 637–646. [Google Scholar]
- Hu R, Kim BR, Chen C, Hebbar V, Kong AN (2003) The roles of JNK and apoptotic signaling pathways in PEITC‐mediated responses in human HT‐29 colon adenocarcinoma cells. Carcinogenesis 24, 1361–1367. [DOI] [PubMed] [Google Scholar]
- Jackson SJ, Singletary KW (2004a) Sulforaphane inhibits human MCF‐7 mammary cancer cell mitotic progression and tubulin polymerization. J. Nutr. 134, 2229–2236. [DOI] [PubMed] [Google Scholar]
- Jackson SJ, Singletary KW (2004b) Sulforaphane: a naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis 25, 219–227. [DOI] [PubMed] [Google Scholar]
- Jakubikova J, Bao Y, Bodo J, Sedlak J (2006) Isothiocyanate iberin modulates phase II enzymes, posttranslational modification of histones and inhibits growth of Caco‐2 cells by inducing apoptosis. Neoplasma 53, 463–470. [PubMed] [Google Scholar]
- Jakubikova J, Sedlak J, Bacon J, Goldson A, Bao Y (2005a) Effects of MEK1 and PI3K inhibitors on allyl‐, benzyl‐ and phenylethyl‐isothiocyanate‐induced G2/M arrest and cell death in Caco‐2 cells. Int. J. Oncol. 27, 1449–1458. [PubMed] [Google Scholar]
- Jakubikova J, Sedlak J, Mithen R, Bao Y (2005b) Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane‐ and erucin‐induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco‐2 cells. Biochem. Pharmacol. 69, 1543–1552. [DOI] [PubMed] [Google Scholar]
- Kelloff GJ, Crowell JA, Steele VE, Lubet RA, Malone WA, Boone CW, Kopelovich L, Hawk ET, Lieberman R, Lawrence JA, Ali I, Viner JL, Sigman CC (2000) Progress in cancer chemoprevention: development of diet‐derived chemopreventive agents. J. Nutr. 130, 467S–471S. [DOI] [PubMed] [Google Scholar]
- Kim GY, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E (2002) The stress‐activated protein kinases p38 alpha and JNK1 stabilize p21 (Cip1) by phosphorylation. J. Biol. Chem. 277, 29792–29802. [DOI] [PubMed] [Google Scholar]
- Komata T, Kanzawa T, Nashimoto T, Aoki H, Endo S, Kon T, Takahashi H, Kondo S, Tanaka R (2005) Histone deacetylase inhibitors, N‐butyric acid and trichostatin A, induce caspase‐8‐ but not caspase‐9‐dependent apoptosis in human malignant glioma cells. Int. J. Oncol. 26, 1345–1352. [DOI] [PubMed] [Google Scholar]
- Kong ANT, Yu R, Hebbar V, Chen C, Owuor E, Hu R, Ee R, Mandlekar S (2001) Signal transduction events elicited by cancer prevention compounds. Mutat. Res. 480, 231–241. [DOI] [PubMed] [Google Scholar]
- Kuang YF, Chen YH (2004) Induction of apoptosis in a non‐small cell human lung cancer cell line by isothiocyanates is associated with p53 and p21. Food Chem. Toxicol. 42, 1711–1718. [DOI] [PubMed] [Google Scholar]
- Kurose A, Tanaka T, Huang X, Traganos F, Darzynkiewicz Z (2006) Synchronization in the cell cycle by inhibitors of DNA replication induces histone H2AX phosphorylation: an indication of DNA damage. Cell Prolif. 39, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Pelling JC, Ju J, Chu E, Brash DE (1998) Antioxidant action via p53‐mediated apoptosis. Cancer Res. 58, 1723–1729. [PubMed] [Google Scholar]
- Miyoshi N, Takabayashi S, Osawa T, Nakamura Y (2004a) Benzyl isothiocyanate inhibits excessive superoxide generation in inflammatory leukocytes: implication for prevention against inflammation‐related carcinogenesis. Carcinogenesis 25, 567–575. [DOI] [PubMed] [Google Scholar]
- Miyoshi N, Uchida K, Osawa T, Nakamura Y (2004b) A link between benzyl isothiocyanate‐induced cell cycle arrest and apoptosis: involvement of mitogen‐activated protein kinases in the Bcl‐2 phosphorylation. Cancer Res. 64, 2134–2142. [DOI] [PubMed] [Google Scholar]
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. [DOI] [PubMed] [Google Scholar]
- Myzak MC, Dashwood RH (2006) Chemoprotection by sulforaphane: Keep one eye beyond Keap1. Cancer Lett. 233, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Kawakami M, Yoshihiro A, Miyoshi N, Ohigashi H, Kawai K, Osawa T, Uchida K (2002) Involvement of the mitochondrial death pathway in chemopreventive benzyl isothiocyanate‐induced apoptosis. J. Biol. Chem. 277, 8492–8499. [DOI] [PubMed] [Google Scholar]
- Payen L, Courtois A, Loewert M, Guillouzo A, Fardel O (2001) Reactive oxygen species‐related induction of multidrug resistance‐associated protein 2 expression in primary hepatocytes exposed to sulforaphane. Biochem. Biophys. Res. Commun. 282, 257–263. [DOI] [PubMed] [Google Scholar]
- Petermann E, Caldecott KW (2006) Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle 5, 2203–2209. [DOI] [PubMed] [Google Scholar]
- Singh SV, Herman‐Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L, Baskaran R (2004) Sulforaphane‐induced G2/M phase cell cycle arrest involves checkpoint kinase 2‐mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 279, 25813–25822. [DOI] [PubMed] [Google Scholar]
- Smith TK, Lund EK, Parker ML, Clarke RG, Johnson IT (2004) Allyl‐isothiocyanate causes mitotic block, loss of cell adhesion and disrupted cytoskeletal structure in HT29 cells. Carcinogenesis 25, 1409–1415. [DOI] [PubMed] [Google Scholar]
- Srivastava SK, Singh SV (2004) Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti‐proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis 25, 1701–1709. [DOI] [PubMed] [Google Scholar]
- Svehlikova V, Wang S, Jakubikova J, Williamson G, Mithen R, Bao Y (2004) Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo‐2 cells. Carcinogenesis 25, 1629–1637. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK‐p38 MAP kinases on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Xiao D, Johnson CS, Trump DL, Singh SV (2004) Proteasome‐mediated degradation of cell division cycle 25C and cyclin‐dependent kinase 1 in phenethyl isothiocyanate‐induced G2‐M‐phase cell cycle arrest in PC‐3 human prostate cancer cells. Mol. Cancer Ther. 3, 567–575. [PubMed] [Google Scholar]
- Zhang Y (2004) Cancer‐preventive isothiocyanates: measurement of human exposure and mechanism of action. Mutat. Res. 555, 173–190. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang L, Gonzalez V (2003) Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol. Cancer Ther. 2, 1045–1052. [PubMed] [Google Scholar]
- Zhao B, Seow A, Lee EJ, Poh WT, Teh M, Eng P, Wang YT, Tan WC, Yu MC, Lee HP (2001) Dietary isothiocyanates, glutathione S‐transferase‐M1‐T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol. Biomarkers Prev. 10, 1063–1067. [PubMed] [Google Scholar]