

Abstract
Objective: Chromosome segregation during mitosis requires a physically large proteinaceous structure called the kinetochore to generate attachments between chromosomal DNA and spindle microtubules. It is essential for kinetochore components to be carefully regulated to guarantee successful cell division. Depletion, mutation or dysregulation of kinetochore proteins results in mitotic arrest and/or cell death. HEC1 (high expression in cancer) has been reported to be a kinetochore protein, depletion of which, by RNA interference, results in catastrophic mitotic exit.
Materials and methods and results: To investigate how HEC1 protein is controlled post‐translation, we analysed the role of anaphase‐promoting complex/cyclosome (APC/C)‐Cdh1 in degradation of HEC1 protein. In this study, we show that HEC1 is an unstable protein and can be targeted by endogenous ubiquitin‐proteasome system in HEK293T cells. Results of RNA interference and in vivo ubiquitination assay indicated that HEC1 could be ubiquitinated and degraded by APC/C‐hCdh1 E3 ligase. The evolutionally conserved D‐box at the C‐terminus functioned as the degron of HEC1, destruction of which resulted in resistance to degradation mediated by APC/C‐Cdh1. Overexpression of non‐degradable HEC1 (D‐box destroyed) induced accumulation of cyclin B protein in vivo and triggered mitotic arrest.
Conclusion: APC/C‐Cdh1 controls stability of HEC1, ensuring normal cell cycle progression.
Introduction
Kinetochores are proteinaceous structures that perform multiple functions in mitosis in control of kMT (kinetochore microtubule) dynamics, attachment of chromosomes to spindle michrotubules, generation of force for chromosome movement and initiation of spindle checkpoint signalling required for the onset of anaphase. Because of the multiple functions, the kinetochore is a focal point for regulation, which occurs through phosphorylation, sumoylation, ubiquitination and methylation of its components (1, 2).
The ubiquitin–proteasome system is responsible for degrading a wide range of mitotic regulators including cyclins, mitotic protein kinases, microtubule‐associated proteins and kinetochore proteins (3, 4, 5). A ubiquitin‐activating enzyme (E1), a conjugating enzyme (E2), and a ubiquitin ligase (E3) are three essential components of this system. Ubiquitin activated via E1 is transferred to E2 and finally conjugated to the substrate by E3. It is only when this happens that the polyubiquitinated proteins can be recognized and degraded by the proteasome complex (6).
Anaphase‐promoting complex/cyclosome (APC/C), a multisubunit E3 ubiqutin ligase, plays an indispensable role in regulation of mitotic exit by regulation of degradation of a series of proteins including TPX2 and Cdc20 (7, 8). APC/C is regulated by mitosis‐specific phosphorylation and by binding of two Fizzy proteins, Cdc20 and Cdh1, which affect APC/C activity in early mitosis and late mitosis/G1 respectively (9, 10). Activity of APC/C‐Cdc20 is responsible for degradation of anaphase inhibitors that trigger chromatid separation (10). APC/C‐Cdh1 is required to target an array of cell cycle regulators, such as cyclin B, to ensure that cells can enter into the next cell cycle successfully (9). APC/C recognizes two motifs in its substrates: destruction box (D‐box: RXXLXXXXN) and KEN‐box (KENXX(DQEN)) (11).
Conserved Ndc80 complex (a key component of the outer kinetochore) plays an essential role in kinetochore functions, including microtubule binding and control of the spindle checkpoint. HEC1 (high expression in cancer) is a functional subunit of NDC80 complex, which is involved in regulation of kMT dynamics and attachment to kinetochores of kMTs. HEC1 protein remains at a high level during prophase to anaphase and decreases markedly at telophase (12, 13). HEC1 protein localized in the cytoplasm of interphase cells and translocated to chromosomes during mitosis, suggesting that both subcellular localization and protein level are regulated during the cell cycle (12, 14). Cells lacking HEC1 carry unstable microtubules and their chromosomes fail to attach to the anaphase spindle (15, 16). Overexpression of HEC1 in an inducible mouse model results in mitotic checkpoint hyperactivation (17). Thus, it may be important to carefully regulate the protein level of HEC1 to ensure normal cell division. Previous reports have indicates that temporal transcriptional regulation of HEC1 expression may also help restrict its activity to a narrow window (18).
In this study, we demonstrate that one mechanism for regulation of HEC1 is through control of its protein stability. Exogenous HEC1 protein levels fluctuate during the cell cycle; depletion of hCdh1 by RNAi results in accumulation of HEC1 proteins, while overexpression of hCdh1 can significantly reduce HEC1 protein levels. Cdh1 overexpression can sharply enhance polyubiquitination of HEC1 protein. Our results also reveal that overexpression of an HEC1 stable mutant results in mitotic arrest, suggesting the importance of HEC1 protein regulation during the cell cycle.
Materials and methods
Cell culture
Human embryonic kidney cells (HEK293T) were kindly donated by Dr Kuai and were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum.
Statistical analysis
To determine the effect of hCDH1 overexpression on fluorescence intensity of HEC1 and difference in mitotic indices between cells transfected with non‐degradable HEC1 and mock vector, values were compared using the Student’s t‐test. P‐value of <0.05 was used as level of significance. Statistical analysis was performed using EXCEL software. Results are reported as mean ± SD.
Plasmid construction
Human HEC1 was amplified by PCR using human foetal liver cDNA as template, and was subcloned into pEGFP‐N1 and pCMV‐myc vector. Deletions of HEC1 were constructed by PCR. PCR products were inserted into pCMV‐myc (Clontech, Palo Alto, CA, USA) and pEGFP‐N1 as needed. Plasmids which encode hCdh1 and Ub‐HA were kindly donated by Dr Michael Brandeis and Dr Yue Xiong respectively (19, 20).
Cell cycle distribution analysis
HEK293T cells were transfected with either pEGFP‐N1‐HEC1 variants or with pEGFP‐N1. Attached cells were harvested, resuspended in 300 μl of PBS (containing 30 μl FBS) and fixed in 2 ml of 70% ice‐cold ethanol. Samples were kept at −20 °C until analysis by flow cytometry. Cells were washed in PBS, treated with 0.1 mg/ml RNase A (Pharmacia‐Biotech, Uppsala, Sweden) for 30 min and stained with 40 μg/ml propidium iodide (Sigma, St Louis, MO, USA). DNA content was analysed by FACS Calibur (BD, San Jose, CA, USA) (21).
Fluorescence microscopy and image analysis
A total of 2 × 105 HEK293T cells were seeded per well in six‐well plates. After 16 h incubation, cells were co‐transfected with hCdh1 and pEGFP‐N1‐HEC1/vector mock or transfected with hCdh1 siRNA and HEC1/vector mock. At 48 h post‐transfection, total fluorescence intensities of each field were calculated using QWIN Software (22).
Western blot analysis
Cells were lysed in lysis buffer [50 mm Tris–HCl (pH 7.5); 150 mm NaCl; 1% Tween 20 (v/v); 0.2% NP40; 10% glycerol] with freshly added protease inhibitor cocktail tablets (Roche, Indianapolis, IN, USA), 1 mm NaF and 0.1 mg/ml phenylmethylsulphonyl fluoride. Samples were separated by 10% SDS–PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) and blotted with anti‐c‐myc (Clontech, Palo Alto, CA, USA), anti‐cyclin B (BD Pharmingen, San Jose, CA, USA) or anti‐actin (Santa Cruz, CA, USA) antibody. Immunostained bands were visualized using Super‐Signal (Pierce, Rockford, IL, USA) western blotting detection system.
In vivo ubiquitination assay
HEK293T cells in 9 cm plates were transfected with combinations of 5 μg of HA‐ubiquitin expression plasmid, 1 μg of plasmid encoding human HEC1‐myc and 3 μg of hCdh1. Thirty‐six hours after transfection, cells from each plate were collected and divided into two aliquots. One aliquot (10%) was used for conventional western blotting to confirm expression and degradation of transfected proteins. The remaining cells (90%) were used for purification of myc‐tagged proteins by protein A/G beads. Cell pellets were lysed in lysis buffer (6 m guanidinium‐HCl, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris–HCl (pH 8.0), 5 mm imidazole, 10 mmβ‐mercaptoethanol] and incubated in anti‐c‐myc for 4 h at 4 °C; this was followed by incubation with protein A/G beads for 6–8 h at 4 °C. Beads were washed in cell lysis buffer. Eluted proteins were analysed by western blotting of conjugated HEC1 using HA antibody (23).
Cell synchronization and RNA interference
HEK293T cells were synchronized using nocodazole as described previously (24). Knockdown of Cdh1 was accomplished by transfecting cells with small interfering RNAs (Shanghai Genepharma Co., Shanghai, China) directed against Cdh1 using Oligofectamine™, as instructed by the manufacturer (Invitrogen, Carlsbad, CA, USA). RNA interference target sequences are Cdh1: 5′‐GGATTAACGAGAATGAGAAGT‐3′ and negative control was provided by Shanghai Genepharma Co. Forty‐eight hours post‐transfection, cells were harvested and whole cell extracts were Western blotted with the indicated antibodies (5).
Evaluation of mitotic index
HEK293T cells transfected with plasmids encoding non‐degradable HEC1‐GFP or GFP were fixed in 4% polymerized formaldehyde at room temperature and stained with DAPI 48 h post‐transfection. Percentages of cells with condensed chromatin were calculated in five fields of view (each >100 cells) using a fluorescence microscope (25).
Results
HEC1 protein turnover was regulated by the ubiquitin–proteasome pathway
Cycloheximide (10 μg/ml, a protein synthesis inhibitor) was applied to detect turnover of exogenous HEC1 in vivo (26). Figures 1a and S1 reveal that protein level of HEC1 decreased significantly after incubation with CHX for 8 and 16 h, indicating that HEC1 protein might be regulated by an endogenous degradation system.
Figure 1.
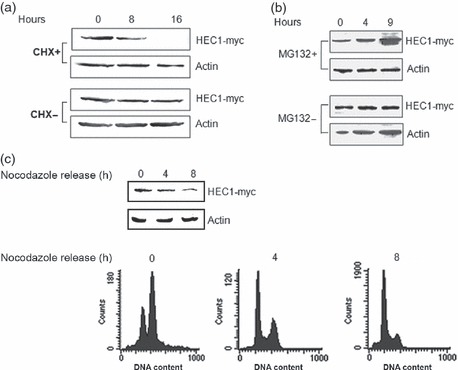
Overexpression of hCdh1 accelerates HEC1 protein turnover. (a) HEC1 could be degraded by the endogenous proteasome system. HEK293T cells transfected with myc‐HEC1 were treated with CHX and harvested at the indicated times. HEC1 was quantified by immunoblotting analysis with anti‐myc (for HEC1) antibodies, and actin was used as loading control. (b) HEC1 protein turnover was regulated by a proteasome pathway. HEK293T cells were transiently transfected with pCMV‐HEC1. After 24 h, MG132 was added and incubated for 0, 4 and 9 h. Total cell lysates were subjected to immunoblotting analysis with anti‐myc (for HEC1) and anti‐actin (loading control) antibodies. (c) HEC1 protein levels fluctuated during mitosis to G1 phase. HEK293T cells transfected with plasmid encoding HEC1‐myc were arrested with nocodazole treatment at metaphase. Cells were harvested at 0, 4 or 8 h after nocodazole release. Bottom: Cell cycle distribution was monitored by flow cytometry. As Table S1 shows, more than 80% cells accumulated at G2/M after nocodazole treatment and a few cells remained with 4N DNA content at 8 hours after release. Top: Protein levels of HEC1 were detected by immunoblotting with anti‐myc antibodies.
To analyse the role of the ubiquitin–proteasome pathway in regulation of HEC1, MG132 (20 μm, inhibitor of the ubiquitin–proteasome pathway) treatment was applied (27). As shown in Figs 1b and S2, protein levels of endogenous and exogenous HEC1 increased rapidly after incubation with MG132 for 9 h, suggesting that HEC1 might be regulated by the ubiquitin–proteasome system.
To understand how HEC1 protein is controlled in cell cycle progression, HEK293 cells were transfected with HEC1‐myc plasmid, arrested at metaphase (more than 80% cells accumulated with 4N DNA content) by nocodazole treatment (100 ng/ml) and then released to allow cells to progress from metaphase to the next G1. Cell cycle profiles of released cells were assayed by flow cytometry (Fig. 1c, Up). Figure 1c shows that HEC1 protein levels decreased markedly by 4 h post‐nocodazole release. This agrees with previous reports that HEC1 protein levels can hardly be detected by telophase (13), suggesting that HEC1 proteins are tightly regulated in cell cycle progression.
Taken together, these results suggest that turnover of HEC1 proteins may be regulated by the endogenous ubiquitin–proteasome system in vivo.
HEC1 protein as substrate of APC/C‐Cdh1
That APC/C can target its substrates by recognition of either KEN box [KENxxx (DQEN)] or destruction box (D box, RxxLxxxxN) in the proteins is well known (28). As shown in Fig. 2a, sequence alignment of HEC1 proteins from different species has revealed presence of a conserved protein D‐box at the C‐terminus of HEC1. This was also consistent with previous results that HEC1 protein levels decreased sharply at 4 h post‐nocodazole release (as Fig. 1c shown), implying a steep level of degradation. We therefore hypothesized that HEC1 might be targeted and degraded by APC/C‐Cdh1.
Figure 2.
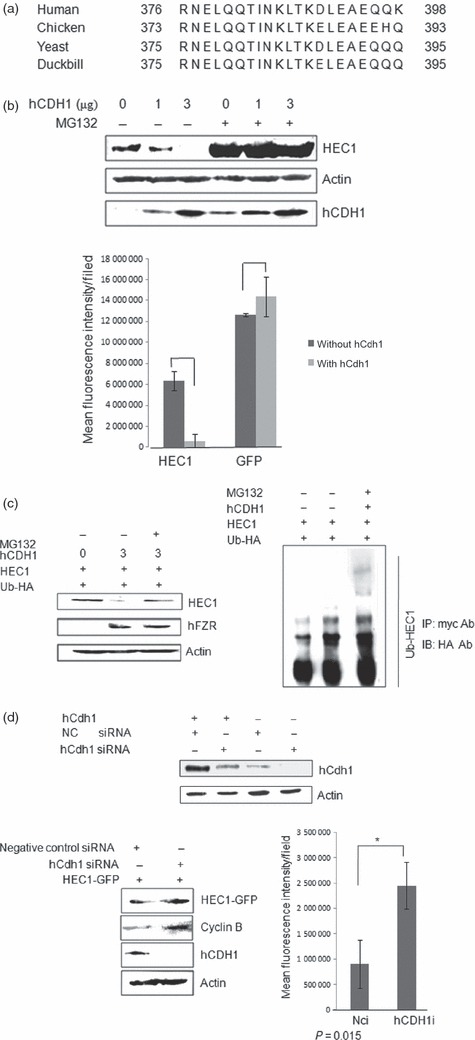
hCdh1 overexpression enhanced HEC1 ubiquitination. (a) Alignment of amino acid sequences of human, chicken, yeast and duckbill HEC1 predicted a conserved D‐box at the C‐terminus of HEC1. (b) HEC1 could be degraded by APC/C‐Cdh1.Top: Plasmids encoding HEC1‐myc and hCdh1 were co‐transfected into HEK293T Cells. MG132 was added 8 h before harvest. Top: HEC1 protein level was analysed by Western blotting. Bottom: Plasmids encoding HEC1‐GFP and hCdh1 were co‐transfected into HEK293T cells. Vector pEGFP‐N1 was used as negative control. Bottom: Protein levels of HEC1 were examined by monitoring fluorescence intensity of GFP (P1 ≈ 0.02 and P2 ≈ 0.28). (c) HEC1 was ubiquitinated by APC/C in vivo. Cells co‐transfected with HEC1‐myc, hCdh1 and HA‐ubiquitin were collected and lysed; 400 μl cell lysates were transferred into a new tube and incubated with anti‐myc antibody. Immunopurified samples were analysed by immunoblotting with anti‐HA antibody to detect levels of ubiquitinated‐HEC1protein (Left). The remainder of the lysate was immunoblotted with anti‐myc and anti‐actin antibodies directly to detect protein levels of HEC1 and actin (Right). (d) Above: Transfection with siRNA against hCdh1 significantly reduced protein level of exogenous and endogenous hCdh1. Middle: Knockdown of hCdh1 by RNA interference resulted in accumulation of HEC1‐myc. siRNA against hCdh1 or negative control siRNA were co‐transfected with HEC1‐myc into HEK293T cells. At 48 h post‐transfection, cells were harvested to analyse protein levels of hCdh1 and HEC1. Cyclin B was used as positive control and actin was used as loading control. Bottom: Knockdown of hCdh1 by RNA interference resulted in increase in fluorescence intensity of cells transfected with GFP‐HEC1 (P‐value = 0.01).
To verify whether HEC1 could be targeted by APC/C‐Cdh1, hCdh1 (an active subunit of APC/C) was overexpressed to activate the endogenous APC/C‐mediated degradation pathway and HEC1 protein level was monitored in two different ways. As expected, a significant reduction in HEC1 protein levels was detected by immunoblotting with anti‐myc antibody. This was consistent with result showing that fluorescence intensity of cells expressing HEC1‐GFP also decreased sharply when hCdh1 was co‐transfected and MG132 treatment abrogated this decline in HEC1‐myc expression (Fig. 2b; Left, lane2 and lane 4).
To analyse the role of APC/C in stability of HEC1, we tested whether HEC1 could be subjected to ubiquitination. Plasmids encoding hFZR were co‐transfected with HEC1‐myc and Ub‐HA to activate endogenous APC/C complex. Part of the cell lysate was immunoprecipitated with anti‐myc antibody and then immunoblotted with anti‐HA antibody, to analyse levels of Ub‐conjugated HEC1. Figure 2c (Right) indicates that the protein level of polyubiquitinated HEC1 increased sharply, when 26S proteasome inhibitor MG132 was applied. Remnant lysates were immunoblotted directly with anti‐myc antibody and anti‐hFZR antibody. As shown in Fig. 2c, co‐expression of HEC1‐myc with hCdh1 resulted in decrease of HEC1‐myc protein level in the cell lysate.
To confirm that APC/C‐Cdh1 was required for destruction of HEC1 in vivo, we inhibited activity of APC/C‐Cdh1 by RNA interference. HEK293T cells were transfected with Cdh1‐specific siRNA (siCdh1) or control siRNA (siCK) (5). Detection of accumulation of HEC1 protein and cyclin B was clear when Cdh1 was significantly knocked down by siRNA transfection, indicating that APC/C‐Cdh1 played an essential role in degradation of HEC1 protein (Fig. 2d, bottom). Cyclin B was used as positive control. We conclude that HEC1 was degraded by the APC/C‐Cdh1 pathway in vivo.
D‐box at C‐terminus functioned as a degron of HEC1 protein
As mentioned above, a potential D‐box was discovered at the C‐terminus of HEC1 protein (Fig. 2a). To test the importance of the D‐box motifs for HEC1 destruction, the D‐box at the C‐terminus was mutated by deletion of three amino acid residues (RNE, deletion mutant). Mutated fragments and truncated HEC1 (truncated mutant 1–220aa) (Fig. 3a, left) were subcloned into pCMV‐myc.
Figure 3.
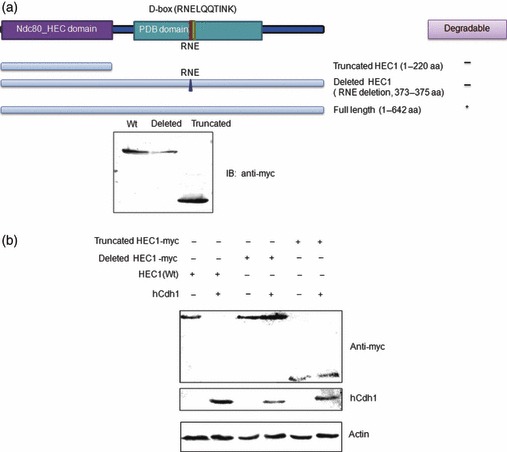
Destruction box at C‐terminus of HEC1 was required for its degradation. (a) Two mutants of HEC1, deleted and truncated HEC1, were subcloned into pCMV‐myc. Constructs derived from HEC1 used in this study are shown; number indicates amino acid position (Above). Expression of myc tagged two mutants and HEC1 verified by immunoblotting with anti‐myc antibody (Bottom). (b) Degradation of two mutated HEC1 by APC/C‐Cdh1. Deleted and truncated HEC1 were co‐transfected with hCdh1 into HEK293T cells and protein levels were detected by western blotting using anti‐myc antibodies.
Figure 3b shows that only wild‐type HEC1 was efficiently degraded when hCdh1 was co‐transfected. The deletion mutant and truncated mutant both displayed resistance to degradation of APC/C. Two mutants lacking D‐box failed to be degraded by Cdh1‐activated APC/C. Therefore, it could be concluded that D‐box in HEC1 was necessary for its degradation by APC/C‐Cdh1.
Expression of non‐degradable mutant of HEC1 resulted in mitotic arrest
To investigate the physiological significance of degradation of HEC1 by APC/C‐Cdh1, mutated HEC1 proteins with RNE deletions were subcloned into pEGFP‐N1. Figure 4a shows that overexpression of hCdh1 failed to decrease protein levels of GFP‐fused deleted HEC1 mutant.
Figure 4.
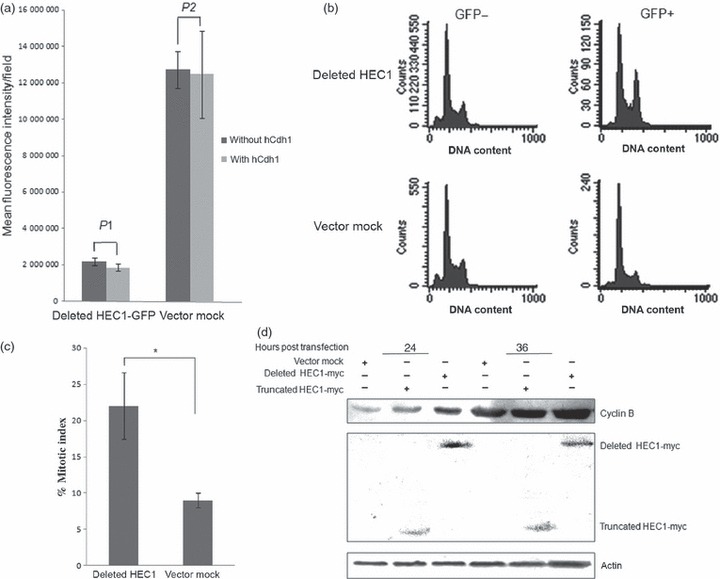
Accumulation of non‐degradable HEC1 results in mitotic arrest. (a) Deleted HEC1‐GFP failed to be degraded by APC/C‐Cdh1. The deleted mutant was inserted into pEGFP‐N1 (Above); this plasmid was co‐transfected with hCdh1 into HEK293T cells. Vector pEGFP‐N1 was used as a negative control. Protein levels of HEC were examined by monitoring fluorescence intensity of GFP (Bottom). P1 and P2 were both greater than 0.05 (P1 ≈ 0.45; P2 ≈ 0.85). (b) Overexpression of HEC1 variant could trigger G2/M delay in HEK293T. A significant increase in G2/M percentage was detected after GFP‐deleted HEC1 transfection. Cells were transfected with plasmids expressing GFP alone or GFP‐deleted HEC1. Cell cycle distribution was analysed according to DNA content of GFP‐positive cells at 36 h post‐transfection, using flow cytometry. (c) Cell population with mitotic chromatin increased in cells expressing deleted HEC1‐GFP protein. Cells transfected with plasmids encoding deleted HEC1‐GFP and GFP were stained by DAPI, at 24 h after transfection. Percentage of mitotic cells in each group was counted using a fluorescence microscope (P‐value is 0.03). (d) Overexpression of HEC1 variant could result in accumulation of cyclin B protein levels. An clear increase in cyclin B protein level was observed after deleted HEC1 transfection. Cells were transfected with plasmids encoding myc‐truncated HEC1, myc‐deleted HEC1 or pCMV‐myc. Cells were harvested and lysed to analyse cyclin B protein level at 24 and 36 h post‐transfection respectively.
HEK293T cells transfected with GFP‐deleted mutant, GFP‐HEC1 (Wt) or vector mock were analysed by flow cytometry to observe relative DNA content in GFP‐positive cells (Fig. 4b). A striking accumulation of GFP‐positive cells with 4N DNA content was observed that had been transfected with GFP‐deleted mutant. In contrast, the GFP‐positive cells from the GFP‐vector mock transfection and GFP‐negative cells from the sample and control both displayed typical asynchronous cell cycle distribution. We speculate that cell accumulation with 4N DNA content is a direct consequence of accumulation of stable HEC1 mutants during the cell cycle. This observation pointed to the importance of controlling levels of HEC1 protein in the cell cycle. However, overexpression of wild‐type HEC1 also induced a slight increase in proportion of G2/M cells (Fig. S3). G2/M arrest induced by wild type HEC1 might be attributed to inadequate ability of the endogenous degradation system to proteolyse excess exogenous HEC1 protein in time. To further understand the mechanism of cell cycle delay induced by deleted mutant accumulation, mitotic index was calculated. Figure 4c shows that elevated mitotic index was observed in cells expressing GFP‐deleted mutant and Fig. S4 showed deleted HEC1 overexpression led to a sharp increase of the phosphorylated Histone H3 protein level, suggesting that deleted mutant overexpression resulted in mitotic arrest. Moreover, a significant increase in cyclin B protein level was detected in cells transfected with deleted mutant (lane 3 and lane 6), compared to cells transfected with deleted mutant and vector mock (lane 1 and lane4) (Fig. 4d). This result provided another piece of evidence indicating that deleted mutant accumulation induced mitotic arrest.
Discussion
Degradation of HEC1 by APC/C‐Cdh1 and its physiological role
Previous studies have shown that protein level of HEC1 peaks in G2 and M phases (13). Its expression pattern is similar to other substrates of APC/C‐Cdh1 (the major E3 ubiquitin ligase complex), such as cyclin B and TPX2, implying that HEC1 degradation activity is tightly regulated in the cell cycle (3, 7). In this study, we have presented a potential molecular mechanism that contributes to regulation of HEC1. We have provided several lines of evidence to support the hypothesis that HEC1 is targeted and ubiquitinated by APC/C‐Cdh1 in vivo. First, treatment with CHX, a protein synthesis inhibitor, significantly reduced levels of HEC1 protein. Second, treatment with the proteasome inhibitor MG132 resulted in HEC1 accumulation in vivo. Third, protein levels of HEC1 decreased sharply in cells 4 h post‐nocodazole release. Fourth, HEC1 contains a degradation signal (D‐box), which could be essential for degradation by APC/C‐Cdh1. Fifth, overexpression of hCdh1 decreased protein levels of ectopically expressed HEC1 and enhanced polyubiquitination of HEC1. Finally, depletion of hCdh1by RNA interference resulted in accumulation of ectopically expressed HEC1. Taken together, HEC1 is shown to be a novel substrate of APC/C‐Cdh1 in cell cycle progression.
Previous reports also indicate that HEC1 can prevent degradation of cyclin B, and overexpression of its C‐terminus fragment results in the death of daughter cells, caused by cyclin B accumulation (29). However, the detailed mechanism of mitotic arrest induced by deleted mutant overexpression remains unclear. A clear accumulation of mitotic cells expressing non‐degradable HEC1 mutant (Fig. 4a) was observed and high concentration of cyclin B was also detected in cells transfected with non‐degradable HEC1 (Fig. 4b). As overexpression of stable cyclin B results in mitotic arrest (30), we speculated that high concentration of cyclin B might contribute to the mitotic arrest induced by stable HEC1 accumulation. However, we could not neglect mitotic arrest and cyclin B accumulation induced by deleted HEC1 because that the mitotic checkpoint was activated by deleted HEC1 accumulation. Although the detailed physical role of HEC1 degradation still needs further investigation, serious consequences of HEC1 degradation failure indicate the importance of spatial and selective regulation of substrate degradation by APC/C to ensure normal cell cycle progression.
Inter‐regulation of APC/C‐26S proteasome and mitotic regulators
To guarantee normal cell division, it is necessary for protein level of mitotic regulators to be controlled by the ubiquitin–proteasome pathway. Yet, activity of the APC/C‐mediated ubiquitin–proteasome system is also closely regulated by mitotic kinases, including Plk1 and Bub1 (31). This implies existence of inter‐regulation between mitotic regulators and APC/C. However, there is little evidence to show how the 26S proteasome complex is controlled in mitosis. Our research on HEC1 will be useful in furthering understanding of these areas.
As discussed above, HEC1 can bind and regulate activity of 26S, and HEC1 is also degraded by APC/C‐mediated proteolysis (29), suggesting that mitotic regulators control activity of the ubiquitin–proteasome system in at least two different ways: via inactivation or activation of the E3 ligase (APC/C) and the 26S proteasome complex. Continuous association between the C‐terminus of HEC1 and MSS1 resulted in death of daughter cells, and non‐degradable HEC1 accumulation induced mitotic arrest, suggesting that inter‐regulation of the ubiquitin–proteasome system and mitotic regulators might play an important role in control of mitotic progression.
In conclusion, we have demonstrated a novel molecular mechanism that contributed to restriction of the protein level of HEC1 during cell cycle progression. HEC1 is an unstable protein, which can be targeted and polyubiquitinated by APC/C‐Cdh1, and D‐box at C‐terminus of HEC1 protein functions as the degron, destruction of which results in degradation failure. Overexpression of stable HEC1 induced mitotic arrest, indicating that degradation of HEC1 by APC/C‐Cdh1 is essential to guarantee normal cell division.
Supporting information
Fig. S1 Endogenous protein level of HEC1 decreased sharply after CHX treatment. CHX was used a inhibitor to stop protein synthesis and detect the fluctuation of endogenous HEC1 protein level. Actin was used as a loading control. Figure showed that HEC1 protein was almost undectable at 16 hours after addition of CHX, suggesting HEC1 protein was regulated by endogenous degradation system.
Fig. S2 MG132 treatment induced accumulation of endogenous HEC1 protein. HEK293Tcells were harvested at indicated time points (0 or 9 hours) after MG132 treatment and lyzed to analyze the protein level of endogenous HEC1. Actin was used as a loading control. As figure shown, endogenous HEC1 protein accumulated significantly after incubation with MG132 for 9 hours, indicating HEC1 protein might be a substrate of ubiquitin‐protesome pathway.
Fig. S3 Overexpression of wild‐type HEC1 induced G2/M arrest slightly. Plasmid encoding GFP‐HEC1 were transfected into HEK293T cells. 36hours post transfection, cells were harvested to analyze the cell cycle distribution of GFP postive cell. As figure shown, HEC1 overexpression resulted in 16.5 percent of cells remaining at G2/M phase, whlie only 5.49 percent of cells expressing GFP maintained at G2/M phase.
Fig. S4 Overexpression of deleted HEC1 mutant induced a sharp increase of phosphorylated histone H3 protein level. Protein level of phosphorylated histone H3 was used a biomarker of mitosis. To verify whether deleted HEC1 mutant arrested cells in mitosis or not, the protein level of phosphorylated histone H3 was detected. As figure shown, overexpression of HEC1 mutant and nocodazole treatment both significantly induced an increase of phosphorylated histione H3 protein level. Histone H3 protein was used as a loading control. Cells incubated with nocodazole for 16 hours were used as positive control.
Table S1 Cell cycle distribution following release from growth arrest by nocodazole treatment.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
We would like to thank Dr Michael Brandeis and Dr Yue Xiong for providing key plasmids. We thank Mrs Qiaoling Zhang and Mr Hongkun Wu for their technical assistance in the FACS analysis.
References
- 1. Asbury CL, Trisha ND (2008) Insights into the kinetochore. Structure 16, 834–836. [DOI] [PubMed] [Google Scholar]
- 2. Yoon HJ, Carbon J (1995) Genetic and biochemical interactions between an essential kinetochore protein, Cbf2p/Ndc10p, and the CDC34 ubiquitin‐conjugating enzyme. Mol. Cell. Biol. 15, 4835–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamamoto TM, Iwabuchi M, Ohsumi K, Kishimoto T (2005) APC/C‐Cdc20‐mediated degradation of cyclin B participates in CSF arrest in unfertilized Xenopus eggs. Dev. Biol. 279, 345–355. [DOI] [PubMed] [Google Scholar]
- 4. Stewart S, Fang G (2005) Destruction box‐dependent degradation of aurora B is mediated by the anaphase‐promoting complex/cyclosome and Cdh. Cancer Res. 65, 8730–8735. [DOI] [PubMed] [Google Scholar]
- 5. Seki A, Fang G (2007) CKAP2 is a spindle‐associated protein degraded by APC/C‐Cdh1 during mitotic exit. J. Biol. Chem. 282, 15103–15113. [DOI] [PubMed] [Google Scholar]
- 6. Hershko A, Ciechanover A (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 7. Stewart S, Fang G (2005) Anaphase‐promoting complex/cyclosome controls the stability of TPX2 during mitotic exit. Mol. Cell. Biol. 25, 10516–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen RH (2007) Dual inhibition of Cdc20 by the spindle checkpoint. J. Biomed. Sci. 14, 475–479. [DOI] [PubMed] [Google Scholar]
- 9. Jaspersen SL, Charles JF, Morgan DO (1999) Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 9, 227–236. [DOI] [PubMed] [Google Scholar]
- 10. Kramer ER, Gieffers C, Hölzl G, Hengstschläger M, Peters JM (1998) Activation of the human anaphase‐promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol. 8, 1207–1210. [DOI] [PubMed] [Google Scholar]
- 11. Pfleger CM, Kirschner MW (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655–665. [PMC free article] [PubMed] [Google Scholar]
- 12. Lin YT, Chen Y, Wu G, Lee WH (2006) Hec1 sequentially recruits Zwint‐1 and ZW10 to kinetochores for faithful chromosome segregation and spindle checkpoint control. Oncogene 25, 1–14. [DOI] [PubMed] [Google Scholar]
- 13. Chen Y, Riley DJ, Zheng L, Chen PL, Lee WH (2002) Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J. Biol. Chem. 277, 49408–49416. [DOI] [PubMed] [Google Scholar]
- 14. Hori T, Haraguchi T, Hiraoka Y, Kimura H, Fukagawa T (2003) Dynamic behavior of Nuf2‐Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J. Cell Sci. 116, 3347–3362. [DOI] [PubMed] [Google Scholar]
- 15. Vorozhko VV, Emanuele MJ, Kallio MJ, Stukenberg PT, Gorbsky GJ (2008) Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma 117, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ciferri C, Deluca J, Monzani S, Ferrari KJ, Ristic D, Wyman C et al. (2005) Architecture of the human ndc80‐hec1 complex, a critical constituent of the outer kinetochore. J. Biol. Chem. 280, 29088–29095. [DOI] [PubMed] [Google Scholar]
- 17. Diaz‐Rodríguez E, Sotillo R, Schvartzman JM, Benezra R (2008) Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc. Natl. Acad. Sci. USA 105, 16719–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen L, Li L, Qiao X, Liu J, Yao X (2007) Functional characterization of the promoter of human kinetochore protein HEC1: novel link between regulation of the cell cycle protein and CREB family transcription factors. Biochim. Biophys. Acta 1769, 593–602. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y, Xiong Y (2001) A p53 amino‐terminal nuclear export signal inhibited by DNA damage‐induced phosphorylation. Science 292, 1910–1915. [DOI] [PubMed] [Google Scholar]
- 20. Bharadwaj R, Yu R (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23, 2016–2027. [DOI] [PubMed] [Google Scholar]
- 21. Normand G, Hemmati PG, Verdoodt B, von Haefen C, Wendt J, Guner D et al. (2005) p14ARF induces G2 cell cycle arrest in p53‐ and p21‐deficient cells by down‐regulating p34cdc2 kinase activity. J. Biol. Chem. 280, 7118–7130. [DOI] [PubMed] [Google Scholar]
- 22. Franks DJ, Mroske C, Laneuville O (2001) A fluorescence microscopy method for quantifying levels of prostaglandin endoperoxide H synthase‐1 and CD‐41 in MEG‐01 cells. Biol. Proced. Online 3, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li L, Deng B, Xing G, Teng Y, Tian C, Yin X et al. (2007) PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc Natl. Acad. Sci. USA 104, 7951–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M et al. (2004) Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 23, 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsai YV, Qi H, Lin CP, Lin RK, Kerrigan JE, Rzuczek SG et al. (2009) A G‐quadruplex stabilizer induces M phase cell cycle arrest. J. Biol. Chem. 284, 22535–22543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao CW, Yan X, Li Y, Reddy SA, Tsang BK (2003) Resistance of human ovarian cancer cells to tumor necrosis factor alpha is a consequence of nuclear factor kappaB‐mediated induction of Fas‐associated death domain‐like interleukin‐1beta‐converting enzyme‐like inhibitory protein. Endocrinology 144, 623–630. [DOI] [PubMed] [Google Scholar]
- 27. Oka M, Yanagewa Y, Asada T, Yoneda A, Hasezawa S, Sato T et al. (2004) Inhibition of proteasome by MG‐132 treatment causes extra phragmoplast formation and cortical microtubule disorganization during M/G1 transition in synchronized tobacco cells. Plant Cell Physiol. 45, 1623–1632. [DOI] [PubMed] [Google Scholar]
- 28. Burton JL, Solomon MJ (2001) D box and KEN box motifs in budding yeast Hsl1p are required for APC‐mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 15, 2381–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y, Sharp ZD, Lee WH (1997) HEC binds to the seventh regulatory subunit of the 26 S proteasome and modulates the proteolysis of mitotic cyclins. J. Biol. Chem. 272, 24081–24087. [DOI] [PubMed] [Google Scholar]
- 30. Rimmington G, Dalby B, Glover DM (1994) Expression of N‐terminally truncated cyclin B in the Drosophila larval brain leads to mitotic arrest at late anaphase. J. Cell Sci. 107, 2729–2738. [DOI] [PubMed] [Google Scholar]
- 31. Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J et al. (2003) Mitotic regulation of the human anaphase‐promoting complex by phosphorylation. EMBO J. 22, 6598–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Endogenous protein level of HEC1 decreased sharply after CHX treatment. CHX was used a inhibitor to stop protein synthesis and detect the fluctuation of endogenous HEC1 protein level. Actin was used as a loading control. Figure showed that HEC1 protein was almost undectable at 16 hours after addition of CHX, suggesting HEC1 protein was regulated by endogenous degradation system.
Fig. S2 MG132 treatment induced accumulation of endogenous HEC1 protein. HEK293Tcells were harvested at indicated time points (0 or 9 hours) after MG132 treatment and lyzed to analyze the protein level of endogenous HEC1. Actin was used as a loading control. As figure shown, endogenous HEC1 protein accumulated significantly after incubation with MG132 for 9 hours, indicating HEC1 protein might be a substrate of ubiquitin‐protesome pathway.
Fig. S3 Overexpression of wild‐type HEC1 induced G2/M arrest slightly. Plasmid encoding GFP‐HEC1 were transfected into HEK293T cells. 36hours post transfection, cells were harvested to analyze the cell cycle distribution of GFP postive cell. As figure shown, HEC1 overexpression resulted in 16.5 percent of cells remaining at G2/M phase, whlie only 5.49 percent of cells expressing GFP maintained at G2/M phase.
Fig. S4 Overexpression of deleted HEC1 mutant induced a sharp increase of phosphorylated histone H3 protein level. Protein level of phosphorylated histone H3 was used a biomarker of mitosis. To verify whether deleted HEC1 mutant arrested cells in mitosis or not, the protein level of phosphorylated histone H3 was detected. As figure shown, overexpression of HEC1 mutant and nocodazole treatment both significantly induced an increase of phosphorylated histione H3 protein level. Histone H3 protein was used as a loading control. Cells incubated with nocodazole for 16 hours were used as positive control.
Table S1 Cell cycle distribution following release from growth arrest by nocodazole treatment.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item