

Abstract
Objectives: Human embryonic stem cells (hESC) are promising for tissue engineering (TE) purposes due to their unique properties. However, current standard mechanical passaging techniques limit rates of possible TE experiments, as it is difficult to obtain high enough numbers of the cells for experimentation. In this study, several dissociative solutions and application methods are tested for their applicability to, and influence on, hESC culture and expansion.
Materials and methods: Expansion of two hESC lines, H1 and VUB01, subjected to different passaging techniques, was evaluated. Four dissociative solutions – TrypLE™ Express, Trypsin‐EDTA, Cell Dissociation Solution and Accutase™– were combined with two application protocols. As reference conditions, manual and bead‐based passaging techniques were used.
Results: Results showed that use of Cell Dissociation Solution in combination with a slow adaptation protocol, generated the best expansion profile for both cell lines. The hESC single cell lines remained pluripotent, had good expansion profiles and were capable of differentiation into representatives of all three germ layers. Reproducibility of the results was confirmed by adaptation for three other hESC lines.
Conclusion: Use of Cell Dissociation Solution, combined with slow adaptation protocol, allows a fast switch from the mechanical passaging technique to a single‐cell split technique, generating stable and robust hESC cell lines, which allow for large scale expansion of hESC for TE purposes.
Introduction
Since the first derivation of human embryonic stem cell (hESC) lines (1), scientists have been researching these cells and discovering their importance, complexity and capabilities. This cell population is characterized by three unique traits. First, the cells are unspecialized, secondly, they can be kept undifferentiated in culture for unlimited time, which is called long‐term self renewal and finally, they retain the capacity to form virtually every cell type present in the adult human body and can be steered into a certain tissue direction (differentiation) using specific culture media, a trait known as pluripotency (2). These remarkable traits turn hESC into one of the most interesting cell types for tissue engineering and cell therapy applications. The scientific world hopes to rapidly resolve some major health issues, such as the organ shortage problem and treatment of currently incurable diseases (diabetes, Parkinson’s disease, heart failure and more) (3, 4) by formation of tissue‐engineered hESC constructs or by hESC cell therapy.
Numerous studies have been performed to develop basic culture requirements and key parameters of undifferentiated and differentiated hESC culture. Although there has been significant progress in establishing culture conditions, many questions and improvement opportunities still remain. Some rate‐limiting steps that delay fast progression of hESC tissue engineering are: need for feeder layers originating from animal or human tissues (5, 6, 7); use of animal derived feeder‐free alternatives (8, 9, 10) and lack of alternatives; and application of ill‐defined media and components (11) with their inherent contamination risks. However, the most important issue is difficulty in obtaining large numbers of undifferentiated hESC. Currently, the manual passaging technique (microdissection using sharp glass cutting pipettes) is one of the most widespread methods for hESC passaging, due to the need for high level of control over differentiation. During this procedure, undifferentiated and differentiated colonies are cut into small pieces, separated based upon their morphology and transferred manually to new feeder plates (6, 12). As can be deducted, this manual procedure is very labour‐intensive, time‐consuming and needs to be performed by highly trained specialists. The technique allows for generation of sufficient numbers of undifferentiated hESC for experiments involving techniques such as gene screening and transfer as well as for drug discovery and in vitro toxicology testing; unfortunately, it is not efficient enough for generating large numbers of hESC (3).
Due to low clonal survival level of hESC (13) at low densities and their dependence on cell–cell interactions and para/autocrine signals, it is not obvious how to alter the passaging technique. Different protocols for ‘cluster passaging’ and ‘single cell passaging’ have been suggested in the literature, using different enzymatic solutions, such as CTK solution (14), TrypLE™ Express (15), Trypsin‐EDTA (16, 17, 18) and Accutase™ (19, 20). The use of these has in some cases, led to the development of chromosomally unstable cell lines that develop CD30 expression (21) or karyotypic alterations (16). CD30, a member of the tumour necrosis factor super family, is a surface marker for malignant cells in Hodgkin’s disease and embryonic carcinomas (22, 23), but a clear link between CD30 expression and chromosomal abnormalities has not been shown (24). In addition, it is unclear if the enzymatic solutions are responsible for these instabilities. Large comparative studies of different passaging techniques and dissociative solutions, focusing on cell number expansion, have not yet been performed using the same hESC lines. However, these studies are of crucial importance for the improvement of hESC large scale culture, as variety between hESC cell lines could be responsible for differences in responses between passaging techniques (25, 26).
In this article, we have examined and compared expansion profiles of two different hESC cell lines – commercially available H1 and VUB01 – with 10 different passaging protocols. We focused on their expansion profiles under different conditions, to discover the most suitable protocol for fast and robust large scale hESC expansion. Meanwhile, we also took into account that such a protocol needed to be easy to perform, not time consuming and cost effective for future applications in tissue engineering and cell therapy. Our most favourable condition was then applied to three other hESC lines to confirm our findings.
Materials and methods
Cell culture
Mouse embryonic fibroblasts. Mouse embryonic fibroblasts (MEFs), originating from MF1 mouse embryos between passage 0 and 4, were used as a feeder layer to sustain the hESC culture. The MEFs were treated with 10 μg/ml mitomycin C (MP Biomedicals, Brussels, Belgium) for 3 h to arrest mitosis, and were seeded at a density of 8500 cells/cm2, on gelatine‐coated tissue culture plates.
hESC culture protocols. The human embryonic stem cell lines used for these experiments were the H1 (p50) (Wicell, Madison, WI, USA) and VUB01 (p260) (27) (VUB, Brussels, Belgium). All cells were cultured on mitomycin‐C‐inactivated MEFs on tissue culture plates at 37 °C and 5% CO2. hESC medium consisted of 80% Dulbecco’s Modified Eagle’s medium (DMEM)‐F12 (Invitrogen, Merelbeke, Belgium) + 20% Serum Replacement (Invitrogen) + 1 mm l‐glutamine (Invitrogen) + 0.1 mm Non Essential Amino Acids (NEAA; Invitrogen) + 0.1 mmβ‐mercapto‐ethanol (Invitrogen) and 4 ng/ml basic fibroblast growth factor (bFGF) (Millipore, Brussels, Belgium). Medium was changed daily and cells grew as dense colonies. Our most favourable strategy was also tested on H9 (p30), BG01 (p30) (Wicell) and 181 (p30) (Karolinska Institutet, Stockholm, Sweden) hESC lines.
An overview of the ten different test protocols can be found in Table 1. Four different dissociative solutions were investigated in this study TrypLE™ Express (Invitrogen), Trypsin‐EDTA (Invitrogen), Cell Dissociation Solution (Sigma, Bornem, Belgium) and Accutase™ (Millipore) and a slow adaptation and fast adaptation protocols were applied. The slow protocol proceeds as follows: during the first three passages, the hESC were incubated with Collagenase IV solution (1 mg/ml) (Invitrogen) for 30–40 min. Detached colonies were collected and centrifuged for 3 min at 90 g. After removal of supernatants, colonies were resuspended in one of the four dissociative solutions, for 5–10 min at 37 °C. At this stage, it was visually confirmed that single cells were obtained. In case of observation of cell clumps or undetached colonies, incubation time was prolonged (up to 20 min) or a cell scraper was used in combination with trituration. Next, culture medium was added and suspensions were centrifuged at 185 g for 5 min. After removal of supernatants and addition of fresh culture medium, hESC were seeded on MEF plates. These first three passages are considered to be the adaptation period for the cells. After three passages, colonies were immediately treated with one of the dissociative solutions and cell mesh was used to ensure achievement of single cells. The fast protocol does not allow for an adaptation period, so colonies are immediately treated with one of the dissociative solutions, as described above. Manual mechanical passaging, which is routinely used for hESC culture, is described in the general open literature and will not be repeated here (6, 12).
Table 1.
Protocol overview
| Dissociative solution | Test protocol | Tools | |
|---|---|---|---|
| 1 | TrypLE™ Express | Slow protocol | Vigorous trituration |
| 2 | Trypsin‐EDTA | ||
| 3 | Cell Dissociation Solution | Occasional trituration | |
| 4 | Accutase™ | ||
| 5 | TrypLE™ Express | Fast protocol | Vigorous trituration |
| 6 | Trypsin‐EDTA | ||
| 7 | Cell Dissociation Solution | Occasional trituration | |
| 8 | Accutase™ | ||
| 9 | / | Manual mechanical passaging | Sharp glass cutting pipettes |
| 10 | Collagenase IV | Bead‐based passaging | Soda‐lime glass beads |
Bead‐based passaging is an enzymatic bulk passaging approach. hESC were incubated for 10 min with Collagenase IV solution (1 mg/ml; Invitrogen) and scraped from tissue culture flasks by application of a small amount of soda lime glass beads. Afterwards, cell suspensions were collected, centrifuged for 3 min at 90 g and plated on fresh feeder plates.
hESC embryoid body formation. Plates with fully grown hESC colonies were selected and cells were detached from the feeder layers by incubation with Collagenase IV solution (1 mg/ml) for 30–40 min. Colonies were pelleted by centrifugation for 3 min at 90 g, resuspended in embryoid body (EB) medium and transferred to shaker flasks. EB medium consisted of 80% DMEM‐F12 (Invitrogen) + 20% foetal bovine serum (Invitrogen) + 1 mm l‐glutamine (Invitrogen) + 0.1 mm NEAA (Invitrogen) and 0.1 mmβ‐mercapto‐ethanol (Invitrogen). EBs were formed at passage 15 of the experimental conditions and cultured for 28 days in these settings.
Evaluation, data collection and statistical analysis
hESC morphological evaluation and data collection. hESC colonies were evaluated and counted at every passage, using an inverted phase‐contrast light microscope (Olympus inverted Research System Microscope IX81) employing a gradual scoring system (28, 29). This system allows for division of all observed colonies into four groups (grade A to D) based on morphological appearance. Grade A colonies – more than 80% undifferentiated; these had uniform compact morphology and clear, sharp edges. Grade B colonies consisted of 50–80% undifferentiated cells; here, colony edges were less sharp and showed mild differentiation. Grade C colonies – more than 50% differentiation, and grade D colonies dead or detached. The morphological scoring system allowed us to observe differences in colony quality and level of differentiation present in each grade, an important factor when comparing different working protocols. This system provided clearer results compared to reporting only total number of colonies present on a plate or the number of single cells, at each passage. For each plate, an uncertainty degree (established after repeated counting experiments of the same plate) of five percent, was taken into account. This was used in uncertainty analysis based on ‘An introduction to error analysis’ (30) – to determine error bars in statistical analysis.
hESC data processing. The following strategy was applied for comparison of the data generated in the first passages of the cultures, obtained by the different protocols and solutions. All conditions in the slow protocol were started from one plate of fully grown hESC colonies, which was divided into equal quantities over the four different test solutions. For the fast protocol, we applied the same strategy, although each condition was started up from a single plate, which was divided into four at the beginning. Mean (μN) of the total amount of generated colonies for the different conditions was calculated and used as reference, to normalize amounts of colonies (N) for each condition. By normalizing the data (N* X), results for different conditions can be compared on a fair basis to determine which solution gave the best start‐up, in combination with distribution of colonies in the first passage.
X = TrypLE™ Express, Trypsin‐EDTA, Cell Dissociation Solution, Accutase™.
To eliminate random variations, results represent a pool of three independent experiments for each dissociation solution tested. Amount of generated colonies over different passages was calculated, based on observed number of total counted colonies on a single plate and applied split ratios of each passage. This number was then normalized by initial numbers of colonies, to eliminate experimental variation. Data are plotted in 3D bar graphs to visually represent expansion differences.
hESC colony measurements. Colonies, formed in the first passage, under the different experimental conditions, were measured to evaluate and compare the mean size of the obtained colonies. Digital images of randomly selected colonies of each experimental condition were recorded. Then, using Excellence® software of the Olympus microscope system (Olympus Inverted Research System Microscope IX81, Brightfield, Objective 4x), colonies were measured. Statistically significant differences were calculated using two‐sided Student’s t‐test (P = 0.01).
hESC statistical analysis. 3D bar graphs indicate cumulative expansion profiles of each experimental condition, logarithmically. To establish whether these observed expansion differences were significantly different, two‐sided Student’s t‐test (P = 0,01) was performed.
Cell characterization
Immunocytochemical analysis of hESC colonies. Human embryonic stem cell pluripotency was determined by immunostaining for SSEA‐4, TRA 1‐60, TRA 1‐81 (ES Cell Marker Sample kit by Chemicon, Brussels, Belgium) and with alkaline phosphatase staining (BCIP/NBT Liquid Substrate System by Sigma, Bornem, Belgium). For SSEA‐4, TRA 1‐60, TRA 1‐81, colonies were fixed in 1% paraformaldehyde for 10 min, washed with PBS and blocked in blocking serum (PBS/5% normal rabbit or goat serum/0,2% Tween) for 30 min. Primary antibodies used were mouse monoclonal anti‐SSEA‐4 (IgG 1/50), mouse monoclonal anti‐TRA 1‐60 (IgM1/50) and mouse monoclonal anti‐TRA 1‐81 (IgM 1/50) (Millipore). After 1h incubation, cells were washed in PBS and incubated in secondary antibody (FITC‐labelled rabbit anti‐mouse IgG or FITC‐labelled goat anti‐mouse IgM 1/80) for 1 h in the dark. After washing, cells were immersed in PBS and evaluated using fluorescence microscopy (Olympus Inverted Research System Microscope IX81, GFP filter, CellM software).
Real time RT‐PCR analysis of hESC colonies. The hESC colonies were detached with Collagenase IV solution (1 mg/ml) (Invitrogen), collected and centrifuged at 90 g, 3 min. After removal of supernatants, TRI Reagent was added to lyse the cells. RNA was isolated using chloroform and further purified with isopropanol and ethanol. It was then transcribed to cDNA using a Reverse Transcriptase Core Kit (Eurogentec, Ougrée, Belgium) on a Thermocycler (Applied Biosystems 2720 Thermocycler). Real time PCR was performed on ABI 7500 Fast Real Time PCR device with ABI Taqman probes (Applied Biosystems, Halle, Belgium) for the following genes: OCT4 (Hs00742896_s1), NANOG (Hs02387400_g1), HAND1 (Hs00231848_m1), COL2A1 (Hs01064869_m1), CD34 (Hs00990732_m1), RUNX2 (Hs00231692_m1), NESTIN (Hs00707120_s1), SOX9 (Hs00165814_m1), PAX6 (Hs01088112_m1), CERBERUS (Hs00193796_m1), AFP (Hs00173490_m1) and GATA4 (Hs00171403_m1). Standard culture H1 and VUB01 reference samples were used for evaluation and GAPDH was selected as endogenous control. Analysis of gene expression was performed using ABI software for gene expression analysis on ABI 7500 Fast Real Time device.
Karyotyping of colonies. Plates with fully grown hESC colonies were selected for G‐band karyotyping (p20). Cells were treated with KaryoMAX Colcemid solution (Invitrogen) and fixed in cold fixer solution (one part acetic acid:three parts methanol). Slide preparation and metaphase spreading were performed at the Department of Paediatrics and Medical Genetics of Ghent University. Metaphases were detected and analysed (minimum of 20 spreads) using Metafer 4 software (MetaSystems, Altlussheim, Germany) on a microscope (Zeiss Axio Imager M1) with high‐resolution, monochrome megapixel CCD camera.
Results
Transfer of hESC to the experimental protocols: first passage
Preceding this study, the hESC lines were passaged every 6–7 days, using the standard mechanical passaging technique. When applying dissociative solutions to the hESC, differences could be seen in the way the cells detached from the plates. TrypLE™ Express and Trypsin‐EDTA had a similar effect: all cells, including the MEF feeder cells, detached rapidly and floated as single cells in suspension. Collected cell solutions needed to be triturated quite vigorously, as remaining MEFs in solution had the tendency to clump together. Cell dissociation solution worked differently: when observing plates microscopically in real‐time after addition of Cell Dissociation Solution, the hESC could be seen detaching as single cells from the centre towards the exterior of colonies. Most differentiated hESC and MEF feeder cells remained on the tissue culture plate bottoms. Using this solution, incubation period sometimes needed to be prolonged if very large or dense colonies were present, and in some cases a cell scraper was used combined with trituration. Accutase™ detached hESC as single cells from plates, but more MEF feeder cells appeared to remain on plates compared to after use of TrypLE™ Express or Trypsin‐EDTA.
Figure 1a,b graphically shows normalized quantity of hESC colonies, generated in the first passage with the different protocols. The most favourable experimental conditions for both cell lines are 3 and 7. These two top conditions had their dissociative solution – Cell Dissociation Solution – in common, but with slow and fast protocols respectively. As the third method demonstrates generation of more colonies than the seventh method, there was indication that the slow protocol had an advantage over the fast protocol.
Figure 1.
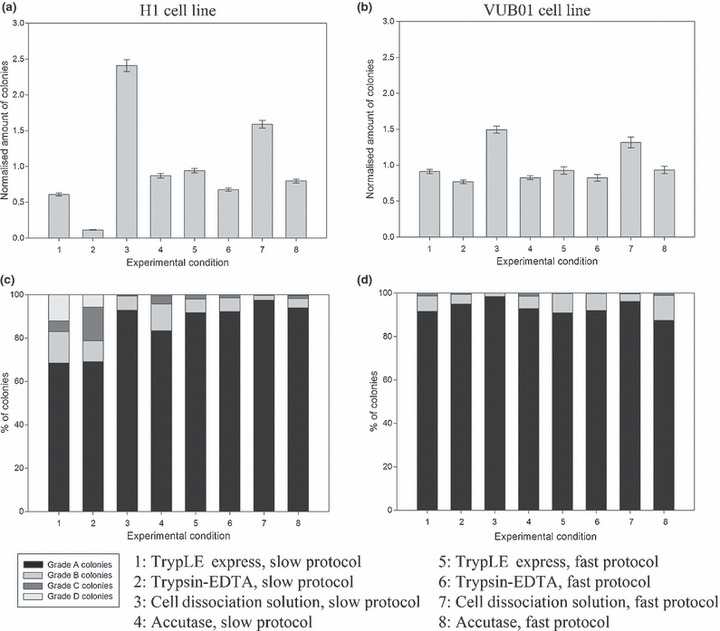
Graphical representation of normalized number of colonies and colony quality, generated in the first passage, for the different experimental conditions. The two best conditions, for both cell lines, were combination of Cell Dissociation Solution with the slow protocol and with the fast protocol. Use of Trypsin‐EDTA results in the formation of fewest colonies. TrypLE Express and Accutase™ generate intermediate amounts of colonies. (a) Bar chart of normalized number of generated colonies, under the different experimental conditions, in the first passage for the H1 cell line. (b) Bar chart of normalized amount of generated colonies, under the different experimental conditions, in the first passage for the VUB01 cell line. (c) Bar chart of colony quality, under the different experimental conditions, in the first passage for the H1 cell line. (d) Bar chart of colony quality, under the different experimental conditions, in the first passage for the VUB01 cell line.
Figure 1c,d depicts morphological quality of hESC colonies in first passages of the experimental conditions. Little difference can be seen in quality of the colonies across all conditions. Conditions 3 (Cell Dissociation Solution, slow protocol) and 7 (Cell Dissociation Solution, fast protocol) had highest quantities of grade A colonies, in both cell lines. H1 line showed more variation in colony gradation than VUB01, indicating that H1 was more sensitive to culture changes than VUB01.
In Fig. 2, start‐up images (passage 1, day 6) of H1 and VUB01 colonies under the different experimental protocols are presented for comparison. When conventional methods were used (conditions 9 and 10), slightly larger colonies were obtained. Using the slow protocol (conditions 1 to 4), less cell debris and differentiated cells were observed compared to the fast protocol (conditions 5 to 8). The largest and highest quantity of colonies was found in conditions 3 and 7, both generated with Cell Dissociation Solution. Colonies generated with TrypLE™ Express (1 and 5) or Trypsin‐EDTA (2 and 6) appeared to be much smaller. Those generated with Accutase™ (4 and 8) were of small to intermediate size.
Figure 2.

Phase contrast microscopy of H1 and VUB01 hESC colonies under the different experimental conditions. Morphologically, there is no difference visible in the first passage between the colonies formed by H1 cell line and VUB01 cell line, under the same experimental conditions. In general, colonies formed with TrypLE™ Express and Trypsin‐EDTA are the smallest. Those formed by Cell Dissociation Solution are comparable to the reference conditions and those formed with Accutase are of an intermediate size.
These visually observed size differences were confirmed by the measurement data of start‐up colonies, in Fig. 3. Here, graphical representation of size measurements can be found in the boxplots. Mean value of measured area (mm2) of colonies is noted above each bar. Data represented are calculated based on VUB01 cell line, but were similar for H1. In general, colonies obtained by application of the slow protocol (1–4) were somewhat larger than those obtained by application of the fast protocol (5–8) and use of TrypLE™ Express, Trypsin‐EDTA or Accutase™, regardless of applied protocol, lead to colonies significantly smaller than those of the reference conditions (P = 0.01). Use of Cell Dissociation Solution and slow protocol resulted in colonies which were (not significantly) different in size from the reference conditions. Application of Cell Dissociation Solution and the fast protocol generated colonies which were significantly different from those of the reference conditions. These differences in size could only be observed in first passages during adaptation of the cells to the dissociative solutions; thereafter (passage 4 and onwards), colonies were relatively comparable in size to each other and statistically significant differences disappeared.
Figure 3.
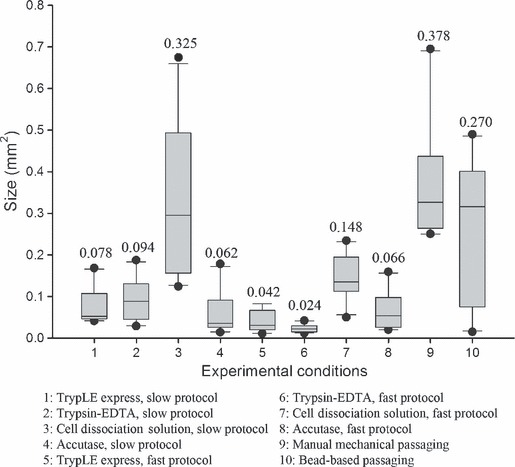
Measurement data of colonies under the different experimental conditions (passage 1) of VUB01 cell line. Measurement data confirm visual observations regarding colony size under the different experimental conditions. In general, colonies formed with the fast protocol are smaller than those with the slow protocol, in the first passage. Colonies formed with TrypLE™ Express and Trypsin‐EDTA are the smallest, regardless of which adaptation protocol was applied. Those formed with Cell Dissociation Solution are of comparable size with the reference conditions, especially when the slow adaptation protocol was applied. Application of Cell Dissociation Solution and the fast protocol, resulted in smaller colonies. Colonies formed with Accutase were of small to intermediate size.
Progression and expansion of the hESC cultures
As should be noted beforehand, not all experimental setups were successful. In some cases, very few colonies were generated in the first passage, subsequently obstructing further culture advancement. However, when large numbers of colonies were obtained in the first passage, progression beyond it was normally not a problem.
Cumulative expansion profile of H1 cell line under the different experimental conditions is plotted in Fig. 4. As can be observed, not all experiment conditions were successful for progression further than a few passages; this was generally due to overgrowth of differentiated cells. In some cases, however, hESC colonies simply stopped growing. As indicated by the data, use of TrypLE™ Express (1 and 4), Trypsin‐EDTA (2 and 6) and Accutase™ (4 and 8) did not result in stable continuous hESC culture progression; these conditions were all lost between three and five passages. Use of Cell Dissociation Solution (3 and 7) resulted in formation of stable expanding hESC lines and obtained cumulative expansion at passage 15 of conditions 3 (Cell Dissociation Solution, slow protocol) and 7 (Cell Dissociation Solution, fast protocol) were also higher than of the two reference conditions (9 and 10). In addition, use of the slow protocol (condition 3) in combination with this solution exhibited better results, not only from quantity perspective but also by quality. At passages 5 and 10 respectively, 92% and 98% Grade A colonies were counted. However, use of the fast protocol with Cell Dissociation Solution resulted in 87% of Grade A colonies at passage 5 and 90% at passage 10. Statistical analysis of the data revealed a significant difference (P = 0.01) in expansion between conditions 3 (Cell Dissociation Solution, slow protocol) and 9 (reference, manual mechanical), starting at passage 9. The same significant difference in expansion was observed between conditions 7 (Cell Dissociation Solution, fast protocol) and 9 (reference, manual mechanical), starting at passage 11.
Figure 4.
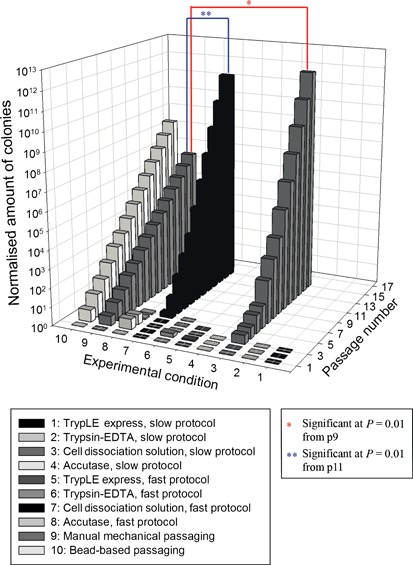
3D bar chart of cumulative expansion profile of H1 cell line under the different experimental conditions. The only two conditions that progressed further than 3 to 5 passages were combination of Cell Dissociation Solution with the slow protocol and the fast protocol.
Cumulative expansion profile of VUB01 line can be found in Fig. 5. All applied experimental conditions were successful with this cell line and resulted in continuously expanding hESC lines. Nevertheless, as clearly can be observed from the plot, conditions 3 and 7 were once more, the best option. Cumulative expansion at passage 15 was for both cell lines, slightly higher when Cell Dissociation Solution combined with the slow protocol was used compared to use of Cell Dissociation Solution with the fast protocol. In addition, as we also observed with the H1 cell line, use of the slow protocol resulted in better morphological profile of the colonies. Here, use of Cell Dissociation Solution combined with the slow protocol (condition 3) resulted in 96% at passage 5 and 98% at passage 10 of grade A colonies. Use of the fast protocol with Cell Dissociation Solution (condition 7) lead to 84% at passage 5 and 92% at passage 10 of grade A colonies. Statistical analysis of the data revealed highly significance difference (P = 0.01) in expansion between conditions 3 (Cell Dissociation Solution, slow protocol) and 9 (reference, manual mechanical), starting at passage 2. The same significant difference in expansion was observed between conditions 7 (Cell Dissociation Solution, fast protocol) and 9 (reference, manual mechanical), starting at passage 4.
Figure 5.
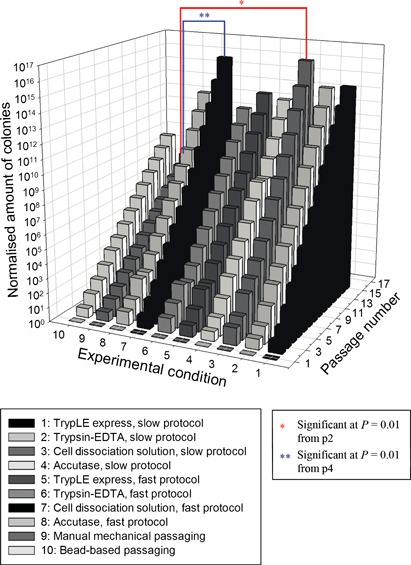
3D bar chart of cumulative expansion profile of VUB01 cell line under the different experimental conditions. All conditions progressed for more than 5 passages and were able to generate stable expanding single passaged hESC lines. However, looking at expansion level, use of Cell Dissociation Solution combined with the slow and fast protocol, were the two best conditions.
Pluripotency, differentiation capacity and karyotype
hESC colonies from our most favourable conditions, Cell Dissociation Solution combined with slow and fast protocols and manual mechanical passaged reference condition were immunocytochemically evaluated after 7 and 15 passages. Results were similar for the three tested conditions (Cell Dissociation Solution + slow and fast and manual mechanical reference). Positive results were obtained for SSEA‐4, TRA 1‐60, TRA 1‐81 and ALP. A representational image is presented in Fig. 6 (Cell Dissociation Solution, slow protocol).
Figure 6.
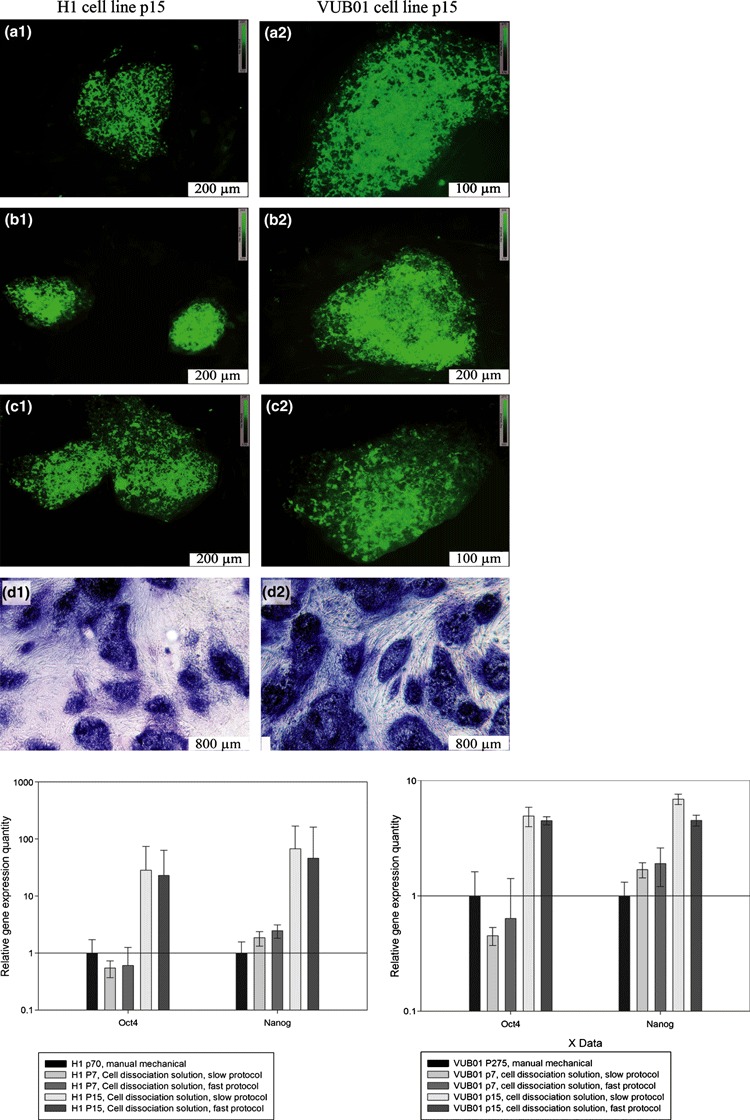
Pluripotency analysis of the hESC cultures. 1: H1 p15, cell dissociation solution, slow protocol A1: SSEA‐4 B1: TRA 1‐60 C1: TRA 1‐81 D1: ALP 2: VUB01 p15, cell dissociation solution, slow protocol A2: SSEA‐4 B2: TRA 1‐60 C2:: TRA 1‐81 D2:: ALP 3: Real‐time PCR data for OCT‐4 and Nanog.
OCT‐4 and NANOG expression were examined using real‐time relative quantitative gene expression PCR. Analysis of the data revealed a slight reduction in OCT‐4 expression at passage 7, compared to the standard mechanically passaged hESC. However, at passage 15, OCT‐4 expression had increased. NANOG expression showed a mild increase at passage 7 and a high increase at passage 15. These results are consistent for both cell lines and were independent of the applied protocol.
Embryoid bodies, formed in experimental conditions with Cell Dissociation Solution (3 and 7) and manually mechanical passaged reference condition (9), were also analysed by real‐time PCR, Table 2 summarizes the results. Embryoid body formation induced differentiation of hESC, resulting in reduced expression of OCT‐4 and NANOG, two pluripotency markers. Several differentiation markers were analysed. There was clear upregulation of ectodermal (Nestin, Pax6, Sox9), endodermal (Cerberus, AFP, GATA4) and mesodermal (Hand1, Col2A1, CD34, Runx2) markers, indicating that differentiation into all three germ layers had occurred. G‐band karyotyping of the experimental conditions using Cell Dissociation Solution (performed at passage 20) revealed no abnormalities of the hESC cultures compared to reference experimental condition 9, for both cell lines.
Table 2.
Overview of real‐time PCR EB analysis
| H1 cell line | ||||||
|---|---|---|---|---|---|---|
| hESC | EB (differentiation) | |||||
| Manual mechanical passaging | Cell Diss Sol Slow prot | Cell Diss Sol Fast prot | Manual mechanical passaging | Cell Diss Sol Slow prot | Cell Diss Sol Fast prot | |
| Cond 9 | Cond 3 | Cond 7 | Cond 9 | Cond 3 | Cond 7 | |
| Oct 4 | +++ | +++ | +++ | + | + | + |
| Nanog | +++ | +++ | +++ | + | + | + |
| Hand1 | +++ | +++ | +++ | |||
| Col2A1 | ++ | ++ | ++ | |||
| CD34 | +++ | +++ | ++ | |||
| Runx2 | ++ | ++ | ++ | |||
| Nestin | ++ | ++ | ++ | |||
| Sox9 | ++ | +++ | +++ | |||
| Pax6 | ++ | ++ | ++ | |||
| Cerberus | + | + | + | |||
| AFP | +++ | +++ | ++ | |||
| GATA4 | ++ | ++ | ++ | |||
| VUB01 cell line | ||||||
|---|---|---|---|---|---|---|
| hESC | EB (differentiation) | |||||
| Manual mechanical passaging | Cell Diss Sol Slow prot | Cell Diss Sol Fast prot | Manual mechanical passaging | Cell Diss Sol Slow prot | Cell Diss Sol Fast prot | |
| Cond 9 | Cond 3 | Cond 7 | Cond 9 | Cond 3 | Cond 7 | |
| Oct 4 | +++ | +++ | +++ | + | + | + |
| Nanog | +++ | +++ | +++ | + | + | + |
| Hand1 | +++ | ++ | ++ | |||
| Col2A1 | ++ | ++ | ++ | |||
| CD34 | ++ | ++ | ++ | |||
| Runx2 | ++ | ++ | ++ | |||
| Nestin | + | + | + | |||
| Sox9 | ++ | ++ | ++ | |||
| Pax6 | ++ | ++ | ++ | |||
| Cerberus | + | + | + | |||
| AFP | +++ | +++ | +++ | |||
| GATA4 | ++ | ++ | ++ | |||
Reproducibility testing
To verify reproducibility, three other hESC lines were tested for their ability to form stable expanding hESC lines when Cell Dissociation Solution and the slow protocol were applied. Results are presented in Fig. 7. All three hESC lines formed stable expanding hESC lines, using this protocol and had good expansive behaviour.
Figure 7.
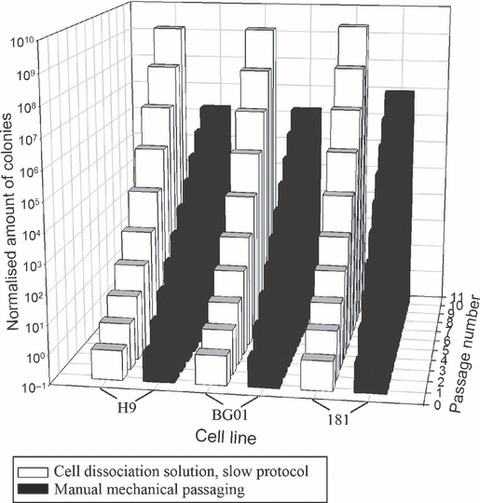
3D bar chart of cumulative expansion profile of H9, BG01 and 181 cell lines. The three cell lines were able to form stable expanding hESC lines when passaged with Cell Dissociation Solution and the slow protocol. They exhibited good expansion profiles.
Discussion
In the last few years, multiple studies have been published regarding alternative passaging methods and solutions for hESC culture. In this article, we have compared ten different passaging strategies for hESC culture, including the standard manual mechanical passaging technique and commonly used bead‐based passaging technique. As our focus is on large scale hESC expansion for future tissue engineering and cell therapy applications, we tried to identify the most suitable option for fast and robust hESC expansion.
Four different solutions were selected for the hESC passaging protocols, each having specific interesting characteristics. First, TrypLE™ Express, an optimized form of Trypsin‐EDTA, was examined. This enzymatic solution had already been successfully tested in hESC culture (15) and has the additional advantages that it does not contain any animal‐ or human‐derived components and that it is stable at room temperature. Secondly, the most commonly used enzymatic cell dissociating solution, Trypsin‐EDTA (16, 17, 18) could not be left out of the comparison. Although this solution is widely used for all types of cultures, its animal origin poses issues for future hESC tissue engineering applications. Thirdly, Cell Dissociation Solution was selected. This mild dissociating solution, also stable at room temperature, is non‐enzymatic and animal‐free. It has not been examined in combination with hESC. Lastly, Accutase™, an enzymatic cell dissociation solution that does not contain any animal‐ or bacteria‐derived proteins and is claimed to be less damaging to hESC than traditionally used Trypsin‐EDTA, was tested. This solution has previously been shown to support hESC culture (19, 20).
The solutions were applied in two different strategies to the hESC, to investigate influence and need for an adaptation period. A short adaptation period of three passages was introduced in the slow protocol. During this adaptation period, hESC colonies were detached from the tissue culture plates using Collagenase IV solution. As Collagenase IV, a very mild enzymatic agent, only detaches entire hESC colonies from the substrate, most MEF feeder cells remained on the plate. This has the additional advantage that the intermediate cell solution is more pure and less dense. In the fast protocol, direct addition of the dissociative solutions on the colonies was applied. Here, many MEFs detached and remained in the intermediate cell solution. These MEFs can reattach to substrate plates and thus increase MEF cell density, which is a crucial factor in maintenance of undifferentiated hESC (31). This implies that the fast protocol inherently carries a higher risk of shock to the hESC, inducing more cell death or differentiation. Results indicated that larger numbers of colonies could be generated in the first passage using the slow protocol compared to the fast protocol for both cell lines (Fig. 1a,b). A longer adaptation period was not tested. Keeping in mind our goal to obtain an easy, robust and economically feasible passaging method for large scale hESC expansion, this would have been counterproductive and our results indicate that longer adaptation is unnecessary as stable expanding hESC lines can be obtained with both the fast and slow protocols (4, 5).
Figure 1 provides an overview of hESC colony quality and quantity during the first passage of experimental conditions. Application of Trypsin‐EDTA resulted in lowest numbers of colonies of the four dissociative solutions tested. TrypLE™ Express and Accutase™ generated intermediate colony numbers. Ellerström et al. (15) also tested Trypsin‐EDTA and TrypLE™ Express as solutions for single‐cell dissociation and obtained similar results. Ellerström et al. observed that use of TrypLE™ Express generated three times more colonies than by use of Trypsin‐EDTA, when applied in their culture settings. In our experiment, such large differences were not observed, but clearly use of TrypLE™ Express allows generation of more hESC colonies. Bajpai et al. compared Trypsin‐EDTA and Accutase™ (19) and again, Trypsin‐EDTA came out as one of the poorest solutions for hESC single‐cell enzymatic passaging.
In Fig. 3, size of hESC colonies under the different experimental conditions can be seen; these findings concur with the observed morphological data. Colony size, generated under almost every experimental condition, was significantly different (P = 0.01) from those of the reference conditions; this means that generated colonies were much smaller than their reference counterparts. After several passages of adaptation (three to four passages), statistically significant differences disappeared and colonies were comparable in size. One exception was condition 3, where Cell Dissociation Solution was used combined with the slow protocol. Size of these colonies was comparable to those of the two reference conditions, from the start of culture.
Cumulative expansion profiles of hESC can be found in 4, 5. Remarkably, not all experimental conditions lead to development of stable expanding hESC lines. Adaptation of H1 to the different conditions appeared to be much more difficult than VUB01. Unexpectedly, no positive outcome could be established with TrypLE™ Express, Trypsin‐EDTA and Accutase™ for the H1 cell line. Only use of Cell Dissociation Solution, a solution previously untested on hESC cultures, resulted in a positive outcome. This was not observed for the VUB01 cell line. We suggest that this might have been caused because the VUB01 cell line had been kept in culture for over 260 passages, and was subsequently well adapted to in vitro cell culture and culture changes. It is also known that there are inherent differences between hESC cell lines, which could also account for these different findings (25).
The two best experimental conditions for both cell lines, conditions 3 and 7, were generated with Cell Dissociation Solution. After several passages, split ratios of 1/20 to 1/40 could easily be applied, without risk of culture loss or decrease in colony quality (>90% Grade A colonies). As a comparison, standard manual mechanically passaged cultures of H1 and VUB01 cell lines have routine split ratios of 1/3 to 1/6 and overall medium high quality of generated colonies (>75% undifferentiated). As can be seen, the proposed single‐cell protocol allows for much faster expansion and accumulation of large quantities of pluripotent hESC for tissue engineering experiments or others. Two sided t‐test revealed a significant difference between conditions 3 and 9 at passage 9 for H1 cell line, and at passage 11 between conditions 7 and 9. Similarly, a significant difference between conditions 3 and 9 for VUB01 cell line was established at passage 2, and between conditions 7 and 9 at passage 4. Predictably, the significant difference was obtained much faster with VUB01 cell line, which adapted easier, compared to H1. The significant difference was also reached two passages earlier with the slow protocol than with the fast protocol, indicating benefit of the short adaptation period. However, adaptation period in the slow protocol can be seen as laborious and unnecessary, as it was possible to obtain stable expanding hESC lines without the adaptation period (by use of the fast protocol). However, as we focussed on obtaining the highest expansion in the shortest period of time, going through the application of the more laborious slow protocol was worthwhile. At passage 10, this resulted in 4‐fold increase in colony number for H1 cell line and 14‐fold increase for VUB01.
We suggest that the observed results could further be improved by use of Rho‐associated kinase (ROCK) inhibitor Y‐27632 during passaging. This inhibitor has beneficial effects on survival of hESC during single‐cell passaging (32, 33). We chose not to add this solution in our study, to investigate basic capacities of each dissociative solution with regard to hESC passaging. We were able to establish stable expanding hESC cell lines without addition of ROCK‐inhibitor, although this was not always the case, and it could explain why it was not possible for us to generate H1 hESC lines with TrypLE™ Express of Accutase™. In addition, our setup was to generate an economically feasible expansion approach. It should be carefully investigated whether benefits of addition of ROCK inhibitor would outweigh the costs.
Immunocytochemical analysis of SSEA‐4, TRA 1‐60, TRA 1‐81 and ALP, and real‐time reverse transcriptase PCR for Oct4 and Nanog (Fig. 5) confirmed the observed undifferentiated status of the colonies. Colonies also retained their differentiation capacity, as indicated by the EB analysis (Table 2) and normal karyotyping.
Our most favourable condition (Cell Dissociation Solution, slow protocol) has been investigated in combination with three other cell lines, H9, BG01 and 181 hESCs. Again, positive results were obtained and stable expanding hESC cell lines were formed. Thus, we conclude that use of Cell Dissociation Solution in combination with a slow adaptation protocol is the best way to generate fast, stable expanding and robust hESC lines. After the adaptation period, large numbers of colonies were obtained with superior morphological characteristics and high split ratios (1/20 to 1/40) could easily be applied. No additional selection procedures were needed to maintain pluripotency, and karyotypic stability remained. This protocol is useful for obtaining large quantities of pluripotent hESC for tissue engineering experiments.
Acknowledgements
This study was conducted as part of a PhD project, funded by BOF (BOF08/24J/063). We express our gratitude to Prof. Dr. Karen Sermon and Dr. Ileana Mateizel for donating the VUB01 line to our research group and for sharing their technical expertise regarding hESC culture. We also thank Nelly François, Johanna Aernoudt and Leen Pieters, lab assistants, for their practical aid in realizing this work and the Department of Paediatrics and Medical Genetics (Prof Franki Speleman, Ghent University) for the assistance with the G‐band karyotyping.
References
- 1. Thomson JA, Itskovitz‐Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS et al. (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- 2. Mountford JC (2008) Human embryonic stem cells: origins, characteristics and potential for regenerative therapy. Transfus. Med. 18, 1–12. [DOI] [PubMed] [Google Scholar]
- 3. Battey JF (2007) Stem cells: current challenges and future promise. Dev. Dyn. 236, 3193–3198. [DOI] [PubMed] [Google Scholar]
- 4. Doss MX, Koehler CI, Gissel C, Hescheler J, Sachinidis A (2004) Embryonic stem cells: a promising tool for cell replacement therapy. J. Cell Mol. Med. 8, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amit M, Margulets V, Segev H, Shariki K, Laevsky I, Coleman R et al. (2003) Human feeder layers for human embryonic stem cells. Biol. Reprod. 68, 2150–2156. [DOI] [PubMed] [Google Scholar]
- 6. Skottman H, Hovatta O (2006) Culture conditions for human embryonic stem cells. Reproduction 132, 691–698. [DOI] [PubMed] [Google Scholar]
- 7. Zhou HS, Yong J, Sun XM, Wang CY, Yang WF, Zhang PB et al. (2008) A human endothelial cell feeder system that efficiently supports the undifferentiated growth of mouse embryonic stem cells. Differentiation 76, 923–930. [DOI] [PubMed] [Google Scholar]
- 8. Amit M, Itskovitz‐Eldor J (2006) Feeder‐free culture of human embryonic stem cells. Methods in enzymology 40, 37–49. [DOI] [PubMed] [Google Scholar]
- 9. Bigdeli N, Andersson M, Strehl R, Emanuelsson K, Kilmare E, Hyllner J et al. (2008) Adaptation of human embryonic stem cells to feeder‐free and matrix‐free culture conditions directly on plastic surfaces. J. Biotechnol. 133, 146–153. [DOI] [PubMed] [Google Scholar]
- 10. Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA (2006) Feeder‐independent culture of human embryonic stem cells. Nat. Methods 3, 637–646. [DOI] [PubMed] [Google Scholar]
- 11. Martin MJ, Muotri A, Gage F, Varki A (2005) Human embryonic stem cells express an immunogenic nonhuman sialic acid. Nat. Med. 11, 228–232. [DOI] [PubMed] [Google Scholar]
- 12. Inzunza J, Gertow K, Stromberg MA, Matilainen E, Blennow E, Skottman H et al. (2005) Derivation of human embryonic stem cell lines in serum replacement medium using postnatal human fibroblasts as feeder cells. Stem Cells 23, 544–549. [DOI] [PubMed] [Google Scholar]
- 13. Pyle AD, Lock LF, Donovan PJ (2006) Neurotrophins mediate human embryonic stem cell survival. Nat. Biotechnol. 24, 344–350. [DOI] [PubMed] [Google Scholar]
- 14. Suemori H, Yasuchika K, Hasegawa K, Fujioka T, Tsuneyoshi N, Nakatsuji N (2006) Efficient establishment of human embryonic stem cell lines and long‐term maintenance with stable karyotype by enzymatic bulk passage. Biochem. Biophys. Res. Commun. 345, 926–932. [DOI] [PubMed] [Google Scholar]
- 15. Ellerstrom C, Strehl R, Noaksson K, Hyllner J, Semb H (2007) Facilitated expansion of human embryonic stem cells by single‐cell enzymatic dissociation. Stem Cells 25, 1690–1696. [DOI] [PubMed] [Google Scholar]
- 16. Chan EM, Yates F, Boyer LF, Schlaeger TM, Daley GQ (2008) Enhanced plating efficiency of trypsin‐adapted human embryonic stem cells is reversible and independent of trisomy 12/17. Cloning Stem Cells 10, 107–117. [DOI] [PubMed] [Google Scholar]
- 17. Hasegawa K, Fujioka T, Nakamura Y, Nakatsuji N, Suemori H (2006) A method for the selection of human embryonic stem cell sublines with high replating efficiency after single‐cell dissociation. Stem Cells 24, 2649–2660. [DOI] [PubMed] [Google Scholar]
- 18. Thomson A, Wojtacha D, Hewitt Z, Priddle H, Sottile V, Di Domenico A et al. (2008) Human embryonic stem cells passaged using enzymatic methods retain a normal karyotype and express CD30. Cloning Stem Cells 10, 89–105. [DOI] [PubMed] [Google Scholar]
- 19. Bajpai R, Lesperance J, Kim M, Terskikh AV (2008) Efficient propagation of single cells accutase‐dissociated human embryonic stem cells. Mol. Reprod. Dev. 75, 818–827. [DOI] [PubMed] [Google Scholar]
- 20. Li XY, Krawetz R, Liu SY, Meng GL, Rancourt DE (2009) ROCK inhibitor improves survival of cryopreserved serum/feeder‐free single human embryonic stem cells. Hum. Reprod. 24, 580–589. [DOI] [PubMed] [Google Scholar]
- 21. Spits C, Mateizel I, Geens M, Mertzanidou A, Staessen C, Vandeskelde Y et al. (2008) Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 26, 1361–1363. [DOI] [PubMed] [Google Scholar]
- 22. Durkop H, Foss HD, Eitelbach F, Anagnostopoulos I, Latza U, Pileri S et al. (2000) Expression of the CD30 antigen in non‐lymphoid tissues and cells. J. Pathol. 190, 613–618. [DOI] [PubMed] [Google Scholar]
- 23. Pera MF, Bennett W, Cerretti DP (1997) Expression of CD30 and CD30 ligand in cultured cell lines from human germ‐cell tumors. Lab. Invest. 76, 497–504. [PubMed] [Google Scholar]
- 24. Mateizel I, Spits C, Verloes A, Mertzanidou A, Liebaers I, Sermon K (2009) Characterization of CD30 expression in human embryonic stem cell lines cultured in serum‐free media and passaged mechanically. Hum. Reprod. 24, 2477–2489. [DOI] [PubMed] [Google Scholar]
- 25. Allegrucci C, Young LE (2007) Differences between human embryonic stem cell lines. Hum. Reprod. Update 13, 103–120. [DOI] [PubMed] [Google Scholar]
- 26. Tavakoli T, Xu X, Derby E, Serebryakova Y, Reid Y, Rao MS et al. (2009) Self‐renewal and differentiation capabilities are variable between human embryonic stem cell lines I3, I6 and BG01V. BMC Cell Biol. 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mateizel I, De Temmerman N, Ullmann U, Cauffman G, Sermon K, Van de Velde H et al. (2006) Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum. Reprod. 21, 503–511. [DOI] [PubMed] [Google Scholar]
- 28. Li T, Zhou CQ, Liu CX, Mai QY, Zhuang GL (2008) Bulk vitrification of human embryonic stem cells. Hum. Reprod. 23, 358–364. [DOI] [PubMed] [Google Scholar]
- 29. Richards M, Fong CY, Tan S, Chan WK, Bongso A (2004) An efficient and safe xeno‐free cryopreservation method for the storage of human embryonic stem cells. Stem Cells 22, 779–789. [DOI] [PubMed] [Google Scholar]
- 30. Taylor JR (1997) An Introduction to Error Analysis. 2nd edition, Sausalito (CA, USA): University Science Books. [Google Scholar]
- 31. Zhou D, Liu T, Zhou X, Lu G (2009) Three key variables involved in feeder preparation for the maintenance of human embryonic stem cells. Cell Biol. Int. 33, 796–800. [DOI] [PubMed] [Google Scholar]
- 32. Gauthaman K, Fong CY, Bongso A (2010) Effect of ROCK inhibitor Y‐27632 on normal and variant human embryonic stem cells (hESCs) in vitro: its benefits in hESC expansion. Stem Cell Rev. 6, 86–95. [DOI] [PubMed] [Google Scholar]
- 33. Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T et al. (2007) A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686. [DOI] [PubMed] [Google Scholar]