

Abstract
Objectives
Mesenchymal stem cells (MSCs) are a reliable resource for tissue regeneration, but their molecular mechanisms of differentiation and proliferation remain unclear; this situation has restricted use of MSCs to a limited number of applications. A previous study of ours found a member of the epidermal growth factor family, epiregulin (EREG), to be involved in regulation of MSC differentiation. In the present study, we have used human dental stem cells from the apical papilla (SCAPs) to investigate the role of EREG on proliferation of MSCs.
Materials and methods
SCAPs were isolated from apical papillae of immature third molars. Retroviral short hairpin RNA (shRNA) was used to silence EREG gene expression, and human recombinant EREG protein was used to stimulate SCAPs. SCAP proliferation was examined using tetrazolium dye colorimetric assay/cell growth curve. Western blotting was performed to detect expressions of extracellular signal‐regulated protein kinases 1 and 2 (Erk1/2), mitogen‐activated protein kinases 1 and 2 (MEK1/2), protein kinase B (Akt), p38 mitogen‐activated protein kinase (p38 MAPK) and c‐Jun N‐terminal kinase (JNK).
Results
Depletion of EREG with shRNA inhibited SCAP proliferation and repressed phosphorylation of Erk1/2 and JNK. Human recombinant EREG protein promoted cell proliferation and enhanced Erk1/2, MEK and JNK phosphorylation in SCAPs. Furthermore, blocking MEK/Erk signalling with specific Erk1/2 inhibitor PD98059, or JNK signalling with specific inhibitor SP600125, abolished effects of EREG on cell proliferation.
Conclusion
These findings indicate that EREG could enhance cell proliferation in dental tissue‐derived MSCs by activating MEK/Erk and JNK signalling pathways.
Introduction
Mesenchymal stem cells (MSCs) were originally isolated from bone marrow; they are multipotent and able to differentiate into a variety of cell types, including osteoblasts, chondrocytes, myocytes and adipocytes. Increasing evidence indicates that MSCs are also present in non‐bone marrow tissues 1, 2. Recently, a new population of MSCs has been isolated from dental and craniofacial tissues (on the basis of their stem‐cell properties), including from the periodontal ligament (PDLSCs), from dental pulp (DPSCs), from apical papilla (SCAPs) and more 3, 4, 5, 6, 7, 8. Although these MSCs derived from dental tissues were of variable origin, pericyte or non‐pericyte origin, they are multipotent, destined for osteo/dentinogenic lineages and further endpoints such as melanocytes, endothelial cells and functionally active neurons; they are capable of self‐renewal 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13. When transplanted into mice, rats, swine or humans, these MSCs generated bone/dentin‐like mineralized tissue and were capable of repairing tooth and mandible defects 7, 8, 14, 15, 16, 17. Although MSCs represent a reliable resource for tissue regeneration, due to only low numbers achieved on harvesting, they need to be further expanded in vitro without biasing future differentiation for optimal utility. This presents a challenge as their molecular mechanisms of differentiation and proliferation remain unclear; thus, use of MSCs has been restricted to a limited number of applications. In addition, in vitro MSC characteristics (including growth, proliferation and viability) might associate with their function in vivo for therapeutic use 18. Thus, elucidation of molecular mechanisms of MSCs involved in growth, proliferation and viability will provide useful information for their therapeutic use.
Previous studies have indicated that epidermal growth factor (EGF) has the potential for enhancing proliferation and/or differentiation of MSCs 19, 20, 21, 22. Soluble EGF has been shown to augment MSC proliferation, but it has preserved early progenitors within the MSCs population, thus didn't induce differentiation; however, a tethered form of EGF has supported osteogenic differentiation 21, 22. One member of the EGF family, epiregulin (EREG), can activate extracellular signal‐regulated protein kinase, mitogen‐activated protein kinase (Erk/MAPK), and protein kinase B (Akt) signalling pathways in biological processes. EREG also acts as a major autocrine/paracrine factor released from Erk and p38 mitogen‐activated protein kinase (p38 MAPK) activated vascular smooth muscle cells, for cell dedifferentiation 23, 24, 25, 26, 27, 28. In addition, epiregulin stimulates cell proliferation through autophosphorylation of the EGF receptor (EGFR) or cross‐induction with other EGF family members 29, 30. A previous study of ours compared gene expressions of SCAPs from healthy individuals and patients with oculo‐facio‐cardio‐dental (OFCD) syndrome by microarray analysis, and found that EREG was highly expressed in SCAPs from OFCD syndrome that had a mutation in BCL6 corepressor (BCOR) 31. BCOR associates with histone demethylase FBXL11 to form a protein complex, this complex directly repressing transcription of EREG target gene by reducing promoter H3K4 and H3K36 methylation implicated in regulation of MSC differentiation 32. However, up to now it has been unknown whether EREG might regulate MSC proliferation.
In this study, we used MSCs derived from dental apical papillae to investigate the function of EREG in their proliferation. Our results showed that EREG enhanced SCAP proliferation by activating MEK/Erk and JNK signalling pathways.
Materials and methods
Cell cultures
All our research involving human stem cells complied with the ISSCR ‘Guidelines for the Conduct of Human Embryonic Stem Cell Research’. Wharton's jelly of umbilical cord stem cells (WJCMSCs) was purchased from Cyagen Biosciences (Guangzhou, China). Five human impacted third molars, with immature roots, were collected from five healthy patients (16–20 years old) under approved guidelines set by Beijing Stomatological Hospital, Capital Medical University (Ethical Committee Agreement, Beijing Stomatological Hospital Ethics Review No. 2011‐02), with informed patient consent. First the wisdom teeth were disinfected in 75% ethanol then were washed in phosphate‐buffered saline (PBS). Both SCAPs and PDLSCs were isolated, cultured and identified, as previously described 3, 6. Briefly, SCAPs were gently separated from the apical papilla of the root; PDLSCs were separated from periodontal ligament in middle one‐third of the root, then digested in a solution of 3 mg/ml collagenase type I (Worthington Biochem, Lakewood, NJ, USA) and 4 mg/ml dispase (Roche, Indianapolis, IN, USA) for 1 h at 37 °C. Single‐cell suspensions were obtained by passing the cells through a 70‐μm strainer (Falcon, BD Labware, San Jose, CA, USA). MSCs were grown in humidified, 5% CO2 atmosphere at 37 °C, in DMEM alpha‐modified Eagle's medium (Invitrogen, Carlsbad, CA, USA), supplemented with 15% foetal bovine serum (FBS; Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen); culture medium was changed every 3 days. MSCs at passages 3–5 were used in subsequent experiments. For human recombinant EREG protein (Abcam, Cambridge, MA, USA), PD98059 (Cell Signaling Technology, Beverly, MA, USA), SP600125 (Merck, Darmstadt, Germany) or Gefitinib (Selleck, Houston, TX, USA) treatment, MSCs were starved for 24 h to synchronize the cells, in DMEM alpha‐modified Eagle's medium without serum, then changed to routine culture medium and treated with the appropriate agents.
Characterization of SCAPs
The human SCAPs were characterized by flow cytometry using fluorescein isothiocyanate‐conjugated or phythoerythrin‐conjugated antibodies. SCAP cultures were harvested using 0.25% trypsin, and cell aliquots (1.0 × 106 cells) were incubated for 1 h at room temperature with monoclonal antibodies specific to CD146 (MCAM, melanoma cell adhesion molecule) (BD Biosciences, San Jose, CA, USA), CD90 (THY‐1, Thy‐1 cell surface antigen) (BD Biosciences), CD31 (PECAM1, platelet/endothelial cell adhesion molecule 1) (BD Biosciences) or HLA‐DR (human leucocyte differentiation antigen class II) (Biolegend, San Diego, CA, USA). Expression profiles were analysed by flow cytometry (Calibur, BD, Franklin Lakes, NJ, USA).
Alkaline phosphatase, alizarin red, oil red O and alcian blue staining analyses
SCAPs were grown in mineralization‐inducing medium using STEMPRO Osteogenesis Differentiation Kit (Invitrogen). For ALP staining, cells were cultured for 7 days, fixed in 4% paraformaldehyde and stained with a solution of 0.25% naphthol AS‐BI phosphate and 0.75% fast red FRV of an ALP staining kit according to the manufacturer's protocol (Sigma‐Aldrich, St. Louis, MO, USA). For detecting mineralization, cells were induced for 2 weeks, fixed in 70% ethanol and stained with 2% alizarin red (Sigma‐Aldrich).
Adipogenic differentiation was induced by using the STEMPRO Adipogenesis Differentiation Kit (Invitrogen). SCAPs were grown in adipose‐inducing medium for 3 weeks. For oil red O staining, cells were fixed in 10% formalin for at least 1 h at room temperature. Next, they were stained in 60% oil red O in isopropanol (as working solution) for 10 min, visualized by light microscopy and images were captured for analysis.
Chondrogenic differentiation was induced using the STEMPRO Chondrogenesis Differentiation Kit (Invitrogen). SCAPs were grown in chondrogenic medium for 2 weeks. For alcian blue staining, cells were rinsed once in PBS and fixed in 4% formaldehyde solution for 30 min. After fixation, wells were rinsed with PBS and cells were stained with 1% alcian blue solution prepared in 0.1 N HCl, for 30 min. Then wells were rinsed three times with 0.1 N HCl and distilled water was added to neutralize the acidity. Blue staining indicated synthesis of proteoglycans by chondrocytes.
Plasmid construction and viral infection
Plasmids were constructed using standard methods; all structures were verified by appropriate restriction digestion and/or sequencing. Short hairpin RNAs (shRNA) with complementary sequences of the target genes were subcloned into pSIREN retroviral vector (BD Clonteh, Mountain View, CA, USA). For viral infections, MSCs were plated overnight, then infected with retroviruses in the presence of polybrene (6 μg/ml; Sigma‐Aldrich) for 6 h. After 48 h, infected cells were selected with 2 μg/ml puromycin for 7 days. Luciferase and EREG were target genes for silencing with shRNAs; thus, complementary sequences were: Luciferase shRNA, 5′‐gtgcgttgctagtaccaac‐3′; and EREG shRNA, 5′‐actactgcaggtgtgaagt‐3′.
MTT assay
Cell proliferation was examined using the MTT (3‐[4, 5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl tetrazolium bromide) assay; this method is dependent on mitochondrial succinate dehydrogenase, in proliferating cells. MTT is reduced to an insoluble purple formazan reaction product which can be determined quantitatively by colorimetric assay. In brief, SCAPs were cultured in 96‐well plates (Costar, Cambridge, MA) at 1 × 104 cells/well initial density. After the indicated culture period, cells were treated with 5 mg/ml of MTT reagent (Sigma‐Aldrich) and incubated at 37 °C for 4 h. Cells were washed twice in PBS, followed by treatment with dimethyl sulphoxide and absorbance at 490 nm was read using an automatic enzyme‐linked immunosorbent assay reader (ELx800; BioTek Instruments Inc. Winooski, VT, USA). MTT results are shown as mean ± SD and represent two experiments performed in triplicate.
Cell growth curve assays
SCAPs were seeded at 1.0 × 104 cells/plate in 60‐mm plates; they were counted 3, 5 and 7 days after seeding. They were then digested using 0.25% trypsin (Invitrogen), resuspended in 1 ml PBS and counted using an automated cell counter (TC10TM; Bio‐Rad Laboratories, Hercules, CA, USA). Equivalent trypan blue was added to the cell suspension to identify non‐viable cells for exclusion. Results represent mean values (±SEM) of three separate experiments.
Western blot analysis
Cells were lysed in RIPA buffer (10 mm Tris‐HCL, 1 mm EDTA, 1% sodium dodecyl sulphate [SDS], 1% NP‐40, 1:100 proteinase inhibitor cocktail, 50 mm β‐glycerophosphate, 50 mm sodium fluoride). Twenty‐five micrograms of total protein from each sample was loaded and samples were separated on 10% SDS polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membranes using semi‐dry transfer apparatus (Bio‐Rad). Membranes were blotted with 5% dehydrated milk for 2 h then incubated with primary antibodies overnight. Immune complexes were incubated with horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse IgG (Promega, Madison, WI, USA) and visualized with SuperSignal reagents (Pierce, Rockford, IL, USA). Primary antibodies used in this study were anti‐Akt (Clone No.11E7, Cat No.4685S; Cell Signaling Technology), anti‐phospho‐Akt (Cat No.9271S; Cell Signaling Technology), anti‐p44/42 MAPK (Erk1/2) (Clone No.137F5, Cat No.4695S; Cell Signaling Technology), anti‐phospho‐p44/42 MAPK (Erk1/2) (Clone No.197G2, Cat No.4377S; Cell Signaling Technology), anti‐MEK1/2 (Clone No.D1A5, Cat No.8727; Cell Signaling Technology), anti‐phospho‐MEK1/2 (Clone No.41G9, Cat No.9154; Cell Signaling Technology), anti‐p38 MAPK (Clone No.D13E1, Cat No.8690; Cell Signaling Technology), anti‐phospho‐p38 MAPK (Clone No.12F8, Cat No.4631; Cell Signaling Technology), anti‐JNK (Clone No.56G8, Cat No.9258; Cell Signaling Technology) and anti‐phospho‐JNK (Clone No.81E11, Cat No.4668; Cell Signaling Technology). We also used a primary monoclonal antibody to detect housekeeping protein, glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Clone No.GAPDH‐71.1, Cat No.G8795; Sigma‐Aldrich).
Reverse transcriptase‐polymerase chain reaction (RT‐PCR) and real‐time RT‐PCR
Total RNA was isolated from MSCs using Trizol reagent (Invitrogen). We synthesized cDNA from 2‐μg aliquots of RNA, oligo(dT) and reverse transcriptase, according to the manufacturer's protocol (Invitrogen). Real‐time PCR reactions were performed using QuantiTect SYBR Green PCR kit (Qiangen, Hilden, Germany) and an Icycler iQ Multi‐colour Real‐time PCR Detection System. Primers for EREG were: forward, 5′‐ttatgggaggctccttcatc‐3′; reverse, 5′‐gccttcgtttaccctagcac‐3′; primers for GAPDH were: forward, 5′‐cggaccaatacgaccaaatccg‐3′; reverse, 5′‐agccacatcgctcagacacc‐3′.
Transplantation into nude mice
Approximately 4.0 × 106 cells were mixed with 40 mg of hydroxyapatite/tricalcium phosphate (HA/TCP) ceramic particles (Engineering Research Center for Biomaterials, Sichuan University, China), then transplanted dorsally subcutaneously into 10‐week‐old immunocompromised beige mice (nu/nu nude mice). These procedures were performed in accordance with specifications of an approved animal protocol. Eight weeks after transplantation, transplanted cells were harvested, fixed in 10% formalin, decalcified with buffered 10% EDTA (pH 8.0) then embedded in paraffin wax. Sections were cut then deparaffinized, hydrated, and stained with haematoxylin and eosin (H&E).
Statistical analysis
All statistical calculations were performed using spss10 statistical software. Student's t‐test or one‐way ANOVA were performed to determine statistical significance; P‐value <0.05 was considered significant.
Results
SCAPs exhibited characteristics of MSCs with multi‐differentiation potential
Using flow cytometry, cell surface marker analysis of SCAPs showed they expressed CD146 and CD90 (Fig. S1a,b); they did not express CD31 or HLA‐DR (Fig. S1c,d).
Next, osteo/dentinogenic differentiation potentials of SCAPs were investigated by culturing them in osteogenic‐inducing medium. Alkaline phosphatase activity, an early marker of osteo/dentinogenic differentiation, was induced (Fig. S2a). Two weeks after culturing SCAPs thus, alizarin red staining revealed mineralization to be significantly induced (Fig. S2b). We then investigated chondrogenic differentiation potentials of the cells. After induction with chondrogenic medium for 2 weeks, alcian blue staining revealed proteoglycans production (Fig. S2c). We also examined adipogenic differentiation potentials of SCAPs. After induction in adipogenic medium for 3 weeks, oil red O staining revealed lipid deposits in the cells (Fig. S2d).
EREG promoted MSC proliferation
First, we treated our SCAPs with 0, 25, 50 or 100 ng/ml human recombinant EREG protein for 24 h. Results of MTT assay showed that OD values were significantly higher with 50 or 100 ng/ml EREG treatment than without it or with 25 ng/ml EREG treatment; there were no differences in results between 50 and 100 ng/ml EREG treatments (Fig. 1a). Next, we added 0, 25 or 50 ng/ml human recombinant EREG protein to SCAPs for 48 h; MTT assay was used to detect cell numbers every 24 h. Results showed that OD values were significantly hgher with 25 or 50 ng/ml EREG compared to no EREG, but there were no differences between 25 ng/ml and 50 ng/ml EREG treatments at 48 h (Fig. 1b). Cell growth curve assay also confirmed that 25 ng/ml human recombinant EREG enhanced SCAP proliferation (Fig. 1c). We had designed shRNA to suppress EREG when it was introduced into the cells by retroviral infection. After selection, knockdown efficiency (80%) was verified by real‐time RT‐PCR (Fig. 2a). Then, we examined whether suppression of endogenous EREG affected SCAP growth and the cell growth curve assay confirmed that knockdown of EREG repressed SCAP proliferation (Fig. 2b).
Figure 1.
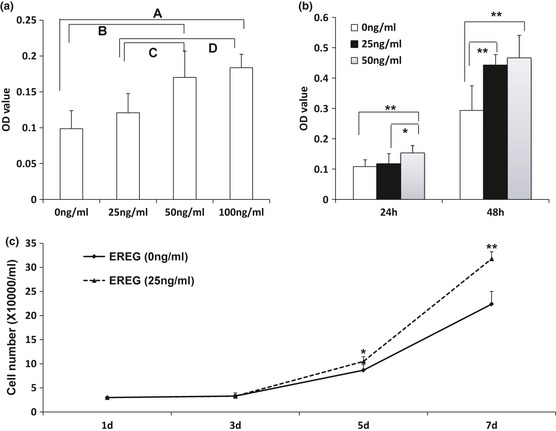
Recombinant human EREG protein promotion of SCAP proliferation. (a) MTT assay showed increased OD value after 24 h with 50 or 100 ng/ml recombinant human EREG protein. SCAPs were treated without or with 25, 50 or 100 ng/ml human recombinant EREG protein for 24 h. Results represent mean ± SD from six independent experiments. One‐way ANOVA was performed to determine statistical significance. (A) 100 ng/ml group >0 ng/ml group, P < 0.01; (B) 50 ng/ml group >0 ng/ml group, P < 0.01; (C) 50 ng/ml group >25 ng/ml group, P < 0.05; (D) 100 ng/ml group >25 ng/ml group, P < 0.05. (b) MTT assay showed increased OD value after 24 or 48 h with 50 ng/ml or 25 ng/ml recombinant human EREG protein. SCAPs were treated without or with 25 or 50 ng/ml human recombinant EREG protein for 24 or 48 h. One‐way ANOVA was performed to determine statistical significance. (c) Cell growth curves indicate that 25 ng/ml recombinant human EREG protein enhanced cell population growth. Results represent mean ± SD from three independent experiments. Student's t‐test was performed to determine statistical significance. All error bars represent SD *P < 0.05, **P < 0.01.
Figure 2.
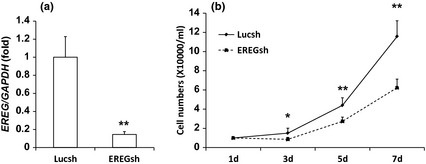
Depletion of EREG inhibited SCAP proliferation. (a) SCAPs were infected with retroviruses expressing shRNA against EREG (EREGsh) or Luciferase (Lucsh, controls). After treatment with 2 μg/ml puromycin for 7 days, EREG expression was determined by real‐time RT‐PCR; GAPDH was used as internal control. EREG was knocked down 80% by EREGsh compared to Lucsh‐infected SCAPs. Results represent mean ± SD from three independent experiments. (b) Cell growth curves showed that depletion of EREG in SCAPs inhibited cell population growth. Cell numbers were counted every 2 days for 1 week. Results represent mean ± SD from three independent experiments. Student's t‐test was performed to determine statistical significance. All error bars represent SD *P < 0.05, **P < 0.01.
To determine whether EREG had similar functions in other MSCs, WJCMSCs and PDLSCs were treated with 25 ng/ml EREG for 48 h. MTT results indicated that OD values were significantly higher after being treated with 25 ng/ml EREG compared to the no EREG group (Fig. S3). There was no abnormality in karyotype of SCAPs (Fig. S4), and no tumour formation when mixed with HA/TCP scaffold and transplanted into nude mice (Fig. S5a). Furthermore, there were no changes in surface markers of the SCAPs including of CD146, CD90, CD31 and HLA‐DR (Fig. S6).
EREG increased Erk1/2, JNK and MEK1/2 phosphorylation in SCAPs
We then investigated how EREG enhanced SCAP proliferation. We detected protein levels of Erk1/2, Akt, JNK, p38 MAPK and MEK1/2 by western blotting and results showed that knockdown of EREG inhibited phosphorylation of Erk1/2 and JNK, but total amount of Erk1/2, Akt, JNK, p38 MAPK and MEK1/2 protein and amounts of phosphorylated Akt and p38 MAPK were not affected; no quantity of phosphorylated MEK1/2 was detected (Fig. 3a). Our SCAPs were then treated with 25 ng/ml of human recombinant EREG protein. Western blotting results indicated that recombinant EREG protein enhanced phosphorylation of Erk1/2 at 24 and 48 h, and level of phosphorylated JNK was higher at 48 h; level of phosphorylated MEK1/2 was higher at 24 h after treatment with 25 ng/ml human recombinant EREG protein. In contrast, no change was observed in total amount of Erk1/2, Akt, JNK, p38 MAPK and MEK1/2 protein and phosphorylated Akt and p38 MAPK (Fig. 3b).
Figure 3.
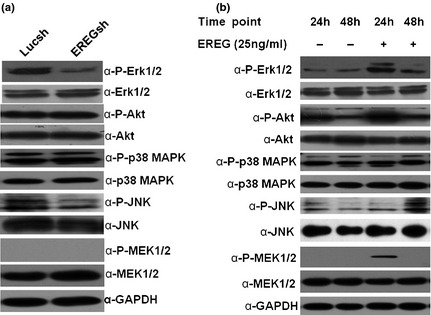
EREG activated MEK/Erk and JNK signallings. (a) Western blot analysis demonstrated that silencing EREG caused reduction in phosphorylated Erk1/2 (phospho‐Erk1/2) and phosphorylated JNK (phospho‐JNK) in SCAPs. (b) Level of phospho‐Erk1/2 was higher at 24 and 48 h after treatment with 25 ng/ml human recombinant EREG protein, and level of phospho‐JNK was higher at 48 h after treatment with 25 ng/ml human recombinant EREG protein; level of phosphorylated MEK1/2 (phospho‐MEK1/2) was higher at 24 h after treatment with 25 ng/ml human recombinant EREG protein. GAPDH served as an internal control.
EREG‐enhanced proliferation of SCAPs was repressed by MEK/Erk, JNK or EGFR inhibition
We used a specific Erk1/2 inhibitor, PD98059, to block MEK/Erk signalling in SCAPs. Western blot analysis indicated that 20 μm PD98059 blocked Erk1/2 signalling efficiently (Fig. 4a). We pre‐treated the cells with 20 μm PD98059 for 1 h to block MEK/Erk signalling, then added 25 ng/ml human recombinant EREG protein. MTT results revealed that MEK/Erk signalling was blocked by the inhibitor and exogenous EREG protein could not enhance SCAP proliferation (Fig. 4b). We then used a specific JNK inhibitor, SP600125, to block JNK signalling. Western blotting showed that 20 μm SP600125 blocked JNK signalling efficiently (Fig. 4c). We pre‐treated cells with 20 μm SP600125 for 48 h to block JNK signalling, then added 25 ng/ml recombinant EREG protein. MTT results showed that SP600125 could suppress EREG‐mediated enhancement of SCAP proliferation (Fig. 4d). We used EGFR special inhibitor, Gefitinib, to block EGFR and MTT assay demonstrated that 0.5 μm Gefitinib suppressed proliferation and EREG‐enhanced proliferation of the cells (Fig. 5).
Figure 4.
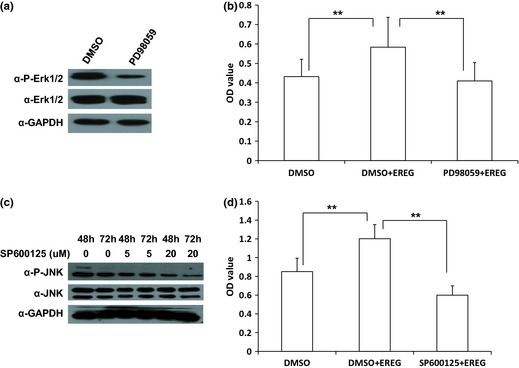
MEK/Erk or JNK inhibition repressed SCAP proliferation. (a) Western blotting showed reduction of phospho‐Erk1/2 in SCAP after being treated with Erk1/2 inhibitor, PD98059 (20 μm in DMSO) for 1 h; GAPDH served as internal control. (b) MTT assay revealed that PD98059 suppressed EREG‐mediated enhancement of cell proliferation. SCAPs were pre‐treated with 20 μm PD98059 or DMSO for 1 h, then cultured with 25 ng/ml EREG protein for 48 h. (c) Western blot results showed that JNK inhibitor, SP600125 (20 μm in DMSO), significantly inhibited phospho‐JNK in the cells after being treated for 48 and 72 h; GAPDH served as an internal control. (d) MTT assays showed that SP600125 suppressed EREG‐mediated enhancement of cell proliferation. SCAPs were pre‐treated with 20 μm SP600125 or DMSO for 48 h, then cultured with 25 ng/ml EREG protein for 48 h. Results represent mean ± SD from six independent experiments. One‐way ANOVA was performed to determine statistical significance. All error bars represent SD **P < 0.01.
Figure 5.
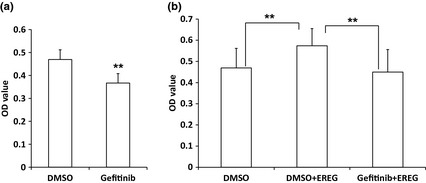
EGFR inhibition repressed SCAP proliferation. (a) MTT assays demonstrated that EGFR inhibitor, Gefitinib, suppressed proliferation of SCAPs. The cells were treated with 0.5 μm Gefitinib or DMSO for 48 h. (b) MTT assays showed that Gefitinib suppressed EREG‐mediated enhancement of cell proliferation. SCAPs were pre‐treated with 0.5 μm Gefitinib or DMSO for 48 h, then cultured with 25 ng/ml EREG protein for 48 h. Results represent mean ± SD from six independent experiments. One‐way ANOVA or Student's t‐test was performed to determine statistical significance. All error bars represent SD **P < 0.01.
Discussion
Currently, MSC‐mediated tissue regeneration has become a hot topic. Use of autologous and allogeneic MSCs has successfully regenerated some tissues in large animal models or humans 15, 16, 17, but although MSC‐mediated tissue regeneration has made good progress, some key issues have remained to be resolved, including molecular mechanisms of directed cell differentiation and proliferation.
With dental MSCs from healthy individuals or from those with OFCD syndrome, previous study of our group has shown that BCOR inhibited MSC proliferation, and mutation in BCOR prompted MSC population growth 31. However, the molecular mechanism was not clear. Using microarray analysis, we identified that with one member of the epidermal growth factor family, EREG, its expression was eight times higher in SCAPs from patients with OFCD than that from healthy individuals 31. Furthermore, we demonstrated that EREG gene was the downstream target of BCOR, and EREG was involved in regulating MSC differentiation 32. Previous reports demonstrated that EREG could regulate cell proliferation 29, 30, so we speculated that EREG might be an important regulator of proliferation of SCAPs. In the present project, with gain‐ and loss‐of‐function studies, we discovered that EREG indeed promoted proliferation of MSCs including SCAPs, PDLSCs and WJCMSCs and identified the possible role of EREG in abnormal growth of SCAPs in OFCD syndrome.
Previous work has reported that EREG is a member of the EGF family and usually associates with EGFR to apply its effect 29, 33, 34. In our study, blocking EGFR by a special inhibitor repressed EREG‐enhanced cell proliferation of SCAPs confirmed that EREG transduced its signalling by associating with EGFR. Other groups have shown that EREG can activate Erk/MAPK and Akt signalling pathways, thereby stimulating proliferation of normal cells, including fibroblasts, hepatocytes, smooth muscle cells and keratinocytes; conversely, these pathways can inhibit expansion of several types of tumour‐derived epithelial cells 23, 34, 35, 36, 37, 38. EREG also plays important roles in reproduction, anti‐viral response, skin inflammation, wound restoration and regeneration of smooth muscle and liver 24, 25, 26, 27, 28. As a growth factor, it can be released from vascular smooth muscle cells, and it acts as a major autocrine/paracrine factor for dedifferentiation via activation of the Erk and p38 MAPK signalling systems 26, 27. On the basis of those studies, we wondered whether Erk, Akt and p38 MAPK signalling were involved in EREG‐mediated cell proliferation of SCAPs. Our results show that knockdown of EREG inhibited phosphorylation of Erk1/2, but not of Akt and p38 MAPK, and that activation of EREG prompted phosphorylation of Erk1/2. Moreover, Erk1/2 inhibitor suppressed EREG‐mediated enhancement of SCAP proliferation. We then detected expression of MEK in the cells, upstream signalling of Erk. Unfortunately, we were not able to detect expression of phosphorylated MEK1/2 after knockdown of EREG, perhaps because the basic level of phosphorylated MEK1/2 is difficult to examine in SCAPs. But human recombinant EREG protein stimulated expression of phosphorylated MEK1/2, indicating that EREG activated MEK then stimulated the Erk signalling. In addition, as mitogen‐activated protein kinases (MAPKs) have three major subfamilies, Erk, JNK and p38 MAPK 39, we further investigated the JNK signalling pathway. Our results showed that knockdown of EREG inhibited phosphorylation of JNK, and that activation of EREG prompted phosphorylation of JNK. Moreover, blocking JNK signalling abolished the effect of EREG on cell proliferation, suggesting that JNK signalling also was involved in EREG‐mediated cell proliferation in the SCAPs. Taken together, our results indicate that EREG enhanced proliferation of SCAPs was through activated MEK/Erk and JNK, not by Akt and p38 MAPK signalling pathways.
Regulation of cell proliferation by external growth factors is a complex process. MAPK pathways involving a series of protein kinase cascades play a critical role in regulation of cell proliferation. Erk has been the best characterized MAPK and the Raf‐MEK‐Erk pathway represents one of the best characterized MAPK signalling pathways. Stimulation of tyrosine kinase receptors (RTKs) provokes activation of MAPKs in a multistep process. For example, essential linkers from epidermal growth factor receptors to MAP kinase include MAPKKes such as MEK1 and MEK2. MEKs ultimately phosphorylate Erk1 and Erk2, thereby increasing their enzymatic activity. Then activated Erk translocates to the nucleus and transactivates transcription factors, changing gene expression to promote cell population growth, differentiation or mitosis 40. Bose and Udupa discovered that both Erk and JNK appear to play key roles in EPO‐enhanced proliferation and suggested that presence of both is required for EPO‐mediated cell proliferation 41. Pedram et al. reported that through a novel Erk to JNK cross‐activation and subsequent JNK action, important events for VEGF‐induced cell proliferation are enhanced. VEGF‐induced Erk was necessary and sufficient for rapid JNK activation and both MAP kinases mediated cell proliferation effects of VEGF. These groups identified that JNK is the final mediator for Erk to stimulate cell proliferation and the role of Erk is mainly to induce JNK, when activated by an endothelial cell growth factor such as VEGF 40, 42. Our results identified both Erk and JNK pathways involved in EREG‐mediated cell proliferation of SCAPs, but the relationship of Erk and JNK signallings in the process are still unclear and need further investigation.
The role of EGF is controversial and some studies indicate that epiregulin is an activator in many tumours and can cause cell dedifferentiation 26, 28, 38. To exclude possible malignant alterations of SCAPs caused by EREG, we treated ours with 25 ng/ml human recombinant EREG protein for 1 week, and some basic characteristics and karyotypes of the cells were examined. Results showed that karyotype of the cells was still normal and no tumour formation was found. Combined with previous results, we speculate that 25 ng/ml human recombinant EREG protein might be safe to stimulate proliferation of SCAPs. Moreover, tests of MSCs in vitro including growth, proliferation and viability will hopefully, accurately predict MSC functions in vivo 18. MSCs with good proliferation and viability abilities have been able to create better vascularized granulation tissue and more long‐term MSC engraftment. Dental tissues derived MSCs, whatever pericyte or non‐pericyte origin, have been shown to differentiate into odontoblasts during tooth growth and in response to damage in vivo. Pericytes have been shown to be capable of acting as a source of MSCs and able to differentiate into cells of mesenchymal origin, alongside MSCs of a non‐pericyte origin; their contribution to MSC‐derived mesenchymal cells is possibly dependent on the extent of their vascularity 9. These discoveries strongly suggest that increasing proliferation and viability outcomes of MSCs could enhance vascularity and tissue regeneration potentials. In the present study, our findings of in vitro cell proliferation function of EREG support that it may have tissue regeneration potential.
In summary, our results indicate that EREG can enhance cell proliferation in dental tissue‐derived MSCs, and we have identified the role of EREG gene in abnormal growth of SCAPs in OFCD syndrome; these provided useful information concerning molecular mechanisms of OFCD. Activation of MEK/Erk and JNK signalling represent discovery of a new function for EREG in SCAPs, and this contributes to molecular mechanisms underlying proliferation of dental SCAPs.
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
Supporting information
Fig. S1. Surface markers of SCAPs.
Fig. S2. Multi‐differentiation potentials of SCAPs.
Fig. S3. Recombinant human EREG protein promoted cell proliferation of WJSMSCs and PDLSCs.
Fig. S4. Karyotype of SCAPs was normal after recombinant human EREG protein treatment. SCAPs were treated with 25 ng/ml human recombinant EREG protein for 1 week, then their karyotypes were analysed.
Fig. S5. Recombinant human EREG protein did not induce tumour formation and enhance the bone‐/dentin‐like tissue regeneration in SCAPs.
Fig. S6. Surface markers of SCAPs after recombinant human EREG protein treatment. SCAPs were treated with 25 ng/ml human recombinant EREG protein for 1 week, and surface markers were detected by flow cytometry.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81271101 to Z. P. Fan, 81141010 to D.S. Xia, 30901692 to S. R. Qi), the National Basic Research Program of China (No. 2010CB944801), the Funding Project to Science Facility in Institutions of Higher Learning Under the jurisdiction of Beijing Municipality (PXM2011_014226_07_000066 to Z. P. Fan), Grant from Beijing Municipal Science and Technology Commission (Z121100005212004 to S. L. Wang) and Beijing Natural Science Foundation (7102067 to S. R. Qi).
References
- 1. Jahagirdar BN, Verfaillie CM (2005) Multipotent adult progenitor cell and stem cell plasticity. Stem Cell Rev. 1, 53–59. [DOI] [PubMed] [Google Scholar]
- 2. Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair – current views. Stem Cells 25, 2896–2902. [DOI] [PubMed] [Google Scholar]
- 3. Seo BM, Miura M, Gronthos S, Bartold PM, Batouli S, Brahim J, et al (2004) Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 364, 149–155. [DOI] [PubMed] [Google Scholar]
- 4. Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 97, 13625–13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG (2003) SHED: stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. USA 100, 5807–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sonoyama W, Liu Y, Yamaza T, Tuan RS, Wang S, Shi S, et al (2008) Characterization of the apical papilla and its residing stem cells from human immature permanent teeth: a pilot study. J. Endod. 34, 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Aquino R, Graziano A, Sampaolesi M, Laino G, Pirozzi G, De Rosa A, et al (2007) Human postnatal dental pulp cells co‐differentiate into osteoblasts and endotheliocytes: a pivotal synergy leading to adult bone tissue formation. Cell Death Differ. 14, 1162–1171. [DOI] [PubMed] [Google Scholar]
- 8. Laino G, D'Aquino R, Graziano A, Lanza V, Carinci F, Naro F, et al (2005) A new population of human adult dental pulp stem cells: a useful source of living autologous fibrous bone tissue (LAB). J. Bone Miner. Res. 20, 1394–1402. [DOI] [PubMed] [Google Scholar]
- 9. Feng J, Mantesso A, De Bari C, Nishiyama A, Sharpe PT (2011) Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proc. Natl. Acad. Sci. USA 108, 6503–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paino F, Ricci G, De Rosa A, D'Aquino R, Laino L, Pirozzi G, et al (2010) Ecto‐mesenchymal stem cells from dental pulp are committed to differentiate into active melanocytes. Eur. Cell Mater. 20, 295–305. [DOI] [PubMed] [Google Scholar]
- 11. Spath L, Rotilio V, Alessandrini M, Gambara G, De Angelis L, Mancini M, et al (2010) Explant‐derived human dental pulp stem cells enhance differentiation and proliferation potentials. J. Cell Mol. Med. 14, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graziano A, D'Aquino R, Laino G, Proto A, Giuliano MT, Pirozzi G, et al (2008) Human CD34+ stem cells produce bone nodules in vivo. Cell Prolif. 41, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang JS, Wang X, Sun ZY, Wang XM, Yang H, Shi ST, et al (2010) Stem cells from human exfoliated deciduous teeth can differentiate into dopaminergic neuron‐like cells. Stem Cells Dev. 19, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang J, Sonoyama W, Wang Z, Jin Q, Zhang C, Krebsbach PH, et al (2007) Noncanonical Wnt‐4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J. Biol. Chem. 282, 30938–30948. [DOI] [PubMed] [Google Scholar]
- 15. Liu Y, Zheng Y, Ding G, Fang D, Zhang C, Bartold PM, et al (2008) Periodontal ligament stem cell‐mediated treatment for periodontitis in miniature swine. Stem Cells 26, 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ding G, Liu Y, Wang W, Wei F, Liu D, Fan Z, et al (2010) Allogeneic periodontal ligament stem cell therapy for periodontitis in swine. Stem Cells 28, 1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. D'Aquino R, De Rosa A, Lanza V, Tirin V, Laino L, Graziano A, et al (2009) Human mandible bone defect repair by the grafting of dental pulp stem/progenitor cells and collagen sponge biocomplexes. Eur. Cell Mater. 18, 75–83. [DOI] [PubMed] [Google Scholar]
- 18. Deskins DL, Bastakoty D, Saraswati S, Shinar A, Holt GE, Young PP (2013) Human mesenchymal stromal cells: identifying assays to predict potency for therapeutic selection. Stem Cells Transl. Med. 2, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X, Wang Y, Gao Y, Liu X, Bai T, Li M, et al (2013) Maintenance of high proliferation and multipotent potential of human hair follicle‐derived mesenchymal stem cells by growth factors. Int. J. Mol. Med. 31, 913–921. [DOI] [PubMed] [Google Scholar]
- 20. Delcroix GJ, Curtis KM, Schiller PC, Montero‐Menei CN (2010) EGF and bFGF pre‐treatment enhances neural specification and the response to neuronal commitment of MIAMI cells. Differentiation 80, 213–227. [DOI] [PubMed] [Google Scholar]
- 21. Tamama K, Kawasaki H, Wells A (2010) Epidermal growth factor (EGF) treatment on multipotential stromal cells (MSCs). Possible enhancement of therapeutic potential of MSC. J. Biomed. Biotechnol. 2010, 795385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Platt MO, Roman AJ, Wells A, Lauffenburger DA, Griffith LG (2009) Sustained epidermal growth factor receptor levels and activation by tethered ligand binding enhances osteogenic differentiation of multi‐potent marrow stromal cells. J. Cell. Physiol. 221, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lindvall C, Hou M, Komurasaki T, Zheng C, Henriksson M, Sedivy JM, et al (2003) Molecular characterization of human telomerase reverse transcriptase‐immortalized human fibroblasts by gene expression profiling: activation of the epiregulin gene. Cancer Res. 63, 1743–1747. [PubMed] [Google Scholar]
- 24. Shirasawa S, Sugiyama S, Baba I, Inokuchi J, Sekine S, Ogino K, et al (2004) Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune‐related responses of keratinocyte and macrophage. Proc. Natl. Acad. Sci. USA 101, 13921–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding X, Wang F, Duan M, Yang J, Wang S (2009) Epiregulin as a key molecule to suppress hepatitis B virus propagation in vitro. Arch. Virol. 154, 9–17. [DOI] [PubMed] [Google Scholar]
- 26. Takahashi M, Hayashi K, Yoshida K, Ohkawa Y, Komurasaki T, Kitabatake A, et al (2003) Epiregulin as a major autocrine/paracrine factor released from Erk‐ and P38MAPK‐activated vascular smooth muscle cells. Circulation 108, 2524–2529. [DOI] [PubMed] [Google Scholar]
- 27. Kim K, Lee H, Threadgill DW, Lee D (2011) Epiregulin‐dependent amphiregulin expression and ERBB2 signaling are involved in luteinizing hormone‐induced paracrine signaling pathways in mouse ovary. Biochem. Biophys. Res. Commun. 405, 319–324. [DOI] [PubMed] [Google Scholar]
- 28. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, et al (2007) Mediators of vascular remodelling co‐opted for sequential steps in lung metastasis. Nature 446, 765–770. [DOI] [PubMed] [Google Scholar]
- 29. Sasaki E, Arakawa T, Fujiwara Y, Kawada N, Fukuda T, Higuchi K, et al (1997) Epiregulin stimulates proliferation of rabbit gastric cells in primary culture through autophosphorylation of the epidermal growth factor receptor. Eur. J. Pharmacol. 338, 253–258. [DOI] [PubMed] [Google Scholar]
- 30. Morita S, Shirakata Y, Shiraishi A, Kadota Y, Hashimoto K, Higashiyama S, et al (2007) Human corneal epithelial cell proliferation by epiregulin and its cross‐induction by other EGF family members. Mol. Vis. 13, 2119–2128. [PubMed] [Google Scholar]
- 31. Fan Z, Yamaza T, Lee JS, Yu J, Wang S, Fan G, et al (2009) BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell Biol. 11, 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du J, Ma Y, Ma P, Wang S, Fan Z (2013) Demethylation of epiregulin gene by histone demethylase FBXL11 and BCL6 corepressor inhibits osteo/dentinogenic differentiation. Stem Cells 31, 126–136. [DOI] [PubMed] [Google Scholar]
- 33. Shelly M, Pinkas‐Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, et al (1998) Epiregulin is a potent pan‐ErbB ligand that preferentially activates heterodimeric receptor complexes. J. Biol. Chem. 273, 10496–10505. [DOI] [PubMed] [Google Scholar]
- 34. Toyoda H, Komurasaki T, Uchida D, Takayama Y, Isobe T, Okuyama T, et al (1995) Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 270, 7495–7500. [DOI] [PubMed] [Google Scholar]
- 35. Draper BK, Komurasaki T, Davidson MK, Nanney LB (2003) Epiregulin is more potent than EGF or TGFalpha in promoting in vitro wound closure due to enhanced ERK/MAPK activation. J. Cell. Biochem. 89, 1126–1137. [DOI] [PubMed] [Google Scholar]
- 36. Taylor DS, Cheng X, Pawlowski JE, Wallace AR, Ferrer P, Molloy CJ (1999) Epiregulin is a potent vascular smooth muscle cell‐derived mitogen induced by angiotensin II, endothelin‐1, and thrombin. Proc. Natl. Acad. Sci. USA 96, 1633–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pastore S, Mascia F, Mariani V, Girolomoni G (2008) The epidermal growth factor receptor system in skin repair and inflammation. J. Invest. Dermatol. 128, 1365–1374. [DOI] [PubMed] [Google Scholar]
- 38. Hu K, Li SL, Gan YH, Wang CY, Yu GY (2009) Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC‐83 through activation of ERK and Akt. Oral Oncol. 45, 156–163. [DOI] [PubMed] [Google Scholar]
- 39. Mishima K, Inoue K, Hayashi Y (2002) Overexpression of extracellular‐signal regulated kinases on oral squamous cell carcinoma. Oral Oncol. 38, 468–474. [DOI] [PubMed] [Google Scholar]
- 40. Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18. [DOI] [PubMed] [Google Scholar]
- 41. Bose C, Udupa KB (2008) Erythropoietin enhancement of rat pancreatic tumor cell proliferation requires the activation of ERK and JNK signals. Am. J. Physiol. Cell Physiol. 295, C394–C405. [DOI] [PubMed] [Google Scholar]
- 42. Pedram A, Razandi M, Levin ER (1998) Extracellular signal‐regulated protein kinase/Jun kinase cross‐talk underlies vascular endothelial cell growth factor‐induced endothelial cell proliferation. J. Biol. Chem. 273, 26722–26728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Surface markers of SCAPs.
Fig. S2. Multi‐differentiation potentials of SCAPs.
Fig. S3. Recombinant human EREG protein promoted cell proliferation of WJSMSCs and PDLSCs.
Fig. S4. Karyotype of SCAPs was normal after recombinant human EREG protein treatment. SCAPs were treated with 25 ng/ml human recombinant EREG protein for 1 week, then their karyotypes were analysed.
Fig. S5. Recombinant human EREG protein did not induce tumour formation and enhance the bone‐/dentin‐like tissue regeneration in SCAPs.
Fig. S6. Surface markers of SCAPs after recombinant human EREG protein treatment. SCAPs were treated with 25 ng/ml human recombinant EREG protein for 1 week, and surface markers were detected by flow cytometry.