

Abstract
Objectives: Mesenchymal stem cells (MSC) are multipotent cells capable of differentiating into adipocytic, chondrocytic and osteocytic lineages on suitable stimulation. We have hypothesized that mechanical loading may influence MSC differentiation and alter their phenotype accordingly.
Materials and methods: Mouse bone marrow‐derived MSC were established in vitro by differential adherence to plastic culture plates and grown in low glucose medium with 10% foetal calf serum and growth factors. Cells grew out and were subcultured up to 20 times. Differentiation protocols were followed for several cell lineages. Clones with trilineage potential were seeded in type I collagen gels and incubated in a tensioning force bioreactor and real‐time cell‐derived forces were recorded. Gels were fixed and sectioned for light and electron microscopy.
Results: Cell monolayers of parent and cloned mouse bone marrow‐derived MSC differentiated into adipocytes, osteocytes and chondrocytes, but not into cardiomyocytes, myotubes or neuronal cells. When cast into type I collagen gels and placed in tensioning bioreactors, MSC differentiated into fibroblast‐like cells typical of tissue stroma, and upregulated α‐smooth muscle actin, but rarely upregulated desmin. Electron microscopy showed collagen and elastin fibre synthesis into the matrix.
Conclusions: These experiments confirmed that MSC cell fate choice depends on minute, cell‐derived forces. Applied force could assist in commercial manufacture of cultured bio‐engineered prostheses for regenerative medicine as it mimics tissue stresses and constitutes a good model for development of tissue substitutes.
Introduction
Ability of bone marrow‐derived mesenchymal stem cells (MSC) to differentiate into other cell types has led to an explosion of interest in their use in regenerative medicine and tissue engineering. In general, a tissue‐engineered living graft would be surgically inserted into the required body site, and gradually, the matrix would be integrated into local tissues by an influx of host cells (1); the exogenous transplanted matrix cells would have to support the graft until this occurred. In vivo, fibroblast‐like cell types have only a finite number of cell cycles available to them (2), and in older patients, the number may not be sufficient for complete repopulation to take place. Thus, we have investigated whether properties of BM‐derived multipotent MSC allow them to differentiate into a fibroblastic stromal lineage, and thus to be able to contribute to the effectiveness of tissue‐engineered grafts.
There is a physical chain of protein events that links extracellular matrix to plasma membranes, intracellular contractile actin cytoskeleton, translation of relevant proteins, their transcription and DNA sequences. The interplay of cells and their matrix has been well studied and it is clear that each affects the other [reviewed by (3)]. Thus, it is probable that cell behaviour can be influenced by externally derived forces. Genes coding for such mechano‐transduction proteins have already been described (4), and certainly, the myofibroblast has been described as being force‐perceptive and force‐transmissive between extracellular matrix adhesions and cytoplasmic α‐smooth muscle actin (5). This is crucial for differentiation of resting fibroblasts into myofibroblasts in healing on the one hand, and in fibrocontractive diseases on the other. In our experiments, external forces on MSC have been produced and measured precisely by using specific bioreactors, and histology and ultrastructure of the stimulated stem cells have been examined, showing that they differentiate and actively produce both collagen and elastin into the extracellular matrix.
Materials and methods
Experimental animals
Young adult C57/Black 6J male mice (6–8 weeks of age) were bred in‐house under strict UK Home Office guidelines using enhanced social environments, and were used for all bone marrow cell (BMC) isolations.
Isolation of bone marrow cells
Mice were killed by CO2 inhalation. Tibias and femurs were dissected, ends of the bones were cut, and marrow was flushed out using 2 ml sterile, ice‐chilled phosphate‐buffered saline (PBS) through a 27‐gauge needle. Pooled BMCs were triturated and then further dispersed by passing through 21‐ and 23‐gauge needles. Cell suspensions were centrifuged at 400 g for 5 min and supernatant discarded; pellets were resuspended in sterile PBS and after sieving through 40 μm mesh (BD Falcon, Oxford, UK), BMCs were resuspended in sterile culture medium as described below.
Culture of MSC
Isolation of MSC was first shown in vitro by Friedenstein et al. (6) who derived fibroblast‐like cells after adherence of bone marrow‐derived cells to plastic tissue culture plates. These cells were subsequently found to differentiate into osteoblasts and adipocytes. In this study, we have used a modification of that method, as described previously (7). In brief, whole BMCs were resuspended in base medium: low glucose (1 g/l) Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Invitrogen, Paisley, UK; 60%) mixed with MCDB‐201 (Sigma, Poole, UK; 40%) and 20% foetal calf serum (FCS; Seralab, Hayward’s Heath, UK) with penicillin and streptomycin (Sigma; 100 U/ml, 0.1 mg/ml respectively). MCDB‐201 was dissolved at concentration of 1.77 g/100 ml, adjusted to pH 7.4 with 8 m NaOH, and sterilized using a 0.2 μm filter (Nalge‐Nunc, Roskilde, Denmark). For initial cell expansion, leukaemia inhibitory factor (Millipore, Watford, UK; 1000U/ml), platelet‐derived growth factor–B chain homodimer (PDGF‐BB) (R and D Systems, Abingdon, UK; 10 ng/ml) and epidermal growth factor (Sigma; 10 ng/ml) were added as described by Jiang et al. (8). Initially, BMC isolates were plated on plasma fibronectin (Sigma)‐coated surfaces (20 μg/ml for 30 min, then air‐dried), but this was found to not affect plating efficiency and was discontinued. Cells were plated at density of 20–40 × 106 cells/25 cm2 culture flasks (BD Falcon) and were incubated at 37 °C in a humidified atmosphere containing 8% CO2. Medium was replaced every 3 days and non‐adherent cells [haematopoietic stem cells (HSCs) and myeloid lineages], were discarded. At approximately 70% confluence, adherent mesenchymal cells were passaged at split ratio of 1:3 using 0.25% trypsin in PBS containing 1 mm ethylene diamine tetraacetic acid. These bone marrow‐derived adherent cell preparations were characterized for their ability to undergo chondrogenic, osteogenic and adipogenic differentiation. One such parent line (JRP73) was then single‐cell cloned using the same culturer conditions, and each of eight successful clones was tested for trilineage differentiation. One clone (JR283) was the most consistent for this MSC characteristic and was chosen for further study. All populations were tested for mycoplasmas and found to be negative.
Differentiation of adherent BMCs
Cells were grown to subconfluence in glass (BD Falcon) or plastic (Permanox, Labtek; Nalge‐Nunc) eight‐chambered microscope slides or 24‐well plates (BD Falcon), then switched to the appropriate cocktails for each differentiation assay (Table 1). Additives were made from sterile 1000× stock solutions. Cells for chondrocytic differentiation were cultured as pellets of 50 000 cells in 1 ml medium plus additives.
Table 1.
MSC differentiation cocktails
| Cell type | Reagent | Supplier | Final concentration |
|---|---|---|---|
| Adipocytic | Hydrocortisone | Calbiochem | 0.5 μm |
| Isobutyl methyl xanthine | Sigma | 0.2 mm | |
| Indomethacin | Sigma | 60 μm | |
| Insulin | Sigma | 5 μg/ml | |
| Osteocytic | Dexamethasone | Sigma | 10 nm |
| Vitamin C Phosphate | Sigma | 0.2 mm | |
| Na β‐glycerophosphate | Sigma | 10 mm | |
| Chondrocytic | Basic FGF ( = FGF2) | Sigma/R and D | 1 ng/ml |
| TGFβ1 | Oncogene | 5 ng/ml | |
| Myocytic | 5‐Azacytidine | Sigma | 3 μm |
| Vit C Phosphate | Sigma | 0.1 mm | |
| Neurogenic | BHA | Sigma | 200 μm |
| Medium A | KCl | BDH | 5 mm |
| Valproic acid | Sigma | 2 μm | |
| Forskolin | Sigma | 10 μm | |
| Hydrocortisone | Calbiochem | 1 μm | |
| Insulin | Sigma | 5 μg/ml | |
| Neurogenic | EGF | Sigma | 10 ng/ml |
| Medium B | bFGF | Sigma/R and D | 20 ng/ml |
Immunocytochemical antibody staining
Cells were stained for presence of a subset of established markers of MSC and HSC lineages, using commercial antibodies as shown in Table 2, using the well‐known 3‐layer method. In brief, cultures were fixed in 4% paraformaldehyde (PFA) in PBS for 20 min, washed in PBS, and endogenous peroxidase was blocked with 0.3% H2O2 in PBS for 30 min then underwent further PBS washing, followed by a blocking step in 5% FCS in PBS for 30 min. All antibodies were diluted in FCS blocking solution. Antibodies and subsequent detecting layers were applied for 60 min, with three PBS washes of 10 min between layers. Detection of mouse monoclonal antibodies was ascertained using biotinylated rabbit anti‐mouse serum (Dako, Glostrup, Denmark; 1:300), followed by streptavidin–peroxidase (Dako; 1:500). Rabbit polyclonal antibodies were detected using a biotinylated swine anti‐rabbit serum (Dako; 1:500), with streptavidin–peroxidase, as before. Sites of antibody binding were detected with diaminobenzidine, 0.5mg/ml in PBS in the presence of 0.03% H2O2. A 30 s counterstain in Harris’ haematoxylin was performed before coverslipping under Glycergel (Dako).
Table 2.
Antibodies used
| Cell type/marker | Antigen | Supplier | Dilution |
|---|---|---|---|
| MSC | CD71 | Dako | 1:100 |
| CD105 | Dako | 1:100 | |
| NGF‐R (p75) | Chemicon | 1:100 | |
| HSC | CD34 | Dako | 1:25 |
| CD45 | Dako | 1:100 | |
| CD133 | BD Pharmingen | 1:100 | |
| Muscle | MyoD | Abcam | 1:200 |
| Myosin Heavy Chain | Dako | 1:10 | |
| Troponin C | Abcam | 1:5000 | |
| Neuron | Gap43 | Sigma | 1:1000 |
| NeuN | Chemicon | 1:100 | |
| Nestin | Chemicon | 1:50 | |
| Neurofilament 200 | EuroPath | 1:50 | |
| TRPV1 | Abcam | 1:100 | |
| Cytoplasmic filaments | α‐smooth muscle actin | Sigma | 1:4000 |
| Vimentin | Dako | 1:200 | |
| Desmin | Dako | 1:100 | |
| Glial fibrillar acidic protein (rabbit) | Dako | 1:100 | |
| Macrophage | Mac3 | Novocastra | 1:500 |
| F4/80 (rat Mab) | Insight Biotech | 1:50 | |
| Endothelial | PECAM‐1 (rat Mab) | Accurate Chem | 1:100 |
Antibodies were mouse monoclonals except where indicated.
Histochemical analyses
Methods for demonstrating elastic and reticular fibres, respectively, were Miller’s [Web reference: Ellis (9)] and the reticulin stains (10). Miller’s method: in brief, dewaxed sections were rinsed in 95% ethanol, stained in Miller’s stain for 90 min, rinsed sequentially in 95% ethanol, distilled water and counterstained in haematoxylin, dehydrated and mounted. Elastic fibres appeared black. Reticulin method: in brief, sections were oxidized in 1% potassium permanganate for 5 min, rinsed in distilled water, bleached in 1% oxalic acid, rinsed again, incubated in 2.5% iron alum for 15 min and rinsed several times. Slides were then immersed in freshly titrated ammoniacal silver nitrate (nominally 1%) for 2 min, washed in several changes of distilled water, reduced in 10% formalin for 10 min, rinsed in distilled water and fixed in 5% sodium thiosulphate for 3 min. Final rinsing was performed before dehydrating and mounting. Reticulin (collagen III) fibres appeared black.
Adipocytic differentiation
Ability of adipocytes to take up the lipophilic dye oil red O (ORO) was used to demonstrate this differentiation lineage (11). Cultures were fixed in freshly thawed 4% PFA in PBS for 20 min, washed in PBS and equilibrated in 60% isopropanol followed by incubation in ORO (1% in 60% isopropanol) for 15 min. After differentiating in 60% isopropanol for 5 s, cells were washed in water, and nuclei were counterstained in haematoxylin for 30 s. Specimens were finally rinsed in water and mounted in Glycergel.
Osteocytic differentiation
This method relies on staining of extracellular calcium deposits with alizarin red (12). Cells were fixed in 4% PFA as above and stained in 2% alizarin red S, pH 4.2, for 3 min, rinsed in ethanol and mounted in Glycergel.
Chondrocytic differentiation
Pelleted cell cultures were fixed in 10% neutral buffered formalin for 30 min, embedded in 1% agarose gel, dehydrated in 70% ethanol and paraffin embedded for sectioning. After dewaxing and rehydrating, sections were stained using the periodic acid–Schiff (PAS) reaction, or alcian blue stain at pH 2.5 for acid mucopolysaccharides (13).
Myocytic differentiation
Cultured monolayers were incubated for up to 3 months to ascertain muscle potentiality, either in azacytidine‐ (14) (3 μm) or in vitamin C phosphate‐ (15) (100 μm) containing media. Immunohistochemical analysis was performed to determine whether stimulated cells would express c‐troponin C, a cardiac and skeletal muscle protein, MyoD, a developmental regulator of skeletal muscle differentiation, or myosin heavy chain, a mature skeletal myocyte marker.
Neurogenic differentiation
Two strategies (media A and B in Table 1) were employed to stimulate cells for up to 2 weeks to express neuronal markers (16). After fixation in 4% PFA, 3‐layer immunohistochemistry (IHC) was performed as described above. Cells were stained with antibodies to several neuronal markers (Table 2).
Electron microscopy
Cells and constructs of all components of the study were collected from culture environments. Routine methods for electron microscopy were used as described previously (17). In brief, samples were fixed in 2% glutaraldehyde for 2 h followed by osmication (1%) for 1 h, and then dehydration through a series of graded alcohols. This was followed by embedding in liquid Araldite (Huntsman Advanced Materials, Basel, Switzerland) before polymerization at 60 °C overnight. They were then cut into 100 nm sections on copper electron microscope grids and stained with saturated aqueous uranyl acetate and 0.4% lead nitrate in 0.2 m sodium citrate, before observing using a Philips CM100 electron microscope (Philips Electronics Nederland BV, Eindhoven, Netherlands).
Methods for mechanical stimulation and contractile force analysis
For mechanical stimulation and contractile force analysis of mesenchymal stem cell differentiation by application of external force only, the stem cells were cultured in base medium as above. Cells were plated at a density of 2–4 × 106 cells/25 cm2 culture flasks (BD Falcon) and were incubated at 37 °C in a humidified atmosphere containing 5% CO2. Measurement of contractile force generated within a three‐dimensional, tethered floating stem cell‐populated collagen lattice was performed as described previously using a culture force monitor (CFM) (18). Briefly, collagen gel was prepared by mixing 4 ml of 1 mg/ml solution of native 10 mm acetic acid‐soluble type I rat tail collagen (First Link Ltd, West Midlands, UK) with 0.5 ml of 10× strength DMEM and neutralizing it by dropwise addition of 1 m NaOH. Cell suspensions (5 × 106) in 0.5 ml of complete DMEM were prepared and added to the neutralized collagen solution. This was poured into the culture well and incubated at 37 °C in a humidified atmosphere containing 5% CO2, and the liquid collagen formed a gel within 5 min, at which stage the cell chamber was removed from the incubator and topped up with a further 15 ml of complete medium. The culture well was now connected to the CFM and repositioned in the incubator at 37 °C; measurements of force generation by the cells themselves over 24 h as cells attached and spread within the collagen matrix were started immediately. The collagen gels were tethered to two flotation bars on either side of their long edges, and in turn attached to a ground point at one end and a force transducer at the other. Minute cell‐generated tensional forces in collagen gels were detected by the force transducer and logged into a personal computer. Data were collected at a rate of one reading every 15 s over a 24 h period as described previously (18). Cultures that were to be mechanically stimulated were prepared in the same manner. Collagen gel/cell lattices were exposed to a cyclic overloading regime as described previously (19). Briefly, the loading cycle, which commenced after 12 h of collagen gel contraction by the resident cell population, consisted of an increase in tension of 130 dynes over a 3 min period, followed by a 15 min resting phase. Unloading over a 3 min time period was followed by a further 15 min resting period. This process was repeated for 22 cycles. Data collected at a rate of one reading every 15 s were post‐processed using Microsoft Excel software.
After the experimental period, gels were fixed in 4% PFA in PBS for 30 min, and samples were processed into paraffin wax with care to retain the known orientation of axes of the force stimuli. Sections were stained routinely with haematoxylin and eosin, type III collagen fibres (reticulin) and elastic van Giesen methods (10). IHC for differentiation antigens shown in Table 2 was carried out as described above.
Results
Cells of the parent mouse BM‐derived cell line, JRP273, had a fibroblastic morphology when viewed using phase‐contrast microscopy, a feature that was shared with the cloned JR283 cells (Fig. 1a,b). Immunohistochemical phenotype of the cells was similar to previously published results (20, 21) for similar lines (Table 3).
Figure 1.
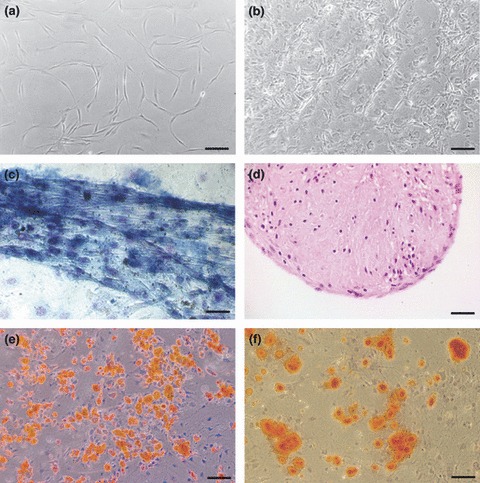
Cloned JR283 mouse MSC in culture. Phase‐contrast cell morphology 2 days post‐plating (a), and at confluence (b). Cells cultured in chondrocytic medium produced and contracted extracellular matrix that was alcian blue‐positive in monolayer culture (c) and PAS‐positive in pellet culture (d; 6 μm section). Adipocytic medium produced many oil red O‐positive cells containing lipid droplets (e). Osteogenic cultures produced calcium‐rich ECM stained with alizarin red (f). Scale bars: a, b, d, e, f = 100 μm; c = 50 μm.
Table 3.
Immunohistochemical phenotypes of parental JR373 and clone JR283 mouse MSC
| Positive | Negative |
|---|---|
| α‐Smooth Muscle Actin | Desmin |
| Vimentin | CD31 |
| CD105 | CD34 |
| NGF‐R (p75) | D45 |
| CD71 | CD117 |
| GFAP | CD133 |
| Mac‐3 | F4/80 |
MSC, mesenchymal stem cells; GFAP, glial fibrillary acid protein.
Thus, parent and cloned cells expressed established MSC markers CD71, CD105 (SH2, endoglin) and NGF‐R (p75), and did not express haematopoietic markers such as CD34, CD45 and CD133. In addition, we found the MSC‐like cells to be positive for α‐SMA, glial fibrillary acid protein and vimentin, as well as one macrophage marker, Mac3, but not another, F4/80. These phenotypic profiles have been maintained for up to 20 passages in vitro.
Parent line JRP73 and its cloned subline JR283 were capable of trilineage differentiation into chondrocytic (Fig. 1c,d), adipocytic (Fig. 1e) and osteocytic (Fig. 1f) cells, as indicated by their positive staining for alcian blue (Fig. 1c) or PAS‐positive (Fig. 1d) material in monolayer or pellet culture respectively, oil red O (Fig. 1e) and alizarin red (Fig. 1f). Monolayer chondrocytic culture resulted in cells making a large amount of extracellular material, which they contracted readily to form small tissue‐like aggregates of cells (Fig. 1c). This phenotype was enhanced by pellet culture (Fig. 1d). Differentiation into muscle cells was not seen (even when cultured up to 3 months), as indicated by negativity for troponin C, MyoD and skeletal muscle myosin staining. Some cells in neuronal differentiation media had nuclear NeuN staining, but other neural markers (TRPV1, Gap43, neurofilament) did not develop (data not shown).
MSC were embedded into type I collagen gels and placed into bioreactors that were developed in our laboratory. Force generation by all mMSC cultures (n = 3) demonstrated a nearly linear increase over 24 h, at a rate of 5 dynes/h. Contractile curves (Fig. 2a,b) did not demonstrate any plateaux, normally indicating the status of tensional homeostasis observed in normal dermal fibroblasts. Cells responded to the manipulation in matrix tension as a result of externally applied loads using the tensioning (t)‐CFM. After loading, release in matrix tension was at a rate of approximately 80 dynes/h (Fig. 2b). Similarly, there was re‐tensioning of the matrix after unloading at a rate of 80 dynes/h. It is interesting to note that after application of 22 loading cycles, rate of matrix contraction increased by a factor of 5–25 dynes/h (Fig. 2b).
Figure 2.
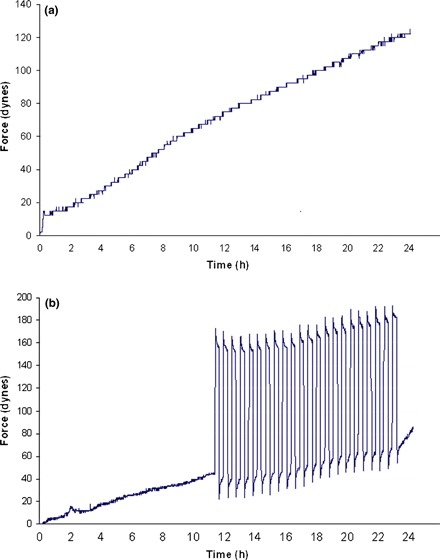
Stress–strain curves over 24 h for cultured murine MSC in type I collagen gels in the tensioning cell force monitor bioreactor (t‐CFM). (a) Typical contractile curve of force generated by MSC over 24 h due to cell attachment and extension of processes in the collagen matrix. (b) 21 cycles of cyclical mechanical stimulation. Note increase in rate of force generated after cyclical loading.
Gels containing MSC subject to tensioning forces were fixed after each experiment and photographed under phase‐contrast illumination. Examples of low (Fig. 3a) and high (Fig. 3b) aspect ratio gels are shown. Sections of gels were stained for markers of stromal and endothelial cells, as well as histochemical stains for elastic and reticular (type III collagen) fibres. We found that the cells quickly aligned to a large extent along the axis of tensioning (horizontal in Fig. 3c), and were histochemically positive for elastic fibres (Fig. 3d), α‐smooth muscle actin (Fig. 3e) and rarely for desmin (Fig. 3f). Cultures were negative for endothelial cell marker CD31 (PECAM1).
Figure 3.
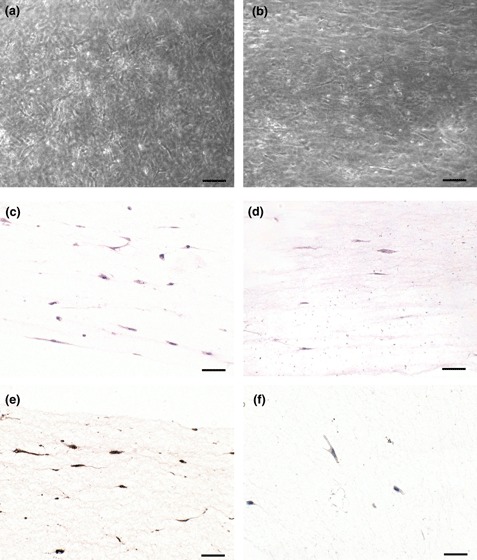
Collagen gels containing JR283 MSC after 24 h force application in the CFM. Low (a) and high (b) aspect ratio gels showing stellate morphology of fibroblasts. Sections of paraffin wax‐embedded gels were stained with haematoxylin and eosin (c), elastic van Giesen for elastic fibres (d), for α‐smooth muscle actin (e) and desmin (f). Faintly positive desmin cells were rare, whereas most cells were α‐SMA positive. Note frequent alignment of cells along the axis of applied force (horizontal). Scale bars = 100 μm.
Electron microscopy of cells that had been subjected to applied loads in the t‐CFM clearly indicated that resultant phenotypic changes had been influenced by specific protein synthesis in the cells. Untensioned control cells retained their original phenotype with no large vacuoles of any nature in the cytoplasm, with only rare plasma membrane‐embedded, exocytosing fibres (Fig. 4a). Cells that had been subjected to a loading regime in the t‐CFM displayed plasma membrane features characteristic of fibroblasts, having synthesized collagen (Fig. 4b) and elastin (Fig. 4c), and we also saw examples of collagen and elastin emerging simultaneously from the same cell (Fig. 4d). Furthermore, force‐stressed cells generally exhibited gap junctions, organelles and electron‐dense granules that were not observed in cells of the undifferentiated population (not shown). Tissue processed for electron microscopy does not conserve lipids that may have been present in life. We noted occasional cells in the t‐CFM which displayed cytoplasmic lipid vacuoles similar to those seen in adipocytes (Fig. 4e, compare to Fig. 1e).
Figure 4.
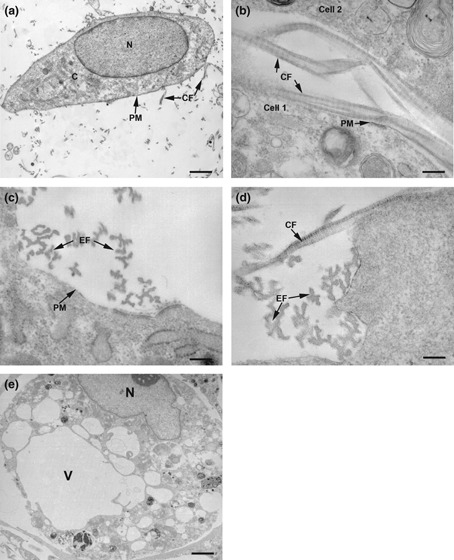
Electron micrographs (EM) of extracellular matrix fibres and MSC in the tCSM bioreactor. (a) Time zero untensioned control cell freshly seeded in collagen, before any forces had been either generated or applied. Control cell morphology was generally oval with a pale, euchromatin‐filled nucleus (N), sparse cytoplasmic organelles (C) and few collagen fibrils (CF, arrows) emanating from the plasma membrane (PM, arrow; scale bar = 1 μm); (b–e) 24 h after tensioning in the tCSM; (b) Two adjacent collagen I‐embedded cells with typical banded collagen fibrils (CF) emerging from them (arrows; scale bar = 25 nm); (c) Plasma membrane with typical elastin fibrils (EF) emerging (scale bar = 15 nm); (d) Edge of matrix‐embedded plasma membrane with adjacent collagen (CF) and elastin (EF) fibrils emerging from the same cell (scale bar = 7.5 nm); (e) Collagen I‐embedded cell with apparent adipocyte morphology, after application of external force, via the t‐CFM (scale bar = 1 μm). Such cells were more spherical, with vacuoles (V) in the cytoplasm, reminiscent of lipid droplets, as seen by light microscopy after staining with oil red O (compare Fig. 1e; scale bar = 1 μm).
Discussion
We have shown that our populations of undifferentiated mouse BM‐derived cells possess several MSC‐specific characteristics without expressing HSC markers. The ability of bone marrow‐derived mMSC to differentiate into the classical three output lineages of fat, bone and cartilage‐like cells has been corroborated here in these studies. That the parent or cloned lines were unable to differentiate into muscle and neuronal cell types suggests that the populations are more lineage‐restricted than some other mesenchymal cells, such as multipotent adult progenitor cell (8) or induced pluripotent stem cells (see reviews by (22, 23). We considered that the ability to produce extracellular matrix material (as in the chondrogenic and osteogenic cultures), and for the former cells to contract this into an organotypic tissue‐like structure both as monolayers or as pellet cultures, indicated that the cells possessed an efficient contractile force‐transducing mechanism. This may have an in vivo utility in tissue engineered for regenerative medicine purposes.
Endogenous cell‐mediated collagen contraction demonstrated in Fig. 2a is similar to that observed in human MSC (Eastwood, unpublished observations) in that there is a linear increase in force as cells attach primarily by action of beta type 1 integrins (24) and migrate through the matrix. The primary difference between these cells and their human counterpart is in overall levels of force generated, human cells generating approximately twice this value. Application of cyclical loading (Fig. 2b) elicited a cellular response both to release matrix tension when loaded, and to regain tension after unloading, to below endogenous levels. This response was evident from the first cycle, demonstrating how exquisitely sensitive these cells are to changes in their physical environment. Mechanical stimulation of a similar nature to normal dermal fibroblasts results in phenotype change from fibroblast to myofibroblast, evidenced by presence of α‐smooth muscle actin (25). During initial stages of normal wound healing, resident fibroblasts are transformed into myofibroblasts due to increased tension caused by wound closure and associated oedema. It is interesting to note that after cyclical mechanical stimulation, rate of contraction demonstrated by the mouse MSC had increased by a factor of five to a rate of 25 dynes/h, a rate of contraction approaching that associated with dermal fibroblasts. We would hypothesize that after mechanical stimulation, we have changed mouse MSCs to a fibroblast phenotype. This leads to the interesting notion that during the process of wound healing, there may be recruitment of MSCs by chemotaxins to a wound site. Cells then attach to connective tissue and are subjected to mechanical stimulation, either from oedema or mediators of inflammation, leading to differentiation into a fibroblastic phenotype. This leads on to the question of whether fibrocytes could be considered to be mesenchymal stem cells.
In addition to responding to stresses and strains induced by the t‐CFM, the cells exhibited particular phenotypic features akin to myofibroblasts, clearly shown by both IHC and electron microscopy. A series of molecules has been shown to convey mechano‐transduction to gene expression (26): for example, the mechanical stimulus‐responsive gene Pip4k2b has been identified as being expressed in osteoblastic cell cultures and in rabbit gastrocnemius muscle and Achilles tendon (4). In addition, short episodes (2 h) of mechanical compression every 5 days using human MSC, which were cultured in 3‐D matrices of a polyurethane scaffold, have been seen to increase their mineralization of calcium, as well as alkaline phosphatase expression and secretion of collagen I into the matrix, over a 24‐day period (27). This clearly indicates that even intermittent mechanical loading of organotypic tissue constructs has an impact on gene expression and results in MSC differentiation towards an osteoblastic phenotype suited to those specific culture conditions.
Recently, there has been a report that mouse embryonic fibroblasts and human corneal fibroblasts both respond to mechanical transduction (within an attached collagen gel) by upregulating α11/β1 integrin, and in the process becoming phenotypically similar to myofibroblasts by induction of α ‐smooth muscle actin expression. This was reversed by siRNA to α11 integrin transfection (28). The mechanism by which fibroblasts differentiate into myofibroblasts and transmit force to collagen gels is thought to reside in part through adherens junction formation and mechanosensitive ion channels, which allow Ca ion oscillations (29). In addition, connexin 43 is thought to bind to actin filaments in contractile avian and human tenocytes subject to cyclical strain, as suggested by their increased colocalization, in contrast to Cos7 cells (30). Ability of cells to contract a collagen gel has been recently confirmed in anterior cruciate ligament fibroblast cultures subjected to 2.5% cyclical load (31). Cell and matrix alignment was seen, in a phenomenon we have also noted in these studies. Henshaw and colleagues concluded that integrin α5/β1 was particularly important for these actions. A further study has looked at the response of MSC seeded on flexible silicone bioreactor substrate on which variable pressure was imposed from the contralateral side, in a manner mimicking vasculature. MSC oriented along the lines of flow and adopted endothelial morphology, as well as upregulated α‐SMA and calponin (32), thus generating a composite phenotype of endothelial and smooth muscle cells.
We confirm and extend some of the above observations by showing that pulsatile external forces acting on a collagen type I matrix containing trilineage‐potential MSC can result in their differentiation into myo‐ or fibroblastic phenotype, with occasional adipocytes. We did not find evidence of osteo‐ or chondrocytic differentiation or their appropriate matrix minerals under tensioning forces. The commonest characteristics of force‐recipient cells were the extruded extracellular fibres of collagen and/or elastin, which mimics the normal process of stromal tissue remodelling that occurs in vivo. This strongly suggests that the usual phenomenon of wound healing by scar formation may be augmented by in situ conversion of MSC into fibroblastic lineages. Our laboratory, among others, has reported that bone marrow‐derived cells frequently engraft into normal tissues and organs forming fibroblasts and myofibroblasts, and that this rises considerably in wounded and inflamed situations [reviewed by (33)]. We can now suggest that the cells directly responsible for these observations are most likely to be BM‐derived MSC, as our MSC were devoid of HSC characteristics and possessed the well‐described abilities of trilineage‐potential MSC [reviewed in (34)].
Under normal conditions in vivo, cells of the connective tissues create and respond to many applied forces. Every tissue and organ has components of connective tissues, and structural fibres such as collagen and elastin permeate all areas of the body. In this study, we have shown that by applying extracellular force alone, we can instigate specific fibre production. This adds to properties that have been already shown in cells that result in a linked pathway from force application to gene expression. There are many mechanosensory motifs such as force‐regulated protein conformational changes, that readily occur in cells under applied force (35), and most normal cell types depend on physical interactions with their extracellular matrix to respond to tissue signals (3).
In conclusion, we have cloned mouse bone marrow mesenchymal stem cells and shown their ability to differentiate into adipocytes, osteoblast‐like cells and chondrocytes by classical differentiation techniques. We have subjected them to cyclical external forces and shown them to differentiate into cells that resemble myo‐ and fibroblast‐like cells. This new technique may be useful in tissue‐engineering novel synthetic living tissue for transplantation in regenerative medicine.
Acknowledgements
The authors would like to thank George Elia for his generous advice on histochemistry, and J Rao for technical assistance with cell culture.
References
- 1. Hench LL, Polak JM (2002) Third‐generation biomedical materials. Science 295, 1014–1017. [DOI] [PubMed] [Google Scholar]
- 2. Baum CL, Arpey CJ (2005). Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatol. Surg. 31, 674–686; discussion 686. [DOI] [PubMed] [Google Scholar]
- 3. Chiquet M, Gelman L, Lutz R, Maier S (2009) From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim. Biophys. Acta 1793, 911–920. [DOI] [PubMed] [Google Scholar]
- 4. Sano S, Okawa A, Nakajima A, Tahara M, Fujita K, Wada Y et al. (2006) Identification of Pip4k2beta as a mechanical stimulus responsive gene and its expression during musculoskeletal tissue healing. Cell Tissue Res. 323, 245–252. [DOI] [PubMed] [Google Scholar]
- 5. Hinz B (2006) Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 85, 175–181. [DOI] [PubMed] [Google Scholar]
- 6. Friedenstein AJ, Gorskaja JF, Kulagina NN (1976) Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 4, 267–274. [PubMed] [Google Scholar]
- 7. Lennon DP, Haynesworth SE, Young RG, Dennis JE, Caplan AI (1995) A chemically defined medium supports in vitro proliferation and maintains the osteochondral potential of rat marrow‐derived mesenchymal stem cells. Exp. Cell Res. 219, 211–222. [DOI] [PubMed] [Google Scholar]
- 8. Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM (2002) Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp. Hematol. 30, 896–904. [DOI] [PubMed] [Google Scholar]
- 9. Ellis R (2007) Miller’s Elastic Staining Protocol. IHC World; http://www.ihcworld.com/_protocols/special_stains/miller%27s_elastic_ellis.htm (accessed 5 August 2010) [Google Scholar]
- 10. Lamar Jones M (2002) Connective tissues and stains In: Bancroft JD, Gamble M, eds. Theory and Practice of Histological Techniques, 5th edn, pp. 139–162. Edinburgh: Churchill Livingstone. [Google Scholar]
- 11. Lamar Jones M (2002) Lipids In: Bancroft JD, Gamble M, eds. Theory and Practice of Histological Techniques, 5th edn, pp. 201–230. Edinburgh: Churchill Livingstone. [Google Scholar]
- 12. Churukian CJ (2002) Pigments and minerals In: Bancroft JD, Gamble M, eds. Theory and Practice of Histological Techniques, 5th edn, pp. 243–267. Edinburgh: Churchill Livingstone. [Google Scholar]
- 13. Totty BA (2002) Mucins In: Bancroft JD, Gamble M, eds. Theory and practice of histological techniques, 5th edn, pp. 163–200. Edinburgh: Churchill Livingstone. [Google Scholar]
- 14. Xu W, Zhang X, Qian H, Zhu W, Sun X, Hu J et al. (2004) Mesenchymal stem cells from adult human bone marrow differentiate into a cardiomyocyte phenotype in vitro. Exp. Biol. Med. (Maywood) 229, 623–631. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi T, Lord B, Schulze PC, Fryer RM, Sarang SS, Gullans SR et al. (2003) Ascorbic acid enhances differentiation of embryonic stem cells into cardiac myocytes. Circulation 107, 1912–1916. [DOI] [PubMed] [Google Scholar]
- 16. Safford KM, Hicok KC, Safford SD, Halvorsen YD, Wilkison WO, Gimble JM et al. (2002) Neurogenic differentiation of murine and human adipose‐derived stromal cells. Biochem. Biophys. Res. Commun. 294, 371–379. [DOI] [PubMed] [Google Scholar]
- 17. Anilkumar TV, Sarraf CE, Hunt T, Alison MR (1992) The nature of cytotoxic drug‐induced cell death in murine intestinal crypts. Br. J. Cancer 65, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eastwood M, McGrouther DA, Brown RA (1994) A culture force monitor for measurement of contraction forces generated in human dermal fibroblast cultures: evidence for cell‐matrix mechanical signalling. Biochim. Biophys. Acta 1201, 186–192. [DOI] [PubMed] [Google Scholar]
- 19. Eastwood M, Mudera VC, McGrouther DA, Brown RA (1998) Effect of precise mechanical loading on fibroblast populated collagen lattices: morphological changes. Cell Motil. Cytoskeleton 40, 13–21. [DOI] [PubMed] [Google Scholar]
- 20. Gregory CA, Prockop DJ, Spees JL (2005) Non‐hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp. Cell Res. 306, 330–335. [DOI] [PubMed] [Google Scholar]
- 21. Quirici N, Soligo D, Bossolasco P, Servida F, Lumini C, Deliliers GL (2002) Isolation of bone marrow mesenchymal stem cells by anti‐nerve growth factor receptor antibodies. Exp. Hematol. 30, 783–791. [DOI] [PubMed] [Google Scholar]
- 22. Graf T, Enver T (2009) Forcing cells to change lineages. Nature 462, 587–594. [DOI] [PubMed] [Google Scholar]
- 23. Shi Y (2009) Induced pluripotent stem cells, new tools for drug discovery and new hope for stem cell therapies. Curr. Mol. Pharmacol. 2, 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sethi KK, Yannas IV, Mudera V, Eastwood M, McFarland C, Brown RA (2002) Evidence for sequential utilization of fibronectin, vitronectin, and collagen during fibroblast‐mediated collagen contraction. Wound Repair Regen. 10, 397–408. [DOI] [PubMed] [Google Scholar]
- 25. Chen Y, Leask A, Abraham DJ, Pala D, Shiwen X, Khan K et al. (2008) Heparan sulfate‐dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 58, 577–585. [DOI] [PubMed] [Google Scholar]
- 26. Hinz B, Gabbiani G (2003) Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 14, 538–546. [DOI] [PubMed] [Google Scholar]
- 27. Sittichokechaiwut A, Edwards JH, Scutt AM, Reilly GC (2010) Short bouts of mechanical loading are as effective as dexamethasone at inducing matrix production by human bone marrow Mesenchymal stem cell. Eur. Cell Mater. 20, 45–57. [DOI] [PubMed] [Google Scholar]
- 28. Carracedo S, Lu N, Popova SN, Jonsson R, Eckes B, Gullberg D (2010) The fibroblast integrin alpha11beta1 is induced in a mechanosensitive manner involving activin A and regulates myofibroblast differentiation. J. Biol. Chem. 285, 10434–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Follonier L, Schaub S, Meister JJ, Hinz B (2008) Myofibroblast communication is controlled by intercellular mechanical coupling. J. Cell Sci. 121, 3305–3316. [DOI] [PubMed] [Google Scholar]
- 30. Wall ME, Otey C, Qi J, Banes AJ (2007) Connexin 43 is localized with actin in tenocytes. Cell Motil. Cytoskeleton 64, 121–130. [DOI] [PubMed] [Google Scholar]
- 31. Henshaw DR, Attia E, Bhargava M, Hannafin JA (2006) Canine ACL fibroblast integrin expression and cell alignment in response to cyclic tensile strain in three‐dimensional collagen gels. J. Orthop. Res. 24, 481–490. [DOI] [PubMed] [Google Scholar]
- 32. O’Cearbhaill ED, Punchard MA, Murphy M, Barry FP, McHugh PE, Barron V (2008) Response of mesenchymal stem cells to the biomechanical environment of the endothelium on a flexible tubular silicone substrate. Biomaterials 29, 1610–1619. [DOI] [PubMed] [Google Scholar]
- 33. Karp JM, Leng Teo GS (2009) Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 4, 206–216. [DOI] [PubMed] [Google Scholar]
- 34. Otto WR, Rao J (2004) Tomorrow’s skeleton staff: mesenchymal stem cells and the repair of bone and cartilage. Cell Prolif. 37, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vogel V (2006) Mechanotransduction involving multimodular proteins: converting force into biochemical signals. Annu. Rev. Biophys. Biomol. Struct. 35, 459–488. [DOI] [PubMed] [Google Scholar]