

Abstract
Objectives: The key to fertility in adult males is production of mature spermatogenic cells. Spermatogonial stem cells (SSC) have the dual capacity of self‐renewal and of differentiation into mature sperm. SSC transplantation may provide potential treatment for specific male infertilities. However, until now, there has been no evidence of offspring produced by transplantation of adult SSC line cells in humans or other mammals.
Materials and methods: A new line of SSCs from adult C57BL/6 mouse was established by using magnetic‐activated cell sorting. The cell line was characterized by immunocytochemistry, karyotype analysis and telomeric repeat amplification protocol (TRAP) telomerase activity assay. Spermatogenic function was examined by allograft into germ cell‐ablated recipient mice.
Results: For more than 14 months with more than 65 maintenance passages, the cell line showed a normal karyotype (40, XY) and high telomerase activity. It represented a Thy‐1+, Oct4+, SSEA‐1‐, c‐kit‐ (99 ± 1%) cell subpopulation. We cryopreserved these SSCs and successfully produced normal offspring after transplanting them into testes of busulphan‐sterilized mice.
Conclusions: We established and long‐term maintained an adult SSC line with normal spermatogenic function, without the need of genetic modification; thus, this study provides a model system for basic research and clinical application.
Introduction
Production of normal, mature sperms in adequate number is the key to male fertility. Spermatogonial stem cells (SSC) maintain lifetime sperm production by self‐renewal and sequential differentiation, a process called spermatogenesis. Impairment of spermatogenesis, either in the SSC itself or in its niche, may cause male infertility. In some cases of infertility (caused by spermatogenic arrest) SSCs are normal or subnormal whereas specific steps in the subsequent mitotic and/or meiotic divisions and differentiation are blocked. Therefore, it is of great importance to study SSC for mechanisms of germ‐line development and possible therapy for infertility. SSC in vitro differentiation and/or transplantation may provide potential treatment for male infertility in these clinical situations. In addition cancer patients, especially prepubertal patients, whose germ cells would be killed in gonadotoxic treatment, can benefit from SSC transplantation; moreover, genetic manipulation of SSC may be applied to treat certain hereditary diseases. Further research requires large numbers of SSCs, but SSCs are extremely rare: 1 in 3000–4000 cells per adult mouse testis (1), 1 in 500 cells in adult rat testis (2), and 1 in 5000 cells in adult human testis (3). Establishment of pure or significantly enriched populations of SSCs (that is, an SSC line that can be stably maintained and efficiently amplified in vitro) is a critical initial step for subsequent applications.
Since long‐term culture of primary SSCs is difficult, SSC lines are generally established using immortalized primary cells from testes of 6‐day‐old mice by genetic modification. It has been reported that an immortalized SSC line by transfection of SV40 large T gene has been established, but this cell line, with a c‐kit‐phenotype, did not differentiate into normal spermatozoa in vivo or in vitro (4, 5). Feng et al. successfully used pLXSN viral particles to transfect mTERT cDNA into SSCs, and established a neonatal mouse SSC line. These c‐kit+ cells could differentiate into round spermatids, but no offspring were reported (6, 7). Other groups long‐term cultured SSCs, but most of them used viral vector‐transfected primary cells or cells from transgenic mice as their starting materials (8, 9). These methods cannot be used for establishment of human SSC lines in future clinical situations, because viral‐based gene transfer poses significant potential risks in the clinic. In most studies, donor cells are derived from neonatal mice in which the highest proportion of total testicular cells are SSCs (10, 11). However in contrast, almost all human counterparts are adult males, as infertility problems become known only in adults. It is thus essential to investigate mechanisms and treatments for infertility using cells derived from human adults. Nevertheless, related methods could help treat prepubertal patients in specific clinical situations. For ethical and safety considerations, human cell lines for the clinic must avoid any genetically modified material. Thus, use of SSCs from adult individuals and successful establishment of a cell line of normal adult SSCs have become a potential alternative. In sexually mature mice, SSCs are present in very small numbers in the fully spermatogenic seminiferous epithelium, as described above and their purification and long‐term culture from adult testes appears to be particularly difficult. To date, there is no evidence to support the production of mice through transplantation of cells of an adult SSC line.
In this proof‐of‐principle study, we used SSCs derived from adult C57BL/6 mice to establish and maintain an adult mouse SSC cell line that does not contain any genetically engineered or artificially modified ingredients. A stem cell‐specific surface antigen (Thy‐1) was used for positive selection, then we characterized these cells. To examine spermatogenic function we transplanted the cells, after cryopreservation, into chemotherapeutically sterilized recipients. Results indicated that these cells underwent normal spermatogenesis in recipient mice, and produced heterozygous progeny. This has important implications for establishment of vector‐free SSC lines from adult humans, and for further applications.
Materials and methods
Animals
C57BL/6 (donor male), CD‐1 (mating female), and C57BL/6 × CD‐1 F1 (recipient male) Mus musculus, 6–8 weeks of age, were used in this study. F1 mice were intraperitoneally injected with busulphan (40 mg/kg) for preparation of recipients and controls (12). All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Shanghai, and were conducted in accordance with the National Research Council Guide for Care and Use of Laboratory Animals.
Isolation and culture of SSCs
Testes of 8‐week‐old C57BL/6 mice were collected and decapsulated and SSCs were isolated essentially according to the methods of Nagano et al. and Wu et al. (two‐step enzymatic digestion) (13, 14, 15, 16). Briefly, decapsulated testes were incubated in approximately 10 volumes of Hanks’ balanced salt solution without calcium or magnesium (HBSS), containing 1 mg/ml collagenase (Type IV, Sigma, St. Louis, MO, USA) at 37 °C with gentle agitation for 15 min. After dispersion of seminiferous tubules, these tubules were then washed two to four times in 10 volumes of HBSS followed by incubation at 37 °C for 5 min in HBSS containing 1 mm EDTA and 0.20% trypsin. When most of the cells were dispersed, trypsin was neutralized by adding 20% volume of foetal bovine serum (FBS; Life Technologies Inc., Gaithersburg, MD, USA). The suspension was centrifuged at 300 g for 5 min, and the supernatant was carefully aspirated. Cell pellets were resuspended and clumps of cells were removed by passing the suspension through a 70‐µm nylon cell strainer. Cells enriched for SSCs were prepared by magnetic‐activated cell sorting (MACS) with magnetic microbeads conjugated to anti‐Thy‐1 antibody (BD Biosciences, Franklin Lakes, NJ, USA), following the manufacturer's instructions (17). Finally, cells were resuspended in SSC medium (see below) at final concentration of 1 × 104 cells/ml. The culture system for SSCs consisted of SSC medium and mitotically inactivated STO (ATCC, mouse SIM embryonic fibroblast cell line) cell feeders (~5 × 104 cells/cm2). STO cells were cultured in Dulbecco's modified Eagle's medium with high glucose (Life Technologies), supplemented with 1% non‐essential amino acids (Life Technologies), 10% FBS, 2 mm glutamine (Sigma), 30 mg/l penicillin and 75 mg/l streptomycin. Cells were treated with 10 µg/ml mitomycin C (Sigma) for 2–3 h for mitotic inactivation. Mitomycin C‐treated STO cells were washed with phosphate‐buffered saline (PBS), and were plated in 0.2% (w/w) gelatin‐coated wells. The culture medium for SSCs consisted of minimum essential medium alpha, 10% FBS, 1 mm sodium pyruvate, 1 mm non‐essential amino acids, 2 mm l‐glutamine, 0.1 mmβ‐mercaptoethanol (Sigma), 10 ng/ml leukaemia inhibitory factor (LIF; Santa Cruz Biotechnology, Santa Cruz, CA, USA), 20 µg/ml transferrin, 5 µg/ml insulin 60 µm putrescine, 10 ng/ml mouse epidermal growth factor (16, 18, 19), 30 mg/l penicillin, and 75 mg/l streptomycin. SSCs were cultured on STO feeders in 24‐well plates with 500 µl culture medium per well. Cells were refreshed every 2–3 days and subcultured at 1 : 2 ratio by enzymatic digestion every 5–7 days. All cultures were maintained at 37 °C in 5% CO2 (20).
Cryopreservation and thawing
After trypsinization, cell suspensions were gently mixed with cryoprotective solution, SSC culture medium with 10% dimethyl sulphoxide, in 2‐ml freezing vials which were then placed in a container and stored at 4 °C for 1 h, –20 °C for 2 h, overnight at –80 °C, and finally preserved in liquid nitrogen (–196 °C). For rapid thawing after long‐term cryopreservation, freezing vials were taken from liquid nitrogen and immediately incubated at 37 °C until ice crystals disappeared; then cells were cultured under the same conditions as described above (21).
Immunofluorescence
Cultured SSCs were fixed with 4% paraformaldehyde (15 min, room temperature). After fixation SSCs were incubated in blocking solution (10% normal goat or rabbit serum in PBS, 60 min, room temperature), followed by rinsing and overnight incubation with appropriate primary antibodies at 4 °C: mouse monoclonal anti‐SSEA‐1 (1 : 100 dilution; Chemicon, Billerica, MA, USA), rabbit polyclonal anti‐Oct4 (1 : 100 dilution; Chemicon), or mouse monoclonal anti‐c‐kit (1 : 100 dilution; Chemicon). Mouse or rabbit serum were paralleled as controls. After extensive rinsing, SSCs were incubated in the dark with fluorescein isothiocyanate‐conjugated secondary antibody (goat anti‐rabbit immunoglobulin G, 1 : 200 dilution, or rabbit anti‐mouse immunoglobulin G), then rinsed, mounted in 4′,6‐diamidino‐2‐phenylindole (DAPI)‐containing medium, and visualized using a Nikon Eclipse E600 microscope equipped with Nikon DXM 1200 digital camera (Tokyo, Japan), with fluorescein optics for fluorescein isothiocyanate and ultraviolet optics for DAPI (16, 17).
Karyotype analysis
Three days after passaging, cells were treated with SSC medium containing colchicine (20 ng/ml) for 4 h, hypotonically treated with 40 mm KCl for 30 min, fixed in methanol–acetic acid (3 : 1), air‐dried, stained with DAPI, visualized and photographed as described above. More than 30 metaphase cells were analysed (22).
Flow cytometry assay
For flow cytometry assay, approximately 2 × 106 cultured SSCs were harvested by trypsinization; testicular cells dissociated from mature testes were used as reference. Cells were fixed in 70% ethanol, incubated at 37 °C for 15 min in PBS containing RNase A (200 µg/ml) plus propidium iodide (20 µg/ml) for DNA staining, and were analysed using a Coulter Elite ESP flow cytometer with software (Beckman Coulter, Fullerton, CA, USA) (23).
TRAP telomerase activity assay
Telomerase activity was tested using the TRAP‐cyber green stain Telomerase Detection Kit (GENMED, Boston, MA, USA) following the manufacturer's instructions (24).
SSC transplantation
For testis microinjection, cells of the adult C57BL/6 SSC line were trypsinized and resuspended in PBS. Recipient mice were anaesthetized by intraperitoneal injection of barbiturate solution (45 mg/kg body weight). Approximately 10 µl of single‐cell suspension containing 2 × 104 cells was introduced into one testis of a 10‐ to 12‐week‐old busulphan‐treated recipient mouse (25). Eight recipient and six control mice were included in the transplantation experiment. Microinjection was performed using the method for efferent ducts of each recipient testis (26, 27).
Histology
For histology, testes of recipient and control mice were fixed in Bouin's fixative at 4 °C overnight, and then were dehydrated through ethanol gradients, vitrificated in xylene, and embedded in paraffin wax. Six‐micrometre sections were mounted on slides. Before staining, sections were dewaxed through xylene and reverse gradients of ethanol. All sections were stained with haematoxylin, and visualized as described above (28).
Intersimple sequence repeat DNA fingerprinting
Genomic DNA was extracted from tail‐tips or cultured SSCs. DNA samples from mating females, recipient males, donor cells and progeny, respectively, were analysed by intersimple sequence repeat (ISSR). The following specific primers were used: (CA)8RY (CA)8RG (AAC)6Y, and (AGC)4Y primers (R = purine; Y = pyrimidine). A 25‐µl reaction system consisted of 50–100 ng genomic DNA, 2.5 µl 10× polymerase chain reaction (PCR) buffer (15 mm MgCl2), 2% formamide, 0.2 mm dNTP, 1 µm primer mix (0.5 µm each), and 1 U Taq polymerase (Promega, Madison, WI, USA). PCRs were performed as follows: 94 °C for 4 min (94 °C for 30 s, 41 °C for 45 s, 72 °C for 2 min) × 30 cycles, and 72 °C for 7 min, executed in PerkinElmer Model 9600 DNA thermal cycler (PerkinElmer Cetus Instruments, Norwalk, CT, USA). PCR products were analysed by 8% nondenaturing polyacrylamide gel electrophoresis (Bio‐Rad, Hercules, CA, USA), and were visualized by silver staining. Gel images were captured using a digital camera (29, 30, 31).
Results
Establishment of a new cell line of SSCs from adult mouse
To obtain a model system for establishment of an SSC line for adult humans, we established a new cell line using SSCs isolated from testes of adult C57BL/6 mice. Because SSCs are comparatively rare in adult testes, we first enriched them by MACS using anti‐Thy‐1 antibody after two‐step enzymatic digestion of adult testes. Enriched SSCs were then maintained on STO feeder layers in SSC culture medium (see Materials and methods), which is different from the medium for neonatal mouse SSC culture (11). After 24 h culture, SSCs showed typical morphology of type A spermatogonia, such as large round or oval cell bodies, spherical nuclei which stained lightly, sparse cytoplasm, and high nucleus/cytoplasm ratio (Fig. 1a) (9). Initially, the SSCs proliferated slowly but following 8–10 passages, they expanded into clusters. Clustered cells further proliferated to form colonies that had unique morphology – compact in the centre but loosely arranged in peripheral regions and spreading from loosely adhered outer regions to form satellite clusters. Figure 1(b) shows cells formed into types of colony surrounded by spread out clusters or single cells, after 4 months culture. As culture time was prolonged, SSCs formed clumps with tight intercellular junctions (or blurred cell boundaries) resulting in convex masses of cells with clear borders, which differ from those of embryonic stem cell colonies (Fig. 1c). These SSCs were stably cultured for more than 14 months showing stem cell activity thus far, and have been passaged more than 65 times (5–7 days per passage) retaining characteristic morphology of the cell line. Moreover, these SSCs have successfully undergone cryopreservation in liquid nitrogen for 1 week, 1 month and 2 months, respectively, and have been successfully used to re‐establish cultures after thawing.
Figure 1.
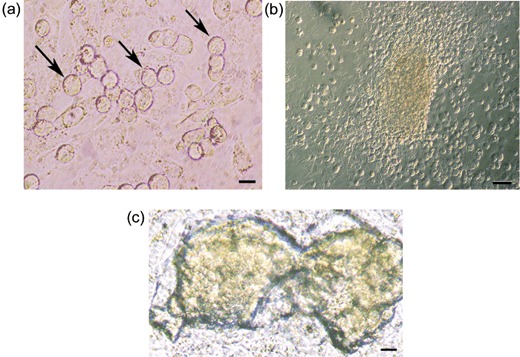
Establishment of a spermatogonial stem cell (SSC) line from adult mouse. (a) Morphology of adult mouse SSCs (arrows) cultured on mitomycin C‐treated STO for 24 h, after purification by magnetic‐activated cell sorting with anti‐Thy‐1‐conjugated magnetic microbeads. (b) Representative morphology of SSC clusters and colonies after 4 months culture. (c) Representative morphology of SSC clumps after more than 1 year of culture. Scale bars: 10 µm in (a), 50 µm in (b) and 25 µm in (c).
Characterization of the cell line of adult mouse SSCs
To characterize this cell line, we first checked for expression of Oct4 (a germ cell‐specific transcriptional factor and multifunctional stem cell marker), SSEA‐1 (embryonic stem cell and primordial germ cell marker), and c‐kit (differentiated spermatogonia marker) (32, 33, 34, 35, 36). Immunocytochemistry showed that these cells expressed Oct4 (Fig. 2a), in contrast, they were completely negative for SSEA‐1. Most cells were also negative for c‐kit with about 1 ± 1% of them expressing it (Fig. 2b). c‐kit + cells were detected by c‐kit assay 1 day after passaging, before clump formation. Results indicated that the cells were likely to have been derived from type A spermatogonia, and the majority of them represented an undifferentiated SSC phenotype (17, 37). Next, karyotype analysis was performed and diploid metaphase of 40 chromosomes was observed for most cells (Fig. 2c,d), suggesting that no discernable cytogenetic abnormalities occurred during long‐term maintenance. Furthermore, flow cytometry analysis showed diploid and tetraploid peaks (Fig. 2e), indicating that these cells proliferated normally. In addition, the cell line showed high telomerase activity (Fig. 2f).
Figure 2.
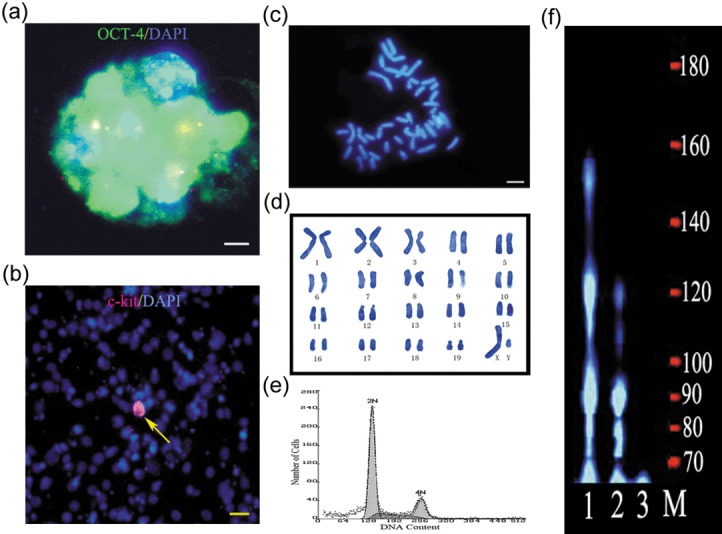
Characteristics of the adult spermatogonial stem cell (SSC) line. (a) Characteristic example of Oct4 expression in an SSC clump. Green indicates Oct4 immunofluorescence, blue indicates DAPI immunofluorescence. (b) Most SSCs were negative for c‐kit (99 ± 1%), the red channel represents c‐kit immunofluorescence and the blue channel represents DAPI immunofluorescence. (c, d) Cytogenetic analysis by DAPI staining showed that these SSCs had a normal karyotype (40, XY). (e) Flow cytometry analysis of SSCs, DNA content was measured to show cell ploidy. Adult mouse testes were used as a reference (data not shown). (f) TRAP telomerase activity assay showed that the cells had high telomerase activity. M, DNA marker; lane 1, F9 cells (positive control); lane 2, SSCs; lane 3, STO (negative control). Data shown are representative of four experiments. Scale bars: 10 µm in (a), 25 µm in (b) and 6 µm in (c).
Mice generated from cryopreserved adult SSCs of the line after transplantation
To examine spermatogenic function of this cell line, cells that had been passaged more than 40 times and had undergone cryopreservation were transplanted into busulphan‐sterilized recipients. Of the eight recipients, one died the next day for an unknown reason, but the other seven were physiologically and behaviourally normal. Six and 12 weeks after transplantation, testis histology were examined, respectively. No germ cell stages were identified in controls (Fig. 3a), but in contrast in the transplanted group, we found that SSCs were located on the basal membrane, differentiating spermatids were present in the epithelium (Fig. 3b), and mature spermatozoa were released in the lumen of some seminiferous tubules (Fig. 3c). Forty‐five days after transplantation, five recipient mice were mated with female CD‐1 mice to determine whether they restored fertility. After transplantation for 86–106 days, progeny were produced from five of seven recipients, and the offspring were all black (Fig. 3d), suggesting that they were not host derived (see Discussion). Therefore, all living recipients had restored fertility. There were no abnormalities in the offspring, and they also were fertile. However, control mice (with busulphan‐treated testes but without cell transplantation) did not produce offspring. Subsequently, histology of testes from recipient and control mice was performed. ISSR DNA fingerprinting was used to further confirm genetic parenthood (29). Because of the genetic comparability of inbred mice, we screened 24 different primers or primer mixtures, and only primer 5‐(CA)8GC‐3 and primer 5‐(AAC)6T‐3 mixtures resulted in specific bands. We observed that there was a specific band (Fig. 3e, arrow) in both the offspring and donor cell lanes, and there was no corresponding band in the mating female or recipient lane. This result indicated that these offspring were not generated from the recipients themselves. As others have reported (8), we were also not successful in implanting SSCs into all seminiferous tubules of mouse testes as evidenced by the observation that some seminiferous tubules were completely devoid of spermatogenic cells similar to all seminiferous tubules in control animals (Fig. 4). This result also suggests that the offspring did not come from spermatogenic cells of the recipients themselves, but from the donor transplanted cells.
Figure 3.
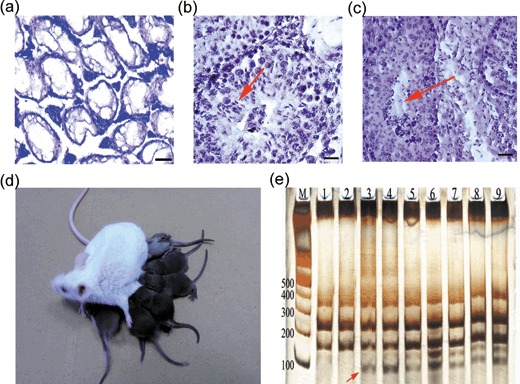
Progenies generated from transplantation of adult spermatogonial stem cells (SSC) after cryopreservation. (a–c) Testis histology of busulphan‐treated recipient mice. (a) Control testes without cell transplantation (note the absence of spermatogenesis). (b) Section of the recipient testes at 6 weeks after transplantation of adult SSCs. Note active spermatogenesis and long spermatids in seminiferous tubules (arrow). (c) Section of recipient testis 12 weeks after transplantation treatment. Sperms are seen (arrow). (d) Progenies from a C57BL/6 × CD‐1 F1 recipient mouse transplanted with C57BL/6 adult SSCs that were passaged more than 40 times and cryopreserved for 1 week, and their mother. The mother is a pure‐blood CD‐1 mouse with pure white coat, and all the offspring are black. (e) ISSR DNA fingerprinting revealed kinship of these mice. M, marker; lane 1, mating female; lane 2, recipient male; lane 3, SSC line; lanes 4–9, offspring. Discriminating bands are marked with arrows. Data shown are representative of three experiments. Scale bars: 100 µm in (a), 25 µm in (b) and 40 µm in (c).
Figure 4.
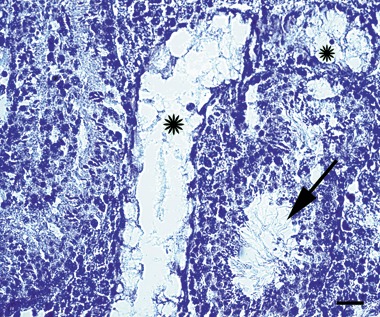
Seminiferous tubules displayed different morphology in recipient testes after spermatogonial stem cell (SSC) transplantation. Some seminiferous tubules show obvious spermatogenesis while others did not (stars). Scale bar: 40 µm.
Discussion
Male infertility is a common condition. According to World Health Organization statistics, about 15% of childbearing age couples are infertile, 50% of which are a result of male infertility. Azoospermia and oligospermia are among the most common symptoms of male fertility disorders in which normal spermatogenesis is impaired. Although modern in vitro fertilization techniques, especially recent developments in intracytoplasmic sperm injection, help to treat many patients, spermatogenic disorder still remains a challenge.
Of note, specific cases of infertility are caused by spermatogenic arrest (that is, SSCs are normal or subnormal where critical step(s) in spermatogenesis may be dysfunctional; for example, meiosis defect) and certain cases of Yq microdeletions (38, 39). This sort of male infertility, including obstructive azoospermia, represents the only ones that can be circumvented by assisted reproductive technology. In specific clinical situations, normal SSCs may be sorted, cultured, differentiated in vitro, transplanted or utilized in in vitro fertilization. Long‐term maintenance and autologous transplantation of SSCs shows particular benefit for cancer patients, especially prepubertal patients undergoing germ cell‐ablative therapies and for whom cryopreservation of mature sperm is unfeasible (40). As compared to in vitro fertilization, autologous SSC transplantation allows natural contraception but on the other hand, maintenance of SSCs keeps an infinite reservoir of germ cell supply, as compared to mature sperm cryobanking. Moreover, SSCs could be genetically modified to treat some otherwise inevitable hereditary diseases (41) and besides its potential applications in reproductive medicine, SSC also pose significant implications for basic research of gamete development. All these utilizations call for stable, safe and scalable sources of SSC. However, limited donors and scarcity of SSC in situ hinder its transplantation and/or differentiation in vitro thus, a well‐characterized, spermatogenically functional SSC cell line will be an attractive source of SSCs for research and application.
To create a mouse model for a human adult SSC cell line, we for the first time established a ‘natural’ cell line using SSCs isolated from testes of adult C57BL/6 mice with no genetic manipulation. Such cell lines can better represent the genuine in vitro state of SSCs in the process of self‐renewal, thus eliminating major uncertainties for further study. Although absence of molecular markers handicapped identifying the parental contributions of offspring generated after transplantation, we used another method to identify the kinships. First, difference in coat colour between SSC donor mice and recipient mice helped to identify offspring of the transplanted SSCs. Second, conventional DNA fingerprinting was performed for the non‐inbred species. In addition, ISSR DNA fingerprinting was performed for the inbred species due to great variability of the simple sequence repeat by mice. Since tested mice were generated by inbreeding (except for CD‐1 mice), conventional DNA fingerprinting cannot distinguish the breeds. As the great variability of SSRs allows high‐resolution fingerprinting, inner genetic relationship among inbred mice and highly similar genotypes can be distinguished. For example, SSR (GT)n used in this study had more than 10 000 copies in the mouse genome (29).
The c‐locus of the mouse is thought to encode a tyrosinase responsible for melanin synthesis in skin and eye melanocytes. The wild‐type allele (C) is dominant over all other alleles, and provides full tyrosinase activity. In contrast, albino mice (c/c) posses a full complement of pigment cells with no tyrosinase activity (40, 42). In this study, SSCs were isolated from pure‐blood C57BL/6 mice with pure black coat whose genotypes are homozygous dominant at the c‐locus (C/C). Mating females were pure‐blood CD‐1 mice with pure white coats, which is homozygous recessive trait at the c‐locus (c/c). Recipient mice were C57BL/6 × CD‐1 hybrids whose coat colour was black with heterozygous C/c at the c‐locus. If the offspring were derived from recipient mice, half of them would appear white with c/c genotype at the c‐locus and if offspring were derived from adult C57BL/6 SSCs, all of them would be black with a C/c genotype. In the event, none of the offspring of SSCs were white, indicating that these offspring were not generated from the recipients themselves. To confirm this, we used ISSR to genotype the offspring. Results were consistent with revealed differences in coat colour.
The cell line established in this study was characterized as Thy‐1+, Oct4+, SSEA‐1‐, and c‐kit‐subpopulation. Thy‐1 (CD90) belongs to the immunoglobulin superfamily, and is expressed on mouse haematopoietic stem cells (43), as well as on cultured embryonic stem cells (44); Thy‐1 is also used as an identification marker of SSCs (17). Oct4 is a unique marker of multipotent stem cells and was found to be expressed in undifferentiated spermatogonia (32, 33, 45), SSCs should therefore positively express Oct4. All cells were SSEA‐1 negative, SSEA‐1 being a specific marker of undifferentiated embryonic stem cells (44) used for primordial germ cell identification (34). The pro‐oncogene c‐kit encodes a cell‐surface receptor of the tyrosine kinase family. Together with its ligand, stem cell factor, activation and expression of c‐kit play a crucial role in maintenance and survival of embryonic stem and primordial germ cells (46, 47) and many studies have used c‐kit as a specific marker for differentiating or differentiated SSCs (35, 36, 48). In our cell line, only a very small proportion of SSCs expressed c‐kit. From these results, we suggest that sorting and culture conditions selected and/or maintained a unique cell population phenotypically different from pluripotent embryonic stem cells or primordial germ cells, mainly derived from undifferentiated type A spermatogonia. Still, we cannot exclude the possibility that these cells, which were artefacts that arose from spontaneous transformation in vitro, do not represent any specific population in vivo. Further stage‐defined expression profiling would help to identify the exact nature of the cells.
After long‐term culture and cryopreservation, this cell line did not show any discernable cytogenetic abnormalities, as revealed by karyotype analysis. Chromosome aberrations frequently occur during long‐term maintenance of embryonic stem cells as well as series of other cell lines, which hinders their clinical use. It has been suggested that progressive adaptation of self‐renewing cells to their culture conditions is associated with chromosomal instability (49). It remains to be examined whether our cell line can retain genomic integrity during further prolonged culture. The cultured cells exhibited (as expected from an actively proliferating diploid cell line) two peaks corresponding to diploid and tetraploid DNA, respectively and TRAP assay revealed high telomerase activity, a feature shared by germ cells and stem/progenitor cells, as well as immortalized cell lines (50). Spermatogenic function remained normal after maintenance and cryopreservation, as tested by transplantation. Although it was shown that donor‐derived offspring were successfully produced, we could not fully trace in vivo fate of these cells after transplantation due to lack of available in vivo markers.
Unlike previous studies, we did not use basic fibroblast growth factor and glial cell line‐derived neurotrophic factor (GDNF) to culture SSCs. Although GDNF is essential for self‐renewal of SSCs in vivo (51), we found that proliferation of C57BL/6 mouse adult SSCs in vitro was GDNF independent but relied on LIF, which was consistent with other studies (52).
In conclusion, our results demonstrate that culture conditions for SSCs in the present study can support long‐term growth of adult mouse SSCs, a procedure devoid of genetic modification. The established cell line was phenotypically similar to undifferentiated SSC. It retained cytogenetic stability and could restore spermatogenic function after transplantation into germ cell‐ablated recipients. This model system may apply to other mammals, including humans, in which mechanisms of spermatogenesis and treatment of infertility could be studied. Up to now, such a human model has yet to be established, and for ethical and safety considerations, subsequent clinical translation should be very prudently evaluated. Our future studies aim to investigate the differentiation potential of this cell line in vitro, and to develop a serum‐free, feeder layer‐free culture system for SSC maintenance and expansion.
Acknowledgements
This work was supported by Key Program of National Natural Scientific Foundation of China (No. 30630012, to J.W.), Shanghai Pujiang Program, China (No. 06PJ14058, to J.W.), and Shanghai Leading Academic Discipline Project (No. B205).
References
- 1. Tegelenbosch RA, De Rooij DG (1993) A quantitative study of spermatogonial multiplication and stem cell renewal in the C3H/101, F1 hybrid mouse. Mutat. Res. 290, 193–200. [DOI] [PubMed] [Google Scholar]
- 2. Huckins C (1971) The spermatogonial stem cell population in adult rats. I. Their morphology, proliferation and maturation. Anat. Rec. 169, 533–557. [DOI] [PubMed] [Google Scholar]
- 3. Desjardins C, Ewing LL (1993) Cell and Molecular Biology of the Testis. New York: Oxford University Press. [Google Scholar]
- 4. Hofmann MC, Narisawa S, Hess RA, Millan JL (1992) Immortalization of germ cells and somatic testicular cells using the SV40 large T antigen. Exp. Cell Res. 201, 417–435. [DOI] [PubMed] [Google Scholar]
- 5. Bertrand S, Patrick V, Claude D, Denise P (1991) Mammalian cell lines can be efficiently established in vitro upon expression of the SV40 large T antigen driven by a promoter sequence derived from the human vimentin gene. Biol. Cell 73, 7–14. [DOI] [PubMed] [Google Scholar]
- 6. Feng LX, Chen YL, Dettin L, Reijo Pera RA, Herr JC, Goldberg E, Dym M (2002) Generation and in vitro differentiation of a spermatogonial cell line. Science 297, 392–395. [DOI] [PubMed] [Google Scholar]
- 7. Hofmann MC, Braydich‐Stollea L, Dettinb L, Johnsona E, Dym M (2005) Immortalization of mouse germ line stem cells. Stem Cells 23, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takehashi M, Kanatsu‐Shinohara M, Inoue K, Ogonuki N, Miki H, Toyokuni S, Ogura A, Shinohara T (2007) Adenovirus‐mediated gene delivery into mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 104, 2596–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanatsu‐Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T (2003) Long‐term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 69, 612–616. [DOI] [PubMed] [Google Scholar]
- 10. Hasthorpe S (2003) Long‐term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 68, 1354–1360. [DOI] [PubMed] [Google Scholar]
- 11. Kanatsu‐Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, Baba S, Kato T, Kazuki Y, Toyokuni S (2004) Generation of pluripotent stem cells from neonatal mouse testis. Cell 119, 1001–1012. [DOI] [PubMed] [Google Scholar]
- 12. Brinster CJ, Ryu B‐Y, Avarbock MR, Karagenc L, Brinster RL, Orwig KE (2003) Restoration of fertility by germ cell, transplantation requires effective recipient preparation. Biol. Reprod. 69, 412–420. [DOI] [PubMed] [Google Scholar]
- 13. Nagano M, Avarbock MR, Leonida EB, Brinster CJ, Brinster RL (1998) Culture of mouse spermatogonial stem cells. Tissue Cell 30, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu J, Jester WF, Laslett AL, Meinhardt A, Orth JM (2003) Expression of a novel factor, short‐type PB‐cadherin, in Sertoli cells and spermatogenic stem cells of the neonatal rat testis. J. Endocrinol. 176, 381–391. [DOI] [PubMed] [Google Scholar]
- 15. Wu J, Jester WF, Orth JM (2005) Short‐type PB‐cadherin promotes survival of gonocytes and activates JAK‐STAT signalling. Dev Biol. 284, 437–450. [DOI] [PubMed] [Google Scholar]
- 16. Wu J, Zhang Y, Tian GG, Zhou K, Lee MC, Yu Q, Yuan Z (2008) Short‐type PB‐cadherin promotes self‐renewal of spermatogonial stem cells via multiple signaling pathways. Cell. Signal. 20, 1052–1060. [DOI] [PubMed] [Google Scholar]
- 17. Kubota H, Avarbock MR, Brinster RL (2003) Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc. Natl. Acad. Sci. USA 100, 6487–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anjamrooz SH, Movahedin M, Tiraihi T, Mowla SJ (2006) In vitro effects of epidermal growth factor, follicle stimulating hormone and testosterone on mouse spermatogonial cell colony formation. Reprod. Fertil. Dev. 18, 709–720. [DOI] [PubMed] [Google Scholar]
- 19. Kubota H, Avarbock MR, Brinster RL (2004) Growth factors essential for self‐renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 101, 16489–16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dym M, Dirami G, Ruttimann JM, Cudicni CB, Pursel VG, Ravindranath N (1998) Spermatogonial stem cell culture. Biol. Reprod. 58, 26. [DOI] [PubMed] [Google Scholar]
- 21. Izadyar F, Matthijs‐Rijsenbit JJ, Den Ouden K, Creemers LB, Woelders H, De Rooij DG (2002) Development of a cryopreservation protocol for type A spermatogonia. J. Androl. 23, 537–545. [PubMed] [Google Scholar]
- 22. Amit M, Carpenter MK, Inokuma MS, Chiu CP, Harris CP, Waknitz MA, Itskovitz‐Eldor J, Thomson JA (2000) Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev. Biol. 227, 271–278. [DOI] [PubMed] [Google Scholar]
- 23. Hong Y, Liu T, Zhao H, Xu H, Wang W, Liu R, Chen T, Deng J, Gui J (2004) Establishment of a normal medakafish spermatogonial cell line capable of sperm production in vitro . Proc. Natl. Acad. Sci. USA 101, 8011–8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomson JA, Itskovitz‐Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- 25. Ogawa T, Dobrinski I, Avarbock MR, Brinster RL (1999) Xenogeneic spermatogenesis following transplantation of hamster germ cells to mouse testes. Biol. Reprod. 60, 515–521. [DOI] [PubMed] [Google Scholar]
- 26. Brinster RL, Zimmermann JW (1994) Spermatogenesis following male germ‐cell transplantation. Proc. Natl. Acad. Sci. USA 91, 11298–11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brinster RL (2002) Germline stem cell transplantation and transgenesis. Science 296, 2174–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagano M, Brinster RL (1998) Spermatogonial transplantation and reconstitution of donor cell spermatogenesis in recipient mice. APMIS 106, 47–55. [DOI] [PubMed] [Google Scholar]
- 29. Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)‐anchored polymerase chain reaction amplification. Genomics 20, 176–183. [DOI] [PubMed] [Google Scholar]
- 30. Julia MB, Nathaniel G, Pierre L (2003) Measurement of genomic instability in preleukemic P190BCR/ABL transgenic mice using inter‐simple sequence repeat polymerase chain reaction. Cancer Res. 63, 4895–4898. [PubMed] [Google Scholar]
- 31. Fernando B, Mónica Z, Mónica F, Marcia RC, Susan EA, Joe MA, Julien L, Gabriel S, Ellen RR, Claudio JC (2002) Application of inter‐simple sequence repeat pcr to mouse models: assessment of genetic alterations in carcinogenesis. Genes Chromosomes Cancer 35, 299–310. [DOI] [PubMed] [Google Scholar]
- 32. Pesce M, Schöler HR (2001) Oct‐4: gatekeeper in the beginning of mammalian development. Stem Cells 19, 271–278. [DOI] [PubMed] [Google Scholar]
- 33. Schöler HR, Ruppert S, Suzuki N, Chowdhury K, Gruss P (1990) New type of POU domain in germ line‐specific protein Oct‐4. Nature 344, 435–439. [DOI] [PubMed] [Google Scholar]
- 34. Solter D, Knowles BB (1978) Monoclonal antibody defining a stage‐specific mouse embryonic antigen (SSEA‐1). Proc. Natl. Acad. Sci. USA 75, 5565–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rossi P, Sette C, Dolci S, Geremia R (2000) Role of c‐kit in mammalian spermatogenesis. J. Endocrinol. Invest. 23, 609–615. [DOI] [PubMed] [Google Scholar]
- 36. Yoshinaga K, Nishikawa S, Ogawa M, Hayashi S, Kunisada T, Fujimoto T, Nishikawa S (1991) Role of c‐kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c‐kit expression and function. Development 113, 689–699. [DOI] [PubMed] [Google Scholar]
- 37. Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL (2006) Identifying genes important for spermatogonial stem cell self‐renewal and survival. Proc. Natl. Acad. Sci. USA 103, 9524–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaganti RS, German J (1979) Human male infertility, probably genetically determined, due to defective meiosis and spermatogenic arrest. Am. J. Hum. Genet. 31, 634–641. [PMC free article] [PubMed] [Google Scholar]
- 39. Foresta C, Moro E, Ferlin A (2001) Y chromosome microdeletions and alterations of spermatogenesis. Endocr. Rev. 22, 226–239. [DOI] [PubMed] [Google Scholar]
- 40. Radford J (2003) Restoration of fertility after treatment for cancer. Horm. Res. 59 (Suppl. 1), 21–23. [DOI] [PubMed] [Google Scholar]
- 41. Kubota H, Brinster RL (2006) Technology insight: in vitro culture of spermatogonial stem cells and their potential therapeutic uses. Nat. Clin. Pract. Endocrinol. Metab. 2, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beermann F, Ruppert S, Hummler E, Bosch FX, Müller G, Rüther U, Schütz G (1990) Rescue of the albino phenotype by introduction of a functional tyrosinase gene into mice. EMBO J. 9, 2819–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62. [DOI] [PubMed] [Google Scholar]
- 44. Ling V, Neben S (1997) In vitro differentiation of embryonic stem cells: immunophenotypic analysis of cultured embryoid bodies. J. Cell. Physiol. 171, 104–115. [DOI] [PubMed] [Google Scholar]
- 45. Buaas FW, Kirsh AL, Sharma M, McLean DJ, Morris JL, Griswold MD, De Rooij DG, Braun RE (2004) Plzf is required in adult male germ cells for stem cell self‐renewal. Nat. Genet. 36, 647–652. [DOI] [PubMed] [Google Scholar]
- 46. Bashamboo A, Taylor AH, Samuel K, Panthier JJ, Whetton AD, Forrester LM (2006) The survival of differentiating embryonic stem cells is dependent on the SCF‐KIT pathway. J. Cell Sci. 119, 3039–3046. [DOI] [PubMed] [Google Scholar]
- 47. Godin I, Deed R, Cooke J, Zsebo K, Dexter M, Wylie CC (1991) Effects of the steel gene product on mouse primordial germ cells in culture. Nature 352, 807–809. [DOI] [PubMed] [Google Scholar]
- 48. Ohta H, Yomogida K, Dohmae K, Nishimune Y (2000) Regulation of proliferation and differentiation in spermatogonial stem cells: the role of c‐kit and its ligand SCF. Development 127, 2125–2131. [DOI] [PubMed] [Google Scholar]
- 49. Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW (2007) Adaptation to culture of human embryonic stem cells and oncogenesis in vivo . Nat. Biotechnol. 25, 207–215. [DOI] [PubMed] [Google Scholar]
- 50. Flores I, Benetti R, Blasco MA (2006) Telomerase regulation and stem cell behaviour. Curr. Opin. Cell Biol. 18, 254–260. [DOI] [PubMed] [Google Scholar]
- 51. Meng X, Lindahl M, Hyvönen ME, Parvinen M, De Rooij DG, Hess MW, Raatikainen‐Ahokas A, Sainio K, Rauvala H, Lakso M, Pichel JG, Westphal H, Saarma M, Sariola H (2000) Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 287, 1489–1493. [DOI] [PubMed] [Google Scholar]
- 52. Guan K, Nayernia K, Maier LS, Wagner S, Dressel R, Lee JH, Nolte J, Wolf F, Li M, Engel W, Hasenfuss G (2006) Pluripotency of spermatogonial stem cells from adult mouse testis. Nature 440, 1199–1203. [DOI] [PubMed] [Google Scholar]