

Abstract
Objectives
While most human adipose tissues, such as those located in the abdomen, hip and thigh, are of mesodermal origin, adipose tissues located in the face are of ectodermal origin. The present study has compared stem cell‐related features of abdomen‐derived adult stem cells (A‐ASCs) with those of eyelid‐derived adult stem cells (E‐ASCs).
Materials and methods
Adipose tissue‐derived cells were maintained in DMEM supplemented with 10% FBS. Before passage 6, cells were analysed using FACS, immunocytochemistry and quantitative real time PCR (qRT‐PCR). To examine multi‐differentiational potential, early passage ASCs were cultivated in each of a commercial Stempro® Differentiation kit.
Results
Unlike fibroblast‐like morphology of A‐ASCs, E‐ASCs had bipolar morphology. Both types of cell exhibited similar surface antigens, and neuronal cell‐related genes and proteins. However, there were differences in mRNA expression levels of CD90 and CD146; neuron‐specific enolase (NSE) and nuclear receptor‐related protein 1 (Nurr1) were different between the two cell types. There was no difference in multi‐differentiational potential between 3 E‐ASCs lines, however, E‐ASCs had higher expression levels of chondrocyte‐related genes compared to A‐ASCs. These cells underwent senescence and maintained normal karyotypes.
Conclusions
Although isolated from similar adipose tissues, both types of cells displayed many contrasting characteristics. Understanding defining phenotypes of such cells is useful for making suitable choices in differing clinical indications.
Introduction
Mesenchymal stromal cells (MSCs) are plastic‐adherent cells isolated from bone marrow and other connective tissues. The International Society for Cellular Therapy (ISCT) recommends that cells can be designated with the description of ‘multipotent mesenchymal stromal cells’ 1 and has proposed three criteria to define them: adherence to plastic surfaces, specific surface antigen expression and multipotential differentiation potential 2.
Bone marrow‐derived MSCs have been widely investigated, and there are various phases of clinical trials currently under way, using these cells 3. Although bone marrow is a reliable source of MSCs, harvesting them from this location is an invasive procedure and numbers of cells isolated is low.
In addition to bone marrow, MSCs can be isolated from many other tissues, for example muscle, adipose tissue, dental pulp, hair follicles, and more 4, 5, 6, 7. One of these – adipose tissue – can be harvested by minimally invasive procedures, which makes it a promising source of adult stem cells. Plastic‐adherent cells isolated from adipose tissues are called ‘adipose‐derived stem/stromal cells’ (ASCs or ADSCs) by the International Fat Applied Technology Society 8.
Many similarities have been found between ASCs and BM‐MSCs, both have a fibroblast‐like morphology and exhibit extensive in vitro proliferation and multilineage differentiation capacity. They also share very similar transcription profiles for stem cell‐related genes 9, 10. Gimble et al. have suggested that stem cells for regenerative medicine applications should ideally meet the following criteria: abundant quantities available (millions to billions of cells), minimally invasive procedure to harvest the cells, differentiation potential development in a regulatable and reproducible manner, safe and effective transplantation into a host and manufacturing in accordance with current Good Manufacturing Practice guidelines 11. Adipose tissue is considered to fulfil these criteria, with its most attractive advantage being its abundance and easy accessibility. In the order of a 500‐fold greater number of MSCs can be obtained from adipose tissues than from bone marrow 12, thus, ASCs are regarded as being a more suitable resource for clinical use.
Adipose‐derived stem cells have been obtained from various types of adipose tissues, but those harvested from different tissue sites have been found to exhibit differences in characteristics; for example, traits of cells isolated from subcutaneous adipose tissue and from omentum are dissimilar. Specifically, stromal cells from subcutaneous adipose tissue proliferate faster than those from the omentum, however, no regional difference in differentiation of the cells has been found 13. While ASC frequency is found to be higher in the abdomen than in the hip/thigh region 14, tissues isolated from the hip yield more stromal cells compared to those isolated from the abdomen 15. Until now, most adipose tissues have been obtained from mesodermal‐origin organs, yet, adipose tissues, particularly facial adipose tissues, have been known to develop from ectodermal‐origin – neural crest cells and ASCs isolated from eyelid adipose tissue have been described. As expected, properties of these ASCs from eyelid tissue were quite different from those of ASCs isolated from elsewhere – A‐ASCs 16. Moreover, the probability that such cells can differentiate into insulin‐secreting cells holds much promise for cell‐based diabetes therapy. In the present study, we have extensively compared characteristics of abdomen‐derived stem cells (A‐ASCs) – one of the more widely investigated types – with newly isolated eyelid‐derived cells (E‐ASCs).
Materials and methods
Culture of adipose‐derived MSCs
Cells were purchased from bcellbio (Seoul, Korea, http://www.bcellbio.co.kr). Cryo‐preserved cells at passage (p) 1 or p2 were thawed and cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 3.7 mg/ml sodium bicarbonate, in 5% CO2 at 37 °C. Medium was changed every 3 or 4 days, and culture periods were divided into early passage (p3–p5), mid passage (p8–p10), and late passage (p13–p15) for the sake of convenience. Donor and cell information are described in Table S1.
RNA extraction and quantitative real‐time PCR (qRT‐PCR)
Total RNA was extracted from cultured cells and 7.5 μg of total RNA was converted into cDNA, using 50 units M‐MLV reverse transcriptase in reaction volume of 50 μl, according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). qRT‐PCR was performed in 96‐well plates with Light Cycler 480 system using SYBR Green I Dye (Roche Applied Science, Indianapolis, IN, USA). qRT‐PCR was performed as follows: in 20‐μl volume containing 2 μl cDNA, 10 μl SYBR Green Master mix (Roche Applied Science), 2 μl primer pairs and 6 μl RNase‐free water. Primer pairs are listed in Table S2. Relative expression levels of each primer set were normalized to expression of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). All samples were run in duplicate for at least five independent experiments.
Immunofluorescence
After culture in Lab‐Tek chamber slides (Nunc, Rochester, NY, USA, http://www.nuncbrand.com), cells were washed in PBS, fixed in 4% paraformaldehyde for 20 min, then permeabilized in 0.2% Triton X‐100, for 20 min. After being washed in PBS, slides were incubated in blocking solution consisting of 2% bovine serum albumin (BSA), for 1 h at room temperature. Subsequently, they were incubated overnight at 4 °C with primary antibody diluted in PBS. Primary antibodies used are listed in Table S3. After being labelled with primary antibody, cells were incubated in fluorescein isothiocyanate (FITC)‐conjugated secondary antibody for 2 h at room temperature; they were then washed three times in PBS, and labelled with 4′‐6‐diamidino‐2‐phenylindole (DAPI) to visualize nuclei (VectaShield; Vector Labs, Burlingame, CA, USA). Cells were then imaged using confocal laser scanning microscopy (Eclipse C1; Nikon, Mississauga, ON, Canada).
Immunophenotype analysis
Early‐passage cells were analysed by flow cytometry, for expression of a number of cell‐surface molecules. Trypsinized cells were washed twice in PBS supplemented with 2% FBS, and resuspended to concentration of approximately 1 × 105 cells/100 μl. They were then incubated in fluorescence labelled antibodies (conjugated to FITC, phycoerythrin (PE), or allophycocyanin (APC) against CD31, CD34, CD44, CD54, CD73, CD90, CD105, CD146, CD166, HLA‐ABC, and HLA‐DR (BD Biosciences San Jose, CA, USA). Control absence of staining was performed using FITC/PE/APC‐conjugated mouse IgG1 isotype antibody. Ratios of fluorescent signals versus background scatter were calculated and histograms were generated using FACS Calibur (Beckman Coulter, Fullerton, CA, USA).
Adipogenic, osteogenic and chondrogenic differentiation
To examine multi‐differentiation potential, early‐passage ASCs were cultured in each of a commercial Stempro® Differentiation kit (A10070, A10071, A10072; Gibco, Carlsbad, CA, USA) consisting of adipogenic, osteogenic and chondrogenic media. For 3 weeks in differentiation culture, medium was replaced every 3–4 days. Adipogenic, osteogenic and chondrogenic differentiation was assessed by two types of method: (i) qualitatively, by specific staining of differentiated cells; and (ii) quantitatively using qRT‐PCR.
Senescence‐associated β‐galactosidase staining
Senescence of cells before and after culture was assessed using SA‐beta‐gal staining kit (ab65351; Abcam, UK) according to the manufacturer's instructions. Cells of early, mid and late passages were fixed, stained overnight and covered with 70% glycerol. Positively labelled cells were imaged using fluorescence microscopy (IX70; Olympus, Japan).
Telomerase activity qRT‐PCR
Telomerase activity of samples was quantified using Quantitative Telomerase Detection Kit (MT3010; Biomax, MD, USA). Extended oligonucleotide substrates for telomerase obtained from samples were subsequently amplified by PCR. Briefly, cells were resuspended in lysis buffer and incubated on ice for 30 min. Supernatants were stored at –80 °C until use. Protein concentration of extracts was assessed using Nanodrop Spectrophotometer (ND 1000; Thermo Fisher Scientific, Waltham, MA, USA). The assay was performed with one extract containing 200 ng of protein and TSR control template to generate a standard curve. As negative controls, each extract was heat‐treated for 10 min at 85 °C. PCR products were visualized using highly sensitive DNA fluorochromes in iQ SYBR Green Supermix (170‐8882; Bio‐Rad, CA, USA); extent of repeated amplification is directly proportional to telomerase activity. Reactions were performed with ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA).
Karyotype analysis
Cytogenetic analysis was conducted on cells from 2 A‐ASCs and 2 E‐ASCs at early, mid and late passages. Before harvest, cells were incubated at 37 °C with colcemid (Irvine Scientific, Santa Ana, CA, USA) at final concentration of 10 μg/ml, for 2 h, to induce mitotic arrest. Cells were fixed and spread, according to standard procedures; those in metaphase were G‐banded and karyotyped in accordance with the International System for Human Cytogenetic Nomenclature recommendations.
Statistical analysis
Data are presented as mean ± SEM, and were analysed using one‐way ANOVA. Differences were considered statistically significant when P < 0.05(*) or P < 0.01(**).
Results
Cell morphology and expansion
To determine growth kinetics, 5 × 104 cells were seeded into 2 T25 flasks. When they achieved around 90% confluence, cells were harvested using 0.125% trypsin/EDTA, then were counted. Cumulative population doubling (PD) numbers were plotted against number of days cultured. Cultures were discontinued when their doubling coefficient reached almost 1. A‐ASCs had typical fibroblast‐like morphology, but E‐ASCs exhibited bipolar morphology and were much smaller than the A‐ASCs (Fig. 1a). Most cell types showed ex vivo self‐renewal capacity, regardless of cell source, until around 20 passages, having gone through 40 PD or more (Fig. 1b).
Figure 1.
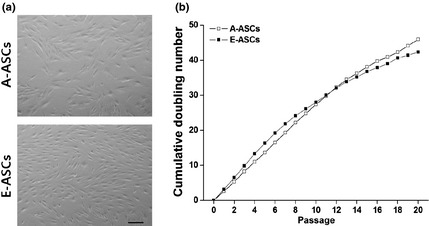
Cell morphology and proliferation rates. (a) Morphological appearance of early‐passage cells. Scale bar: 200 μm. (b) Long‐term growth curves of the A‐ASCs (open) and the E‐ASCs (closed). Cell numbers were determined at the end of every passage and cumulative doubling numbers were calculated in relation to the cell numbers at the first passage.
Gene and protein expression profile
FACS analysis showed that both A‐ASCs and E‐ASCs had many cell surface markers in common. Both types of cell expressed CD44, CD73, CD105, CD166 and HLA ABC. Each half of both cells expressed CD54 antigen. Both types of cell had little expression of CD31, CD34 and HLA DR antigens, but two antigens were differently expressed. While one‐third of A‐ASCs expressed CD146, few E‐ASCs expressed this antigen and whereas 92.2% A‐ASCs expressed CD90 antigen, only 49.4% E‐ASCs expressed it (Fig. 2a). When expression of CD90 was examined by qRT‐PCR, however, both types of cell expressed similar amounts of CD90 mRNA regardless of passage number (Fig. S1). Interestingly, immunocytochemical analyses showed that only few E‐ASCs expressed CD90 at early passage, but most E‐ASCs had it by late passage. A‐ASCs presented similar amounts of CD90 antigen on their surfaces whether they were in early or late passage (Fig. 2b).
Figure 2.
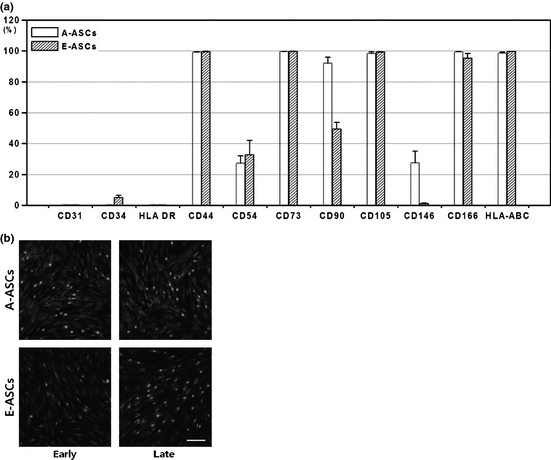
Cell surface antigen expression. (a) Flow cytometric characterization of the cells. For statistical analyses, 4 A‐ASCs and 4 E‐ASCs donors were assessed in pairs. The expression of CD31, CD34 and HLA‐DR was negative, whereas that of others was positive. (b) CD90 expression was visualized by immunohistochemical staining. Cells were stained with anti‐humanCD90 antibody (green) and the nuclei were stained with DAPI (blue). Scale bar: 100 μm.
There was no significant difference in expression of other proteins/genes including ALCAM, ENG, FOXA2, KITLG, Nanog, Nestin, NT5E, SOX2, SOX17 and VCAM1 (Fig. S1). When neural cell‐related gene expression was examined, qRT‐PCR results indicated that both A‐ASCs and E‐ASCs expressed similar amounts of mRNA species including tubulin β3, NSE, Nurr1, GalC, CNPase, FGF5, GFAP, GAP43 and MAP2 (Fig. S2a). Immunocytochemical studies revealed that both types of cell expressed distinct localization of neural proteins including β‐tubulin, GABA, GalC, GFAP, serotonin, nestin and NSE (Fig. S2b).
Differentiation into mesodermal lineages
To compare their differentiational capabilities, early‐passage A‐ASCs or E‐ASC cells were separately cultured in each adipogenic, chondrogenic or osteogenic differentiation medium. When cells were treated with tissue‐specific stains following differentiation culture, both types of cell were distinctly stained with each (Fig. 3a). Nevertheless, qRT‐PCR analyses showed that expression profile of A‐ASCs was a little different from that of E‐ASCs. While expression of Adipsin and PPAR genes among adipocyte‐related genes examined, in A‐ASCs was stronger than that in E‐ASCs, Leptin and particularly LPL gene were more strongly expressed in E‐ASCs than in A‐ASCs (Fig. 3b); expression level of CEEP was similar in both types of cell. Expression levels of chondrocyte‐related genes including Col II, Biglycan, Decorin and COMP in E‐ASCs were higher than in A‐ASCs. Following osteogenic differentiation, CEFA gene expression was stronger in A‐ASCs than in E‐ASCs, however, expression levels of PTHR, Col I, OC and ON genes were similar in both cell types.
Figure 3.
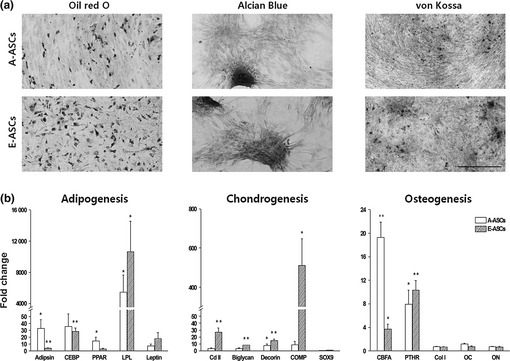
In vitro differentiation of ASCs into mesodermal lineages. (a) A‐ASCs and E‐ASCs were cultivated in adipogenic, chondrogenic or osteogenic differentiation medium. After culture, adipogenic differentiation of cells was assessed by intracellular accumulation of neutral lipids following staining with Oil red O solution. Chondrogenic differentiation was assessed by staining with Alcian blue. Formation of a mineralized matrix after osteogenic differentiation was assessed by von Kossa staining. Scale bar: 500 μm. (b) Quantification of mesodermal lineage‐specific gene expression. Total RNA was extracted from differentiated cells, and was analysed by qRT‐PCR. Gene expression levels were normalized to GAPDH expression to yield 2Ct values, and were calculated relatively to the controls.
Influence of long‐term culture
As subculture was repeated, cell morphology and sizes of cells, changed. Fibroblast‐like morphology and cell size of A‐ASCs did not differ significantly throughout the culture period, whereas E‐ASCs developed typical characteristics of senescence, such as larger cell size and flattened morphology (Fig. 4a); we confirmed cell senescence by β‐gal staining assay (Fig. 4b). As cells were able to proliferate for more than 20 passages over approximately 3 months, we studied karyotypes of long‐term cultured cells, most of which were normal. Some abnormalities were detected more frequently in A‐ASCs than in E‐ASCs (Fig. S3). Additionally, we checked PD time of cells throughout the culture period. As seen in Fig. 4c, E‐ASC cells had a much longer doubling time than A‐ASCs after mid passages. Difference in PD times corresponded to results of telomerase activity assay (Fig. 4d), that is, E‐ASCs had significantly higher telomerase activities, thus had shorter PD time in early passages than A‐ASCs.
Figure 4.
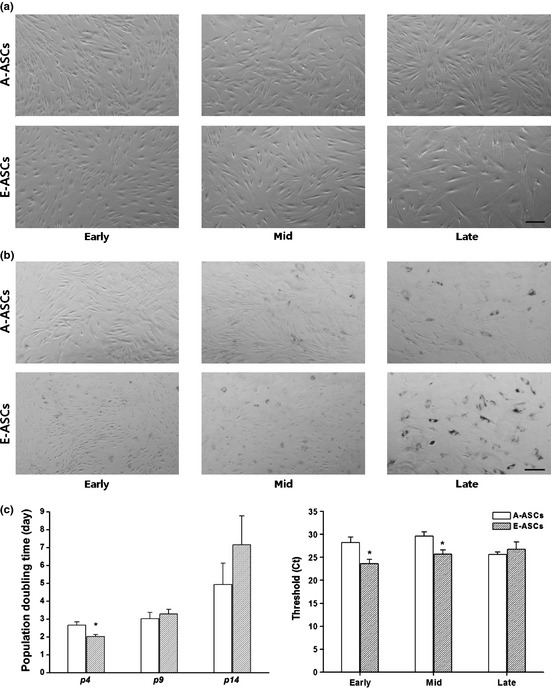
Characterization of the A‐ASCs and E‐ASCs during long‐term culture in vitro. (a) Morphological change due to repetitive subculture. Scale bar: 200μm. (b) Senescence‐associated β‐galactosidase activity of both ASCs at early‐, mid‐, and late‐passage. Scale bar: 200 μm. (c) Population doubling (PD) time of the early‐, mid‐, and late‐passage cells. The PD time of the early‐passage (p4) E‐ASCs was shorter than that of A‐ASCs (P < 0.05) at the same age; however, the time of the late‐passage (p14) E‐ASCs became longer than that of the A‐ASCs at the same age. (d) Telomerase activity of ASCs at the early‐, mid‐, and late passage by qRT‐PCR. Ct values of E‐ASCs at early‐ and mid‐passage were significantly lower than Ct values of A‐ASCs at the same ages.
Discussion
In this study, we compared characteristics of A‐ASCs with those of E‐ASCs. Although isolated from similar adipose tissues, both types of cell displayed many different characteristics in terms of expression profile of surface antigens, proliferation capability, differentiation potential and telomerase activity.
Our findings show that both A‐ASCs and E‐ASCs were by nature adherent, and could be harvested then expanded in vitro. However, surface marker expression levels differed between the two cell types. In particular, the cell population expressing CD90, a typical marker, was significantly lower in E‐ASCs than in A‐ASCs. Similar results were obtained with expression of CD146 – another cell surface marker. CD90 is a membrane‐bound glycoprotein and is expressed by 90% in a variety of tissues 17; some workers have suggested that its function is related to angiogenic stimuli 18, 19. From this point of view, cell responses to angiogenic factors would be different when they were injected into the human body. Additionally, CD146 (also known as melanoma cell adhesion molecule, MCAM) is a typical pericyte marker; it is one of the commonly reported surface markers of MSCs 20, 21, 22, 23; in this study, we used CD146 as a positive surface marker for ASCs. Some studies have suggested that expression of CD146 varies depending on the donor or the cell passage number 24, 25, but although there were donor‐specific or passage‐dependent variations, level of CD146 expression by E‐ASCs was significantly low. However, unlike the flow cytometry data, there were no significant differences between the two cell types when assessed by qRT‐PCR. From these data, the suggested CD marker panel designated by ISCT minimal criteria was not sufficient to define the MSCs when identified by flow cytometry.
In line with earlier reports, A‐ASCs and E‐ASCs could be readily induced into all three mesenchymal lineages 26, 27, 28, 29, 30. In addition to staining differentiated cells, we observed expression levels of lineage‐specific genes, to quantify differences between differentiation potentials of A‐ASCs and E‐ASCs. Interestingly, expression levels of chondrogenesis‐related genes were significantly higher in E‐ASCs than in A‐ASCs. These results show that E‐ASCs were more effective than A‐ASCs, in terms of chondrogenesis. However, there were few changes in levels of osteoblast marker genes after differentiation. This seemed to be the case as differentiation conditions for osteogenesis seemed not to be sufficient.
ASCs have the potential to differentiate into neuronal precursors under neurogenic conditions 31, 32, 33. However, Kang et al. have suggested that undifferentiated E‐ASCs have characteristics of neuronal cells, thus in this study, we compared expression levels of neuronal cell‐specific markers between A‐ASCs and E‐ASCs. Differences in expression levels of most markers, except NSE and Nurr1, were not significant, but as there are only a small number of functional studies concerning NSE and Nurr1 in MSCs, further investigations are needed.
Finally, we investigated alterations caused by long‐term in vitro culture. All somatic cells that can be cultured in vitro go through cell senescence 34, but, in vitro expansion of cell number is mandatory for cell therapy. Here, A‐ASCs and E‐ASCs showed typical cell senescence phenotypes, such as flattening and larger cell sizes, as subculture was repeated. In the cytogenetic study, A‐ASCs displayed some abnormalities, such as trisomy and marker chromosomes; these phenomena were not observed in E‐ASCs. Karyotyped cells had initially been harvested using different surgical procedures, A‐ASCs by tumescent liposuction and E‐ASCs by resection. However, the relationship between these surgical procedures and karyotypes abnormalities will require further investigation, althoug most cells had normal karyotype. Additionally, E‐ASCs had shorter PD time and smaller Ct values in the telomerase assay during early passages, that is, E‐ASCs exhibited higher proliferation rates due to their higher telomerase activity. It will be advantageous to conduct studies using more abundant cell numbers with greater capacities for cell proliferation.
In conclusion, our study has revealed both similarities and differences between A‐ASCs and E‐ASCs. To make better use of cell‐based therapies and tissue engineering, it is most important to understand characteristics of each cell type, thus to ensure safety, these characteristics must be considered when cells are to be used clinically.
Supporting information
Fig. S1 mRNA expression profiles in the A‐ASCs and E‐ASCs.
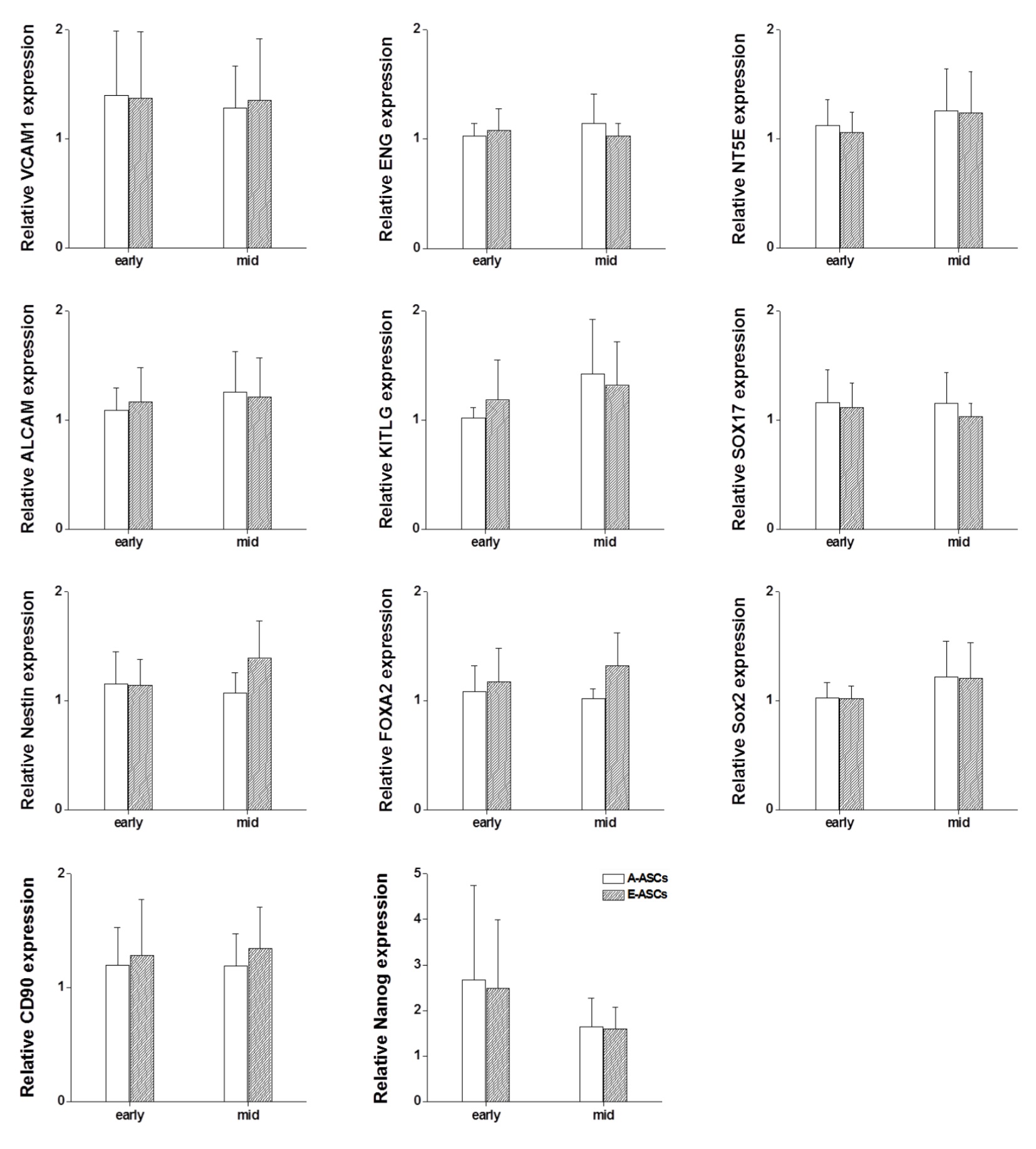{kind=link}
Fig S2 Neuronal cell properties of undifferentiated ASCs.
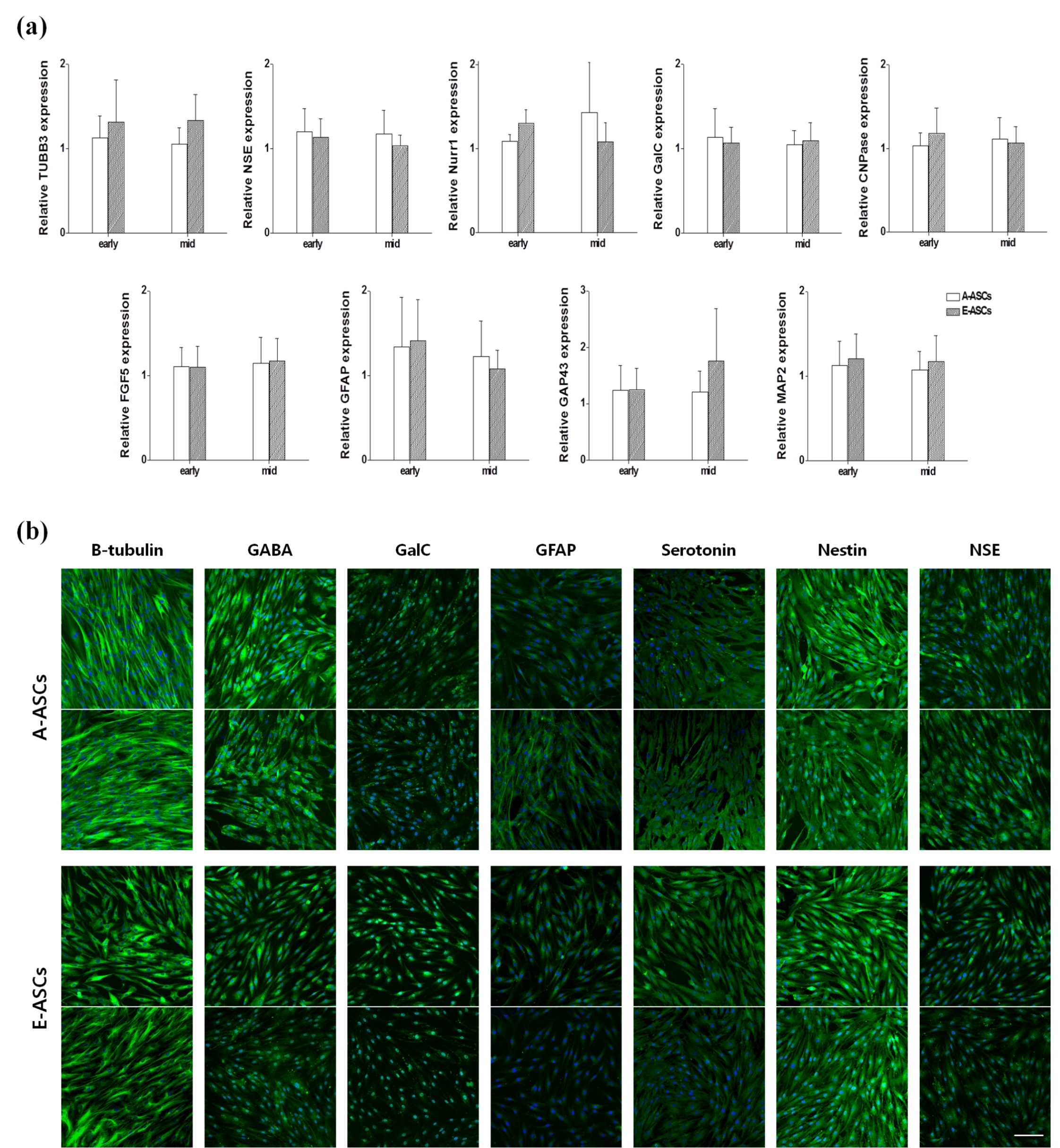{kind=link}
Fig S3 Karyotyping of early‐, mid‐, and late‐passage cells.
{kind=link}
Table S1 Patient description for cell sources.
Table S2 List of primer sequences for the quantitative real time PCR.
Table S3 List of antigens examined for the immunophenotyping of cells.
Acknowledgements
This study was supported by a grant (11171KFDA351) from the Korea Food & Drug Administration in 2011.
References
- 1. Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper‐Cortenbach I, Marini FC et al (2005) Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy 7, 393–395. [DOI] [PubMed] [Google Scholar]
- 2. Dominici M, Le Blanc K, Mueller I, Slaper‐Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- 3. Trounson A, Thakar RG, Lomax G, Gibbons D (2011) Clinical trials for stem cell therapies. BMC Med. 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei Y, Li Y, Chen C, Stoelzel K, Kaufmann AM, Albers AE (2011) Human skeletal muscle‐derived stem cells retain stem cell properties after expansion in myosphere culture. Exp. Cell Res. 317, 1016–1027. [DOI] [PubMed] [Google Scholar]
- 5. Baglioni S, Francalanci M, Squecco R, Lombardi A, Cantini G, Angeli R et al (2009) Characterization of human adult stem‐cell populations isolated from visceral and subcutaneous adipose tissue. FASEB J 23, 3494–3505. [DOI] [PubMed] [Google Scholar]
- 6. Nam H, Lee G (2009) Identification of novel epithelial stem cell‐like cells in human deciduous dental pulp. Biochem. Biophys. Res. Commun. 386, 135–139. [DOI] [PubMed] [Google Scholar]
- 7. Ohyama M, Terunuma A, Tock CL, Radonovich MF, Pise‐Masison CA, Hopping SB et al (2006) Characterization and isolation of stem cell‐enriched human hair follicle bulge cells. J. Clin. Invest 116, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakagami H, Morishita R, Maeda K, Kikuchi Y, Ogihara T, Kaneda Y (2006) Adipose tissue‐derived stromal cells as a novel option for regenerative cell therapy. J. Atheroscler. Thromb. 13, 77–81. [DOI] [PubMed] [Google Scholar]
- 9. Peroni D, Scambi I, Pasini A, Lisi V, Bifari F, Krampera M et al (2008) Stem molecular signature of adipose‐derived stromal cells. Exp. Cell Res. 314, 603–615. [DOI] [PubMed] [Google Scholar]
- 10. Egusa H, Iida K, Kobayashi M, Lin TY, Zhu M, Zuk PA et al (2007) Downregulation of extracellular matrix‐related gene clusters during osteogenic differentiation of human bone marrow‐ and adipose tissue‐derived stromal cells. Tissue Eng. 13, 2589–2600. [DOI] [PubMed] [Google Scholar]
- 11. Gimble JM, Katz AJ, Bunnell BA (2007) Adipose‐derived stem cells for regenerative medicine. Circ. Res. 100, 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraser JK, Wulur I, Alfonso Z, Hedrick MH (2006) Fat tissue: an underappreciated source of stem cells for biotechnology. Trends Biotechnol. 24, 150–154. [DOI] [PubMed] [Google Scholar]
- 13. Van Harmelen V, Rohrig K, Hauner H (2004) Comparison of proliferation and differentiation capacity of human adipocyte precursor cells from the omental and subcutaneous adipose tissue depot of obese subjects. Metabolism 53, 632–637. [DOI] [PubMed] [Google Scholar]
- 14. Jurgens WJ, Oedayrajsingh‐Varma MJ, Helder MN, Zandiehdoulabi B, Schouten TE, Kuik DJ et al (2008) Effect of tissue‐harvesting site on yield of stem cells derived from adipose tissue: implications for cell‐based therapies. Cell Tissue Res. 332, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fraser JK, Wulur I, Alfonso Z, Zhu M, Wheeler ES (2007) Differences in stem and progenitor cell yield in different subcutaneous adipose tissue depots. Cytotherapy 9, 459–467. [DOI] [PubMed] [Google Scholar]
- 16. Kang HM, Kim J, Park S, Kim H, Kim KS, Lee EJ et al (2009) Insulin‐secreting cells from human eyelid‐derived stem cells alleviate type I diabetes in immunocompetent mice. Stem Cells 27, 1999–2008. [DOI] [PubMed] [Google Scholar]
- 17. Sakaguchi Y, Sekiya I, Yagishita K, Muneta T (2005) Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum. 52, 2521–2529. [DOI] [PubMed] [Google Scholar]
- 18. Campioni D, Lanza F, Moretti S, Ferrari L, Cuneo A (2005) Loss of Thy‐1 (CD90) antigen expression on mesenchymal stromal cells from hematologic malignancies is induced by in vitro angiogenic stimuli and is associated with peculiar functional and phenotypic characteristics. Cytotherapy 10, 69–82. [DOI] [PubMed] [Google Scholar]
- 19. Campioni D, Moretti S, Ferrari L, Punturieri M, Castoldi GL, Lanza F (2006) Immunophenotypic heterogeneity of bone marrow‐derived mesenchymal stromal cells from patients with hematologic disorders: correlation with bone marrow microenvironment. Haematologica 91, 364–368. [PubMed] [Google Scholar]
- 20. Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM (2001) Surface protein characterization of human adipose tissue‐derived stromal cells. J. Cell. Physiol. 189, 54–63. [DOI] [PubMed] [Google Scholar]
- 21. Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG et al (2003) SHED: stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad Sci. U. S. A. 100, 5807–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buhring HJ, Treml S, Cerabona F, de Zwart P, Kanz L, Sobiesiak M (2009) Phenotypic characterization of distinct human bone marrow‐derived MSC subsets. Ann. N. Y. Acad Sci. 1176, 124–134. [DOI] [PubMed] [Google Scholar]
- 23. Vishnubalaji R, Al‐Nbaheen M, Kadalmani B, Aldahmash A, Ramesh T (2012) Comparative investigation of the differentiation capability of bone‐marrow‐ and adipose‐derived mesenchymal stem cells by qualitative and quantitative analysis. Cell Tissue Res. 347, 419–427. [DOI] [PubMed] [Google Scholar]
- 24. Bieback K, Hecker A, Schlechter T, Hofmann I, Brousos N, Redmer T et al (2012) Replicative aging and differentiation potential of human adipose tissue‐derived mesenchymal stromal cells expanded in pooled human or fetal bovine serum. Cytotherapy 14, 570–583. [DOI] [PubMed] [Google Scholar]
- 25. Mitchell JB, McIntosh K, Zvonic S, Garrett S, Floyd ZE, Kloster A et al (2006) Immunophenotype of human adipose‐derived cells: temporal changes in stromal‐associated and stem cell‐associated markers. Stem Cells 24, 376–385. [DOI] [PubMed] [Google Scholar]
- 26. Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ et al (2001) Multilineage cells from human adipose tissue: implications for cell‐based therapies. Tissue Eng. 7, 211–228. [DOI] [PubMed] [Google Scholar]
- 27. Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H et al (2002) Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 13, 4279–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ogawa R, Mizuno H, Watanabe A, Migita M, Hyakusoku H, Shimada T (2004) Adipogenic differentiation by adipose‐derived stem cells harvested from GFP transgenic mice‐including relationship of sex differences. Biochem. Biophys. Res. Commun. 319, 511–517. [DOI] [PubMed] [Google Scholar]
- 29. Halvorsen YD, Franklin D, Bond AL, Hitt DC, Auchter C, Boskey AL et al (2001) Extracellular matrix mineralization and osteoblast gene expression by human adipose tissue‐derived stromal cells. Tissue Eng. 7, 729–741. [DOI] [PubMed] [Google Scholar]
- 30. Huang JI, Zuk PA, Jones NF, Zhu M, Lorenz HP, Hedrick MH et al (2004) Chondrogenic potential of multipotential cells from human adipose tissue. Plast. Reconstr. Surg. 113, 585–594. [DOI] [PubMed] [Google Scholar]
- 31. Zavan B, Vindigni V, Gardin C, D'Avella D, Della Puppa A, Abatangelo G et al (2010) Neural potential of adipose stem cells. Discov Med 10, 37–43. [PubMed] [Google Scholar]
- 32. Wrage PC, Tran T, To K, Keefer EW, Ruhn KA, Hong J et al (2008) The neuro‐glial properties of adipose‐derived adult stromal (ADAS) cells are not regulated by Notch 1 and are not derived from neural crest lineage. PLoS ONE 3, e1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shakhbazau AV, Goncharova NV, Kosmacheva SM, Kartel NA, Potapnev MP (2009) Plasticity of human mesenchymal stem cell phenotype and expression profile under neurogenic conditions. Bull. Exp. Biol. Med. 147, 513–516. [DOI] [PubMed] [Google Scholar]
- 34. Smith JR, Pereira‐Smith OM (1996) Replicative senescence: implications for in vivo aging and tumor suppression. Science 273, 63–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 mRNA expression profiles in the A‐ASCs and E‐ASCs.
Fig S2 Neuronal cell properties of undifferentiated ASCs.
Fig S3 Karyotyping of early‐, mid‐, and late‐passage cells.
Table S1 Patient description for cell sources.
Table S2 List of primer sequences for the quantitative real time PCR.
Table S3 List of antigens examined for the immunophenotyping of cells.