

Abstract
Objectives
The posterior lateral line (PLL) system in zebrafish has recently become a model for investigating tissue morphogenesis. PLL primordium periodically deposits neuromasts as it migrates along the horizontal myoseptum from head to tail of the embryonic fish, and this migration requires activity of various molecular mechanisms. Histone deacetylases (HDACs) have been implicated in numerous biological processes of development, by regulating gene transcription, but their roles in regulating PLL during embryonic development have up to now remained unexplored.
Material and methods
In this study, we used HDAC inhibitors to investigate the role of HDACs in early development of the zebrafish PLL sensory system. We further investigated development of the PLL by cell‐specific immunostaining and in situ hybridization.
Results
Our analysis showed that HDACs were involved in zebrafish PLL development as pharmacological inhibition of HDACs resulted in its defective formation. We observed that migration of PLL primordium was altered and accompanied by disrupted development of PLL neuromasts in HDAC inhibitor‐treated embryos. In these, positions of PLL neuromasts were affected. In particular, the first PLL neuromast was displaced posteriorly in a treatment dose‐dependent manner. Primordium cell proliferation was reduced upon HDAC inhibition. Finally, we showed that inhibition of HDAC function reduced numbers of hair cells in PLL neuromasts of HDAC inhibitor‐treated embryos.
Conclusion
Here, we have revealed a novel role for HDACs in orchestrating PLL morphogenesis. Our results suggest that HDAC activity is necessary for control of cell proliferation and migration of PLL primordium and hair cell differentiation during early stages of PLL development in zebrafish.
Introduction
The lateral line is a mechanosensory organ in zebrafish that detects directional water flow. This helps fish avoid obstacles and predators, and also facilitates prey capture 1. The lateral line system is comprised of a set of sense organs, called neuromasts, located on the surface of the head (anterior lateral line) and the body (posterior lateral line, PLL) in species‐specific patterns. The PLL system is generated by a sensory primordium, comprised of around 100 cells, found along the horizontal myoseptum from head to tip of the tail. During its embryological journey, the primordium deposits five or six regularly spaced neuromasts and two or three terminal neuromasts at the tail‐tip at 48 hours post‐fertilization (hpf) (Fig. 1a) 2, 3. Early studies have elucidated complex signalling pathways that regulate primordium migration. Fibroblast growth factor (Fgf) signalling is active in the trailing primordium and regulates several aspects of lateral line morphogenesis. In the leading region, the Wnt/β‐catenin pathway is active and is responsible for inducing and restricting Fgf signalling to the trailing zone, which in turn restricts Wnt/β‐catenin signalling to the leading edge (LE) of the migrating primordium. The Wnt/β‐catenin pathway not only induces proneuromast formation by facilitating interaction with activity of Fgf signalling, but also controls primordium migration by regulating cxcr4b and cxcr7b proteins required for normal migration of PLL primordium 4, 5, 6, 7, 8. Recently, several genes have been shown to be expressed in the migrating PLL primordium cell population, including kal1a, lymphoid enhancer factor 1 and ladybird homeobox homologous gene 2 responsible for PLL formation during embryonic development 9, 10. However, there is currently little understanding regarding effects of epigenetic mechanisms on migration of PLL primordium and periodic deposition of neuromasts during lateral line morphogenesis.
Figure 1.
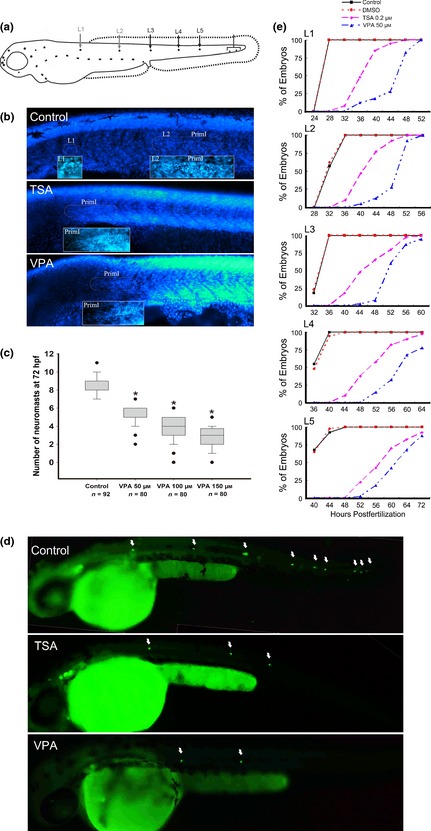
Effects of histone deacetylases ( HDAC ) inhibitors on the migration of posterior lateral line ( PLL ) primordium. (a) A schematic drawing of the PLL along the horizontal myoseptum at 48 hours post‐fertilization (hpf). (b) The effect of HDAC inhibitors on PLL primordium migration was assessed by DAPI staining in 32 hpf wild‐type embryos. (c) Different doses of valproic acid (VPA) were added to embryos from 6 to 72 hpf. Number of PLL neuromasts assessed using DAPI staining in wild‐type embryos at 72 hpf. n, total number of embryos. (d) The process of primordium migration was significantly delayed in transgenic line (Brn3c:mGFP) embryos by HDAC inhibitor treatment from 6 to 48 hpf. The positions of the neuromasts are indicated by arrows. (e) The deposition process of the first five PLL neuromasts (L1–L5). The percentages of wild‐type embryos developing the corresponding neuromasts are shown with black squares (Control), red dots (DMSO), purple diamonds (0.2 μm trichostatin A) and blue triangles (50 μm VPA).
Epigenetic mechanisms such as DNA methylation, histone post‐translational modification and non‐coding RNA‐mediated post‐translational regulation have been shown to play roles in development and fate of cells. Histone modification is a key mechanism of eukaryotic gene transcriptional regulation due to its ability to post‐translationally modify N‐terminal tails of core histones 11, 12. Acetylation and deacetylation of histones are the most widespread histone modifications and are crucial in modulating chromatin structure and transcriptional activity 13. Histone acetylation is usually associated with transcriptional activation and leads to diminished chromatin compaction by relaxing DNA coiling from histone cores and increasing accessibility of transcription factors to interact with regulatory regions. Conversely, histone deacetylation is associated with transcriptional repression 13, 14. Histone acetylation and deacetylation are governed by opposing effects of two classes of enzyme, histone acetyltransferases and histone deacetylases (HDACs) that acetylate and deacetylate specific lysines in tail residues of histones, respectively 15.
The HDAC family is subdivided into four major classes: Class I (HDAC1, ‐2, ‐3, and ‐8), Class II (HDAC4, ‐5, ‐6, ‐7, ‐9, and ‐10), Class III (Sir2 family of NAD+‐dependent enzymes), and Class IV (HDAC11). Each class is essential for multiple biological processes 16, 17. HDAC inhibition has been shown to promote hyperacetylation of nucleosomal histones, and this appears to be an important mechanism that regulates gene expression and many aspects of cell behaviour, including proliferation, apoptosis, differentiation and migration 18, 19, 20, 21, 22. Several studies have suggested that HDACs are critically involved in regulation of developmental processes including neurogenesis, liver and exocrine pancreas development, and heart valve formation in zebrafish embryos 23, 24, 25. HDAC activity has specific functions in neural progenitor differentiation. For example, HDAC1 is required for transformation of neural stem cells into mature neurons and glia during development of the central nervous system in zebrafish and in mice 25, 26, 27, 28. Conditional deletion of both HDAC1 and HDAC2 in mouse neuronal precursors results in severe brain abnormalities 28. In addition, some reports indicate that HDAC inhibitors prevent maturation of oligodendrocytes from progenitor cells and suggest that histone deacetylation is a critical component in oligodendrocyte differentiation 29, 30, 31.
Compared to significant progress that has been made in the study of epigenetics in many biological processes, little research has focused on epigenetics in hearing research. Epigenetics, however, plays an essential role in hearing‐related processes 32. A role of HDACs in regulation of cell proliferation and differentiation in regenerating sensory epithelia from the avian utricle has been reported 33. It has been shown that histone deacetylation is a positive regulator of regenerative proliferation, and inhibition of HDACs is sufficient to prevent entry into the cell cycle of supporting cells. Furthermore, HDAC inhibition does not affect hair cell differentiation during the regenerative process. However, there is little understanding concerning whether or how HDAC activity is connected to production of hair cells in the development process.
The zebrafish is an excellent model for studying hair cell differentiation. The study described here represents an important step in validating use of zebrafish PLL as a powerful model system for studying effects of HDACs on PLL formation and for studying hair cell differentiation. We show that inhibition of HDAC activity by valproic acid (VPA) or trichostatin A (TSA) not only impairs migration of PLL primordium but also interferes with the position of neuromasts. We also demonstrate that treatment with HDAC inhibitors dramatically reduces cell proliferation in the migrating primordium. Finally, we show that HDAC inhibition reduces numbers of hair cells in the neuromast during PLL development. On the basis of these observations, we conclude that HDAC activity is necessary for control of proliferation and migration of PLL primordium during development, ongoing deposition of neuromasts and hair cell differentiation.
Materials and methods
Zebrafish strains and maintenance
Fish and embryos were maintained in our facility according to standard procedures. Brn3c:mGFP transgenic line was obtained from Zhengyi Chen, our collaborator at Harvard University, and all other embryos were obtained from natural spawning of wild‐type adults. Zebrafish were staged according to the method of Kimmel et al. 34 and raised at 28.5 °C in Petri dishes. Ages of embryos and larvae are described as hpf.
Pharmacological treatments
Valproic acid (2‐propyl‐pentanoic acid) and TSA were dissolved as stock concentrations of 200 mm and 500 μm, respectively, then diluted to the indicated concentrations in culture medium. Embryos were soaked in VPA‐ or TSA‐ containing culture medium until an appropriate stage. VPA and TSA were obtained from Sigma‐Aldrich (Buchs, Switzerland).
FM1‐43FX labelling of lateral line HCs
For staining of functional hair cells within neuromasts, a mechanotransduction marker was applied by exposing live 3‐day post‐fertilization (dpf) larvae to 3 μm FM1‐43FX (Invitrogen; F‐35355) for 45 s under darkened conditions. After quickly rinsing three times in culture medium, larvae were anesthetized and killed in MS‐222 (Sigma‐Aldrich) then fixed in 4% paraformaldehyde (PFA) for subsequent analysis.
Cell proliferation and analysis
For analysis of S‐phase cells in migrating primordia, embryos were manually dechorionated and transferred into 10 mm 5‐bromo‐2‐deoxyuridine (BrdU; Sigma‐Aldrich) for 1 h at 28.5 °C. BrdU incorporation was detected by immunocytochemistry. Larvae were anesthetized and killed in MS‐222 (Sigma‐Aldrich) then and fixed in 4% PFA for 2 h at room temperature (RT). Fixed larvae were washed three times in PBS in 0.5% Triton X‐100 (PBT‐2) and placed into 2N HCl for 0.5 h at 37 °C. They were then washed again in PBT‐2, and non‐specific binding was blocked with 10% normal goat serum for 1 h at RT followed by incubation in mouse monoclonal BrdU antibody (1:200; Cat. no. sc‐32323; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4 °C. The next day, larvae were washed three times in PBT‐2 and incubated in secondary antibody for 1 h at 37 °C. BrdU labelling index was calculated as ratio of BrdU‐labelled nuclei to total DAPI‐stained nuclei.
Immunohistochemistry
Embryos fixed in 4% PFA were rinsed three times in PBS and permeabilized with PBT‐2 for 30 min. They were then blocked in 10% normal goat serum for 1 h and incubated in primary antibodies overnight at 4 °C. The following primary antibodies were used: anti‐acetylated histone H4 (1:400); anti‐acetylated histone H3 (1:400); and anti‐cleaved caspase‐3 (1:200). Embryos were washed three times in PBS and incubated in secondary antibodies to detect location of primaries. Nuclei were labelled with DAPI for 20 min at RT.
Western blotting
Total protein was isolated from embryos at 72 hpf (per sample) using AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Protein concentrations were measured using BCA protein kit, and samples of 50 μg each were separated by SDS‐PAGE on 12% gels. After electrophoresis, proteins were transferred to PVDF membranes (Immobilon‐P; Millipore, Bedford, MA, USA) blocked with 5% non‐fat dried milk in TBST (50 mm Tris‐HCl (pH 7.4), 150 mm NaCl, 0.1% Tween‐20) for 1 h at RT. After washing, primary rabbit anti‐acetyl H3 (1:1000), rabbit anti‐acetyl H4 (1:1000), rabbit anti‐histone H3 (1:1000), and rabbit anti‐histone H4 (1:1000) antibodies were added to blocking buffer overnight at 4 °C. Membranes were washed three times (10 min each) in TBST. Antibodies to acetyl H3 and acetyl H4 histones were purchased from Upstate Biotechnology (Upstate Biotechnologies Inc., Lake Placid, NY, USA) and total H3 and H4 histones were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA).
Whole‐mount in situ hybridization
The probe for cxcr7b was amplified from cDNA from 32 hpf embryos and subcloned into pGEMTeasy vector (cat. no. A1360; Promega Corporation, Madison, WI, USA). Forward and reverse primers were 5′‐GTTGACCAGGAACGTCGAAT‐3′ and 5′‐AATATCCCCCTCCGTTTCAC‐3′. The probe was transcribed in vitro using SP6 RNA polymerase (cat. no. P1460; Promega) and prepared with a digoxigenin labelling kit. Whole‐mount RNA in situ hybridization was performed following standard procedures. Briefly, embryos were depigmented with 1‐phenyl‐2‐thiourea (cat. no. P7629; PTU, Sigma‐Aldrich), euthanized in MS‐222, and fixed overnight in 4% PFA at 4 °C. Fixed embryos were washed in PBS with 0.1% Tween‐20 (PBST) and placed in 100% methanol at −20 °C to dehydrate. Prior to use, they were rehydrated in series of graded methanols and washed three times for 5 min in PBST. To permeabilize the embryos, proteinase K (15 μg/ml in PBST) was added for 30 min and the embryos were re‐fixed in 4% PFA for 20 min. After washing in PBST, embryos were prehybridized at 65 °C for ≥2 h in hybridization buffer [50% formamide (Fluka, Buchs, Switzerland), 5× SSC, 0.1% Tween‐20, and 1 M citric acid]. For hybridization, labelled probes were added to the hybridization buffer (1:20) at 65 °C overnight. After washing for 10 min in 75%, 50%, and 25% HB, 2× SSC and for 30 min twice in 0.2× SSC at 65 °C, embryos were rinsed with 75%, 50%, and 25% 0.2× SSC and PBST for 5 min. They were then blocked for at least 2 h at 4 °C in blocking buffer (5% sheep serum in 2 mg/ml bovine serum albumin in PBST) and were incubated with preabsorbed sheep anti‐digoxigenin‐AP Fab fragments (cat. no. 11093274910; Roche Diagnostics, Indianapolis, IN, USA) at a 1:2000 dilution in blocking buffer overnight at 4 °C. The next day, embryos were washed 8 × 15 min in 2 mg/ml bovine serum albumin in PBST and 3 × 5 min in staining buffer (100 mm Tris (pH 9.5), 50 mm MgCl2, 100 mm NaCl, and 0.1% Tween‐20). Afterwards, they were stained in NBT/BCIP staining solution (cat. no. 11681451001; Roche) in the dark for 30–60 min at RT. Finally, the colour reaction was stopped by adding PBST and embryos were observed using a bright field microscope (Leica, Heerbrugg, Switzerland).
Statistical analysis
Comparisons between HDAC treatments and controls were analysed using Student's t‐test with spss version 19.0 (SPSS Inc., Chicago, IL, USA). Multiple samples were statistically analysed for significance using one‐way analysis of variances (one‐way ANOVA). Results were expressed as mean ± SE (standard error). P‐values less than 0.05 were considered to be statistically significant.
Results
Defective formation of zebrafish PLL
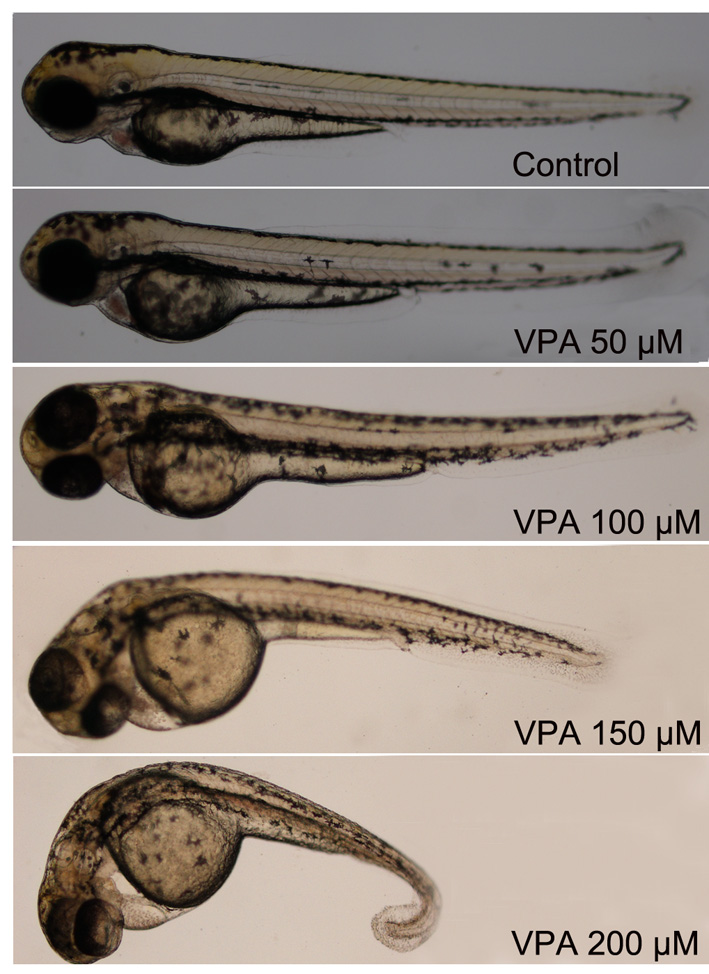
To study the function of HDACs in early zebrafish development, embryos were treated with HDAC inhibitor VPA, a potent compound that preferentially inhibits Class I HDACs. Concentration of VPA used in our experiments was determined empirically to produce visible phenotypes and concentrations of 50, 100, 150 and 200 μm administered from 6 to 72 hpf resulted in morphologically different ones (Fig. S1). General development of embryos treated with 50 μm VPA appeared similar to control embryos with no apparent developmental anomalies nor retardation. As VPA concentration increased, a curved body phenotype became more obvious and this was associated with lower survival and hatching rates. In embryos treated with 100 and 150 μm VPA, hatching rates were in the order of 65.9% (n = 120) and 16.2% (n = 120) at 72 hpf, respectively, compared to nearly 100% in controls and 93.2% (n = 120) in embryos treated with 50 μm VPA. Moreover, at 150 μm VPA, 50.5% hatched embryos had curved bodies. At 200 μm, VPA treatment was lethal and induced severe morphological deformation including pericardiac oedema and curved tails (Fig. S1). These embryos did not reach the age of 120 hpf regardless of being hatched or not. Thus, we used VPA concentration of 50 μm in most following our experiments.
To determine whether HDAC activity is required for organogenesis of the PLL, we treated the embryos with VPA and investigated its effect on PLL development by DAPI staining. As shown in Fig. 1b, VPA treatment reduced numbers of PLL neuromasts at 32 hpf in contrast to normal PLL pattern of control embryos. Similar results were obtained from embryos treated with TSA, the further HDAC inhibitor (Fig. 1b). In 72 hpf control embryos, average number of PLL neuromasts per side was 8.34 ± 1.01 (n = 92). However, for embryos treated with VPA at 50, 100 and 150 μm from 6 to 72 hpf, average number of PLL neuromasts was 5.1 ± 0.12 (n = 80; P < 0.001 versus control), 3.95 ± 0.16 (n = 80; P < 0.001 versus control) and 2.58 ± 0.14 (n = 80; P < 0.001 versus control), respectively (Fig. 1c). We used Tg(Brn3c:mGFP) line that expresses GFP in hair cells of PLL neuromasts, to examine formation of PLL neuromasts during migration of the primordium 35. Tg(Brn3c:mGFP) transgenic zebrafish embryos were treated with HDAC inhibitors starting from 6 hpf. As shown in Fig. 1d, HDAC inhibitor treatment reduced numbers of GFP‐positive PLL neuromasts at 48 hpf in contrast to normal PLL pattern of control embryos. Similar results were also obtained from embryos treated with TSA.
To monitor deposition of PLL neuromasts, embryos were treated with VPA (50 μm) or TSA (0.2 μm) from 6 hpf and numbers of DAPI‐labelled neuromasts spaced along the body axis were recorded starting from 24 hpf (Fig. 1e). No deposited neuromasts were observed in 32 hpf VPA‐treated embryos, in clear contrast to appearance of first (L1) and second (L2) neuromasts in untreated embryos of the same age. At 48 hpf, most control embryos had completed PLL development with deposition of 7–9 neuromasts, but only 27.5% (11/40) VPA‐treated embryos had deposited L1 and L2 neuromasts. At 72 hpf, in the order of 87.5% (35/40) VPA‐treated embryos had deposited L5 neuromast (Fig. 1e). If VPA treatment continued to 5 dpf, PLL primordium was able to finish its migration and deposit the terminal neuromasts at the tip of the tail (Fig. S2). The effect of VPA on PLL morphogenesis was also confirmed by expression of cxcr7b, present in the trailing region (TR) of migrating primordium and freshly deposited neuromasts (Fig. 2) 7. In embryos treated with 0.2 μm TSA, PLL primordium migration was also impaired with delayed cxcr7b expression, and only 7.5% (3/40) of embryos had an L1 neuromast at 32 hpf, and only 65% (26/40) had L1, L2, and L3 neuromasts at 48 hpf. These results indicate that the process of PLL primordium migration can be significantly altered with low concentrations of HDAC inhibitors but cannot be permanently blocked. Taken together, these observations suggest that HDACs were required for migration of PLL primordium.
Figure 2.
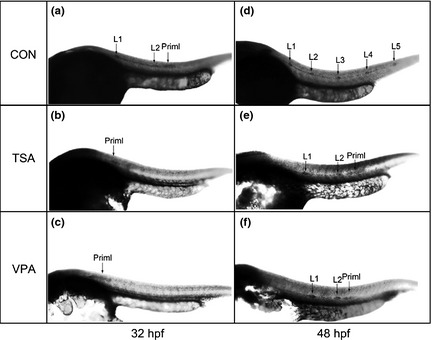
Expression of cxcr7b in the developing posterior lateral line ( PLL ) with whole‐mount in situ hybridization in 32 and 48 hours post‐fertilization ( hpf ) embryos. In 32 hpf control embryos, cxcr7b expression was present in the trailing half of the migrating primordium and in freshly deposited neuromasts L1 and L2 (arrows). As a comparison, cxcr7b expression was only detected in the region of the primordia in 0.2 μm trichostatin A (TSA)‐treated and 50 μm valproic acid (VPA)‐treated embryos (arrows). In 48 hpf control embryos, cxcr7b expression was present in all L1–L5 neuromasts along the trunk (arrows). Meanwhile, the 0.2 μm TSA‐treated and 50 μm VPA‐treated embryos showed fewer neuromasts as indicated by the expression of cxcr7b. The neuromasts are numbered and the primordia are marked with ‘Priml'.
Development of zebrafish PLL can be divided into two stages. During the early stage (less than 20 hpf), initial primordium cells are developed. During the later stage (from 20 to 48 hpf), previously formed sensory primordium travels along the horizontal myoseptum from the head to the tip of the tail. To determine specific function of HDACs over different periods of development, we treated Tg(Brn3c:mGFP) transgenic fish with HDAC inhibitors at various times and analysed development of neuromasts at 72 hpf (Table 1). There was reduction in numbers of neuromasts deposited following inhibitor treatment from 6 to 14 hpf, but the effect was only moderate compared to samples with continuous treatment (VPA‐treated fish from 6 to 14 hpf had 7.59 ± 0.087 neuromasts per side, n = 110, versus 72 hpf control fish had 8.34 ± 0.105 neuromasts per side, n = 92; P < 0.001. VPA treatment from 6 to 14 hpf versus VPA‐treated fish from 6 to 72 hpf had 5.14 ± 0.112 neuromasts per side, n = 90; P < 0.001. TSA‐treated fish from 6 to 14 hpf had 7.21 ± 0.108 neuromasts per side, n = 100, versus 72 hpf control fish had 8.22 ± 0.075 neuromasts per side, n = 90; P < 0.001. TSA treatment from 6 to 14 hpf versus TSA‐treated fish from 6 to 72 hpf had 4.68 ± 0.101 neuromasts per side, n = 100; P < 0.001). However, if HDAC inhibitor treatment was applied from 14 to 20 hpf, PLL neuromasts appeared at the normal developmental time (VPA‐treated fish from 14 to 20 hpf had 8.14 ± 0.079 neuromasts per side, n = 110 versus 72 hpf control fish; P = 0.237. TSA‐treated fish from 14 to 20 hpf had 8.05 ± 0.074 neuromasts per side, n = 112 versus 72 hpf control fish; P = 0.067). This observation suggests that the period from 6 to 14 hpf was an important stage for formation of the initial primordium. When inhibitors were present from 20 to 72 hpf, we also observed fewer neuromasts in the Tg(Brn3c:mGFP) transgenic fish indicating that HDACs had an effect on migration of PLL primordium (VPA‐treated fish from 20 to 72 hpf had 6.15 ± 0.083 neuromasts per side, n = 100 versus 72 hpf control fish; P < 0.001. TSA‐treated fish from 20 to 72 hpf had 5.79 ± 0.092 neuromasts per side, n = 100 versus 72 hpf control fish; p < 0.001).
Table 1.
Time dependence of HDAC inhibitor effects on PLL neuromast formation in Tg(Brn3c:mGFP) zebrafish embryos. GFP‐positive neuromasts can readily be observed at 3 dpf in the Tg(Brn3c:mGFP) line. For short‐term treatments, VPA (50 μm) or TSA (0.2 μm) was removed after 6 or 8 h of treatment and embryos were grown in normal culture medium up to 3 dpf.
| Treatment start time (hpf) | Treatment end time (hpf) | TSA 0.2 μm | VPA50 μm | ||
|---|---|---|---|---|---|
| Number of embryos | Number of neuromasts | Number of embryos | Number of neuromasts | ||
| 6 | 14 | 100 | 7.21 ± 0.108 | 110 | 7.59 ± 0.087 |
| 14 | 20 | 112 | 8.05 ± 0.074 | 110 | 8.14 ± 0.079 |
| 20 | 72 | 100 | 5.79 ± 0.092 | 100 | 6.15 ± 0.083 |
| 6 | 72 | 100 | 4.68 ± 0.101 | 90 | 5.14 ± 0.112 |
| Control | 72 | 90 | 8.22 ± 0.075 | 92 | 8.34 ± 0.105 |
HDAC, histone deacetylases; PLL, posterior lateral line; VPA, valproic acid; TSA, trichostatin A; hpf, hours post‐fertilization.
To further determine the basis for PLL defects observed in embryos treated with HDAC inhibitors, functional (mechanotransductively active) hair cells were stained with vital dye FM1‐43FX (Fig. S3) 36. VPA treatment from 6 hpf or 14 to 72 hpf led to a delay in functional PLL neuromast formation with fewer stained neuromasts appearing at 72 hpf compared to control embryos. Average number of FM1‐43FX–stained PLL neuromasts in control embryos was 7.93 ± 0.077 per side (n = 86). In contrast, average number of functional neuromasts was reduced to 3.79 ± 0.136 (n = 82) (P < 0.001 versus control) and 4.74 ± 0.124 (n = 98) (P < 0.001 versus control) when VPA was present from 6 to 72 hpf or from 14 to 72 hpf, respectively. For treatment from 14 to 72 hpf, we also observed a moderate effect on functional neuromast development compared to embryos with VPA treatment from 6 to 72 hpf (P < 0.001). This observation further suggests that the time from 6 to 14 hpf is an important period for development of the functional PLL neuromast. Similar results were also observed in samples treated with 0.2 μm TSA where the average number of stained neuromasts was 3.12 ± 0.152 (n = 52) (P < 0.001 versus control) and 4.63 ± 0.118 (n = 82) (P < 0.001 versus control) when TSA was present from 6 to 72 hpf or from 14 to 72 hpf, respectively (TSA treatment from 6 to 72 hpf versus TSA treatment from 14 to 72 hpf, P < 0.001).
To confirm that the PLL defect seen in VPA‐ and TSA‐treated embryos was caused by inhibition of HDACs, changes in acetylation of histone H3 and H4 proteins were analysed by immunocytochemistry and western blotting. As shown in Fig. S4, VPA and TSA treatment induced increase in levels of both acetyl H3 and acetyl H4. These results indicated that TSA and VPA might affect PLL formation by inhibiting normal activity of HDACs.
Abnormal pattern of PLL neuromast deposition
To examine effects of HDAC inhibition on spatial distribution of PLL neuromasts, we analysed average positions of the first four neuromasts. In embryos treated with 50 μm VPA, spatial distribution of neuromasts within the PLL organ was clearly altered compared to untreated control embryos at 48 hpf (Fig. 3). In particular, position of the first PLL neuromast (L1) in VPA‐treated larvae was displaced significantly posteriorly (Fig. 3a–c). The effect of VPA on position of L1 neuromast also was dose dependent (Fig. 3d). Average position of L1 neuromast in control embryos was 5.23 ± 0.1 somites (n = 70). In contrast, at VPA concentrations of 50, 100 and 150 μm, average positions of L1 neuromasts were 9.36 ± 0.34 (n = 70), 11.1 ± 0.21 (n = 102), and 11.4 ± 0.19 (n = 102) somites, respectively (Fig. 3d). Similar results were also observed in embryos treated with 0.2 μm TSA in which the L1 position was displaced posteriorly to 8.7 ± 0.25 somites (n = 70) (P < 0.001 versus control) (Fig. 3a–c).
Figure 3.
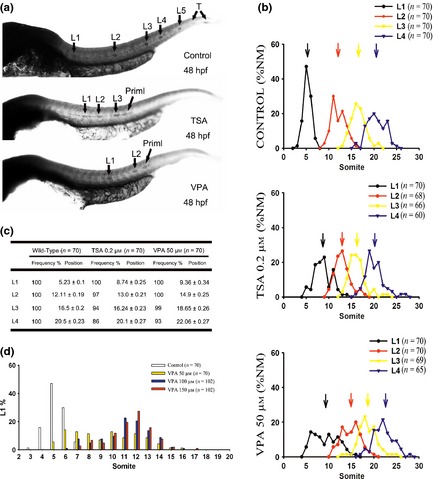
Spatial distribution of neuromasts. (a) Expression of cxcr7b in the developing posterior lateral line (PLL) by whole‐mount in situ hybridization in 48 hours post‐fertilization (hpf) larvae. Displacements of PLL neuromasts were observed in 0.2 μm trichostatin A (TSA)‐treated and 50 μm valproic acid (VPA)‐treated embryos. (b) Spatial distribution of the first four neuromasts (L1–L4). The arrows indicate the average positions (NM, neuromasts). (c) Frequency and positions of the first four neuromasts at 72 hpf. The spatial distribution of the L1–L4 neuromasts in VPA‐treated embryos was clearly altered compared with untreated embryos at 72 hpf. (Position of L1: control versus VPA, P < 0.05; control versus TSA, P < 0.05. Position of L2: control versus VPA, P < 0.05; control versus TSA, P < 0.05. Position of L3: control versus VPA, P < 0.05; control versus TSA, P > 0.05. Position of L4: control versus VPA, P < 0.05; control versus TSA, P > 0.05). (d) The distribution of L1 positions (somite number) in 72 hpf embryos during VPA treatment with various dosages of inhibitor.
Reduced proliferation in migrating primordium
Cell proliferation is crucial for formation of a complete and normal PLL system. To determine whether modulation of HDAC activity could regulate cell proliferation in migrating primordia, we measured ratios between BrdU‐positive cells and total nuclei in migrating primordia at various developmental stages. Figure 4 illustrates that VPA‐treated primordia had significantly lower BrdU index compared to control primordia (Fig. 4a,b; at the developmental stage before deposition of any PLL neuromasts, BrdU index for control primordia was 0.43 ± 0.025, n = 10, and BrdU index for VPA‐treated primordia was 0.17 ± 0.016, n = 10; P < 0.001). Similar results were also observed in embryos treated with TSA (Fig. 4a,b; at the developmental stage before deposition of any neuromasts, BrdU index for control primordia was 0.42 ± 0.025, n = 10, and BrdU index for TSA‐treated primordia at the same stage was 0.18 ± 0.02, n = 10; P < 0.001). We further examined BrdU incorporation index in both LE and TR cells. LE/TR BrdU index was calculated as ratio of BrdU incorporation index in LE to TR in control and HDAC inhibitor‐treated primordia. Our results showed that there was no difference between leading/TRs in HDAC inhibitor‐treated and control embryos. At the developmental stage before deposition of any PLL neuromasts, LE/TR BrdU index for VPA‐treated primordia was 1.79 ± 0.17, n = 10, compared to LE/TR BrdU index for control primordia of 1.88 ± 0.25, n = 10 (P = 0.734). LE/TR BrdU index for TSA‐treated primordia was 1.77 ± 0.29, n = 10, compared to LE/TR BrdU index for control primordia of 1.98 ± 0.14, n = 10 (P = 0.212). We then compared LE/TR BrdU index of the primordium at different developmental stages, including after deposition of neuromasts L1, L2, L3 and L4, and found that LE/TR BrdU index in treated and untreated samples did not differ significantly.
Figure 4.
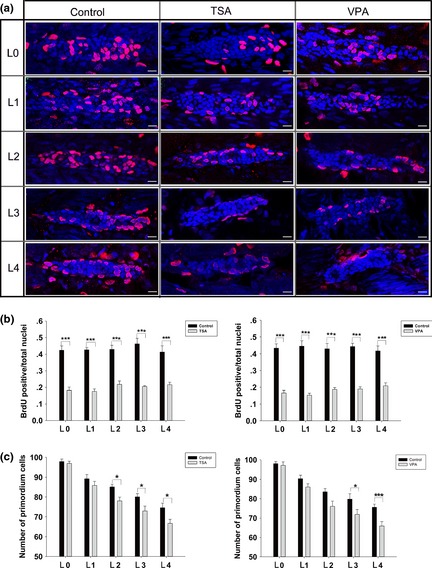
Effect of histone deacetylases ( HDAC ) inhibitors on cell proliferation. (a) Cell proliferation analysis in control, trichostatin A (TSA)‐treated, and valproic acid (VPA)‐treated embryos. After the BrdU pulse, embryos were fixed before (L0; the first row) and after deposition of neuromast L1 (the second row), L2 (the third row), L3 (the fourth row) or L4 (the fifth row). In these images, nuclei are labelled with DAPI and red spots represent BrdU signal. Scale bars = 10 μm. (b) The ratio of BrdU‐positive cells to total cells in control (black bars) and HDAC inhibitor‐treated (grey bars) embryos from 0 to 4 deposited neuromasts. Total cells were counted by DAPI staining. (c) Summary of HDAC inhibitor effects on the number of primordium cells. Primordium cell analysis in control (black bars) and HDAC inhibitor‐treated (grey bars) embryos from 0 to 4 deposited neuromasts, *P < 0.05; ***P < 0.001.
Because initial primordium cells are crucial for formation of a complete and normal PLL, one possible explanation for delay in neuromast development is that HDAC inhibitors reduce initial cell numbers in the primordium. To address this possibility, we counted DAPI‐labelled nuclei in primordia of both VPA‐treated and control embryos at the stage before deposition of the first PLL neuromast (30 hpf for treated and 24 hpf for untreated embryos). Our analysis revealed that there was no significant difference in primordium cell counts (primordium cell count in control embryos at 24 hpf was 98.05 ± 1.13, n = 20 and primordium cell count in VPA‐treated embryos at 30 hpf was 97.15 ± 1.79, n = 20; P = 0.673). Similar results were also obtained with TSA‐treated embryos (primordium cell count in control embryos was 97.85 ± 1.27, n = 20 and primordium cell count in TSA‐treated embryos was 96.9 ± 1.15, n = 20; P = 0.377). These results suggest that HDAC inhibitors did not affect initial numbers of cells in the early PLL primordium. However, during migration, reduction in primordium cell numbers in embryos treated with HDAC inhibitors became more evident when compared to controls (P < 0.05) (Fig. 4c). We also measured cell numbers in the first few deposited neuromasts. Our results showed that neuromasts deposited from treated primordia contained the same number of cells as control neuromasts (Fig. 5).
Figure 5.
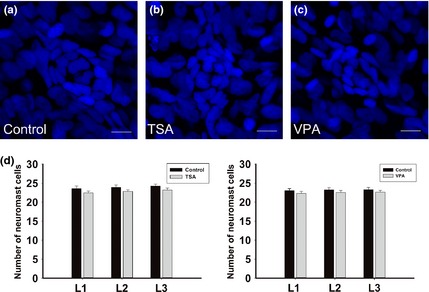
Effect of of histone deacetylases ( HDAC ) inhibitors on the number of the deposited neuromast cells. (a–c) Neuromast cell analysis in control (a), trichostatin A‐treated (b) and valproic acid‐treated (c) embryos. Embryos were fixed after deposition of neuromast L1. In these images, nuclei are labelled with DAPI. Scale bars = 10 μm. (d) Neuromast cell analysis in control (black bars) and HDAC inhibitor‐treated (grey bars) embryos from L1 to L3 neuromasts. n = 20 for all the groups.
We further tested whether cell death was responsible for defective PLL neuromast formation. We detected only limited apoptosis in both control and inhibitor‐treated embryos, and this was confirmed by cleaved caspase‐3 immunolabelling (Fig. S5). Thus, cell death was ruled out as a major cause of PLL defect in embryos treated with HDAC inhibitors.
Fewer hair cells developed in the neuromast
To examine effects of HDAC inhibition on differentiation of hair cells in the developing lateral line neuromast, we treated the transgenic zebrafish embryos with HDAC inhibitor at 6 hpf, fixed them at 72 hpf, and measured numbers of hair cells in the neuromast by FM1‐43FX staining and GFP expression (Fig. 6). We found that treatment with VPA resulted in reduction in hair cells (Fig. 6; 72 hpf control fish had 4.47 ± 0.13 FM1‐43FX–stained hair cells in the L1 neuromast, n = 30 and 72 hpf VPA‐treated fish had 2.67 ± 0.18 FM1‐43FX–stained cells in the L1 neuromast, n = 30; P < 0.001. Seventy‐two hours post‐fertilization control fish had 6.33 ± 0.11 GFP‐positive cells in the L1 neuromast, n = 30 and 72 hpf VPA‐treated fish had 4.0 ± 0.11 GFP‐positive cells in the L1 neuromast, n = 30; P < 0.001). Similarly, treatment with TSA also caused reduction in hair cells (Fig. 6; 72 hpf control fish had 4.6 ± 0.15 FM1‐43FX–stained hair cells in the L1 neuromast, n = 30, and 72 hpf TSA‐treated fish had 2.23 ± 0.09 FM1‐43FX–stained cells in the L1 neuromast, n = 30; P < 0.001. Seventy‐two hours post‐fertilization control fish had 6.23 ± 0.21 GFP‐positive cells in the L1 neuromast, n = 30, and 72 hpf TSA‐treated fish had 4.17 ± 0.07 GFP‐positive cells in the L1 neuromast, n = 30; P < 0.001). Thus, HDAC inhibition results in reduction in numbers of hair cells during initial differentiation processes.
Figure 6.
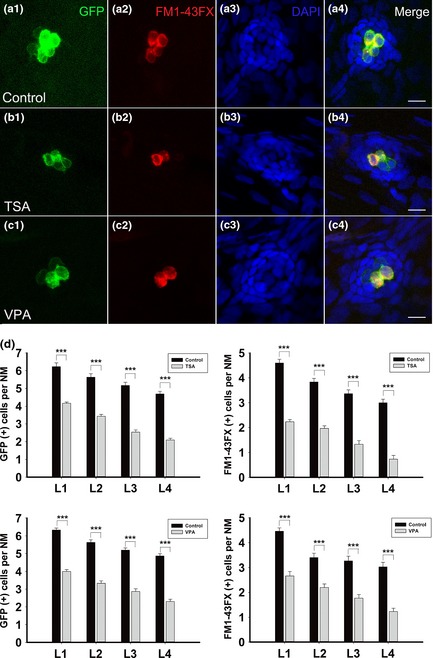
Effect of histone deacetylases ( HDAC ) inhibitors on the number of hair cells. (a–c) FM1‐43FX‐stained functional hair cell analysis in control (a1–a4), trichostatin A‐treated (b1–b4) and valproic acid‐treated (c1–c4) neuromasts. Embryos were fixed at 72 hours post‐fertilization. Scale bars = 10 μm. (d) Hair cell analysis in control (black bars) and HDAC inhibitor‐treated (grey bars) embryos from L1 to L4 neuromasts. n = 30 for all the groups, ***P < 0.001.
Discussion
Histone deacetylases belong to a vast family of enzymes that are known to have roles in numerous biological processes during development. It has been demonstrated that global deletion of Class I HDACs leads to early lethality in mice, suggesting that HDACs play a critical role in control of gene expression programs 15. Early studies have demonstrated that all Class I HDACs are highly expressed in the developing brain, specially in forebrain regions, during early embryogenesis in chickens and mice. Robust Class I HDAC expression can also be observed in optic and otic vesicles, nose placodes, limb buds, neural crest cells, and developing liver, lung and heart 37. Until now, however, the role of HDACs in zebrafish PLL development had remained unclear.
In this study, HDACs were pharmacologically inhibited to investigate their effect on PLL development. A significant reduction in numbers of DAPI, cxcr7b, GFP and FM1‐43FX–stained neuromasts was seen in treated embryos. Our data showed that HDAC inhibitor treatment resulted in defective formation of zebrafish PLL. HDAC inhibition did not disturb migration of the primordium, but affected both differentiation and functionality of PLL neuromasts. It is imperative to note that the overall number of cells in the primordium has an impact on rate of primordium migration. Here, we have compared total number of cells in primordia at different developmental stages. We observed no evident differences in initial numbers of primordium cells in treated and control embryos at a developmental stage just before deposition of the L1 neuromast. However, after migration starts, numbers of primordium cells in treated embryos reduced significantly. One obvious interpretation of these results is that more cells were deposited during primordium migration. We have addressed this possibility by measuring cell number in the first few deposited neuromasts. Our results indicate that control and HDAC inhibitor‐treated primordia deposit neuromasts that are not significantly different in cell number. This observation suggests that reduction in primordium cell numbers in treated embryos was not due to more cells being dropped off.
Previous studies have shown that deposition of neuromasts is largely regulated by cell proliferation within the migrating primordium 38, 39, 40. Here, we have analysed proliferation of migrating primordium cells and observed significantly fewer BrdU‐stained cells in HDAC inhibitor‐treated embryos. This result is consistent with previous studies showing that HDAC inhibitors can hinder cell proliferation in both transformed and normal cells 41, 42, 43. The lower cell number is largely accounted for by reduced cell proliferation within migrating primordia. In the proliferation study, it might be informative to divide the primordium into LE and trailing zone, but it is difficult to tell whether proliferation is different in these two functionally distinct compartments of the primordium. In our experiment, we observed that that there was no difference between changes in leading/TRs in both HDAC inhibitor‐treated and control embryos. This suggests that HDAC activity was required for proliferation of both leading and TR cells of the primordium. Studies by Aman et al. 38 have demonstrated that Wnt/β‐catenin and Fgf signalling pathways act in concert to induce cell proliferation in the primordium. The phenotype that results from inhibition of HDAC activity might be due to disruption of signalling pathways. Future studies will aim to examine the exact nature of how HDAC activity affects proliferation of the primordium and nature of the different underlying signalling pathways.
One previous study has demonstrated that proliferation defects in the primordium lead to depletion of cells in the leading zone and to collapse of the primordium 40. In the present study, we did not observed any collapse of the primordium in embryos treated with HDAC inhibitors, at least not at the 50 μm dose used in most of our experiments. Development of PLL primordium in treated embryos can be completed by reaching the end of the animal at around 5 dpf and depositing 2 or 3 terminal neuromasts at the tip of the tail. This suggests that reduction in number of deposited neuromasts in HDAC inhibitor‐treated embryos could result from reduction in rate of migration of the primordium. Furthermore, we observed that HDAC inhibition induced displacement of PLL neuromasts along the bodies of the embryos. In particular, the first PLL neuromast (L1) was displaced posteriorly in a dose‐dependent manner. This phenotype did not recover over time. We presently have no interpretation for the abnormal somite distribution of PLL neuromasts in the treated embryos. We speculate, however, that these changes are possibly the result of defects in cell proliferation or in other molecular mechanisms associated with morphogenesis.
To further clarify the specific effects of HDAC inhibitors on different stages of PLL development, we divided treatment time from 6 to 72 hpf into the subintervals of 6–14, 14–20 and 20–72 hpf. It has previously been shown that proliferation behaviour in lateral line precursors mainly occurs between 6 and 10 hpf and precedes formation of the PLL placode 39. In our study, transient VPA treatment from 6 to 14 hpf led to a delay in PLL neuromast formation. It seems that HDACs are probably required in specification of PLL primordium before the PLL placode, from the underlying basement membrane. We hypothesize, therefore, that the defect observed in treated embryos from 6 to 14 hpf is possibly due to an effect of HDAC inhibitors on proliferation of primordium precursors. However, our data show that there was no significant difference in initial primordium cell counts. It seems that the migration defect in treatment from 6 to 14 hpf is possibly the result of defects in other molecular signalling mechanisms associated with primordium morphogenesis. We also found that HDAC activity was crucial during developmental stages between 20 and 72 hpf. HDAC inhibitor treatment from 20 to 72 hpf reduced the number of neuromasts, and this was associated with migration of the primordium. Altogether, our results suggest that HDACs might have effects on both development of primordium precursors and migration of the primordium.
In an early study, Slattery et al. reported regulatory effects of HDACs on regenerative proliferation and differentiation in the avian utricle 33. Importantly, these workers suggested that HDAC inhibition did not appear to directly affect hair cell differentiation during the regenerative process. However, there is little understanding about whether or how HDAC activity is required for production of hair cells during embryonic development. In the present study, we found that pharmacological inhibition of HDACs resulted in reduction in numbers of hair cells in neuromasts during the initial differentiation process. This suggests that HDAC activity is required not only for migration and proliferation of the PLL primordium but also for hair cell differentiation.
Several studies have demonstrated that Wnt/β‐catenin signalling controls proliferation of progenitors of the PLL primordium leading zone and are critical for stable morphogenesis of the lateral line 5, 38, 40, 44. Wnt/β‐catenin signalling has been shown to play a role in regulating migration of PLL primordia, proliferation of lateral line progenitors during development, neuromast formation and hair cell regeneration 38, 45. HDACs can participate in numerous developmental signalling pathways, including Notch and Wnt, by interacting with transcriptional repressors or activators to form transcriptional complexes 25, 46. An earlier study found that HDAC1 antagonized Wnt signalling to promote cell cycle exit of retinoblasts 25. Anti‐proliferative function of HDAC in that study was unexpected as it was generally accepted that HDAC inhibitors efficiently suppress cell proliferation in both transformed and normal cells 41, 42, 43. Furthermore, HDAC1‐deficient mice have severe proliferation defects 47. Yamaguchi et al. demonstrated that hyperproliferation occurs exclusively in the neural retina, but not in the brain, in the add mutants that encode HDAC1. They explained that this result was because the roles of HDACs are diverse among species and cell types. In our study, we have no interpretation for PLL neuromast developmental defects in HDAC inhibitor‐treated embryos. We speculate that these changes are possibly the result of defects in cell proliferation or in other molecular mechanisms associated with morphogenesis.
Given the importance of Wnt/β‐catenin signalling in control of proliferation at the leading zone of PLL primordia and its role in proliferation of lateral line progenitors during development, neuromast formation and hair cell regeneration, we hypothesize that roles of HDACs in PLL neuromast formation might depend on the Wnt signalling pathway. Kim et al. have reported that HDAC activity has an essential role in vertebrate heart tube formation by activating Wnt/β‐catenin signalling 24. Here, we speculate that HDACs may play similar key roles in controlling formation of PLL neuromasts in zebrafish by activating Wnt signalling. It would, therefore, be interesting to look at expression of Wnt targets in the PLL primordium following HDAC inhibition. Further investigations could also be directed towards identifying specific genes as possible targets for HDAC inhibitor regulation. This will be a future focus of our research and might provide better understanding of the link between epigenetic regulation and zebrafish embryonic PLL development.
In this study, we demonstrate a functional role for HDACs in zebrafish PLL development. We show that HDACs are important for primordium migration and neuromast development. Treatment with HDAC inhibitors caused significant reduction in numbers of both proliferating cells in the primordium and sensory hair cells in the neuromast during PLL development. Further studies will aim to explore specific types of HDACs that have effects on PLL development and will investigate relationships between the signalling pathways (such as Wnt/β‐catenin signalling) and the role of HDACs in primordium migration.
Conflict of interests
Jianyong Shou is an employee of China Novartis Institutes for Biomedical Research (CNIBR). This does not alter the author's adherence to all the Cell Proliferation policies on sharing data and materials.
Supporting information
Figure S1. Dose dependence of VPA's effect on the early development of zebrafish embryos.
{kind=link}
Figure S2. Imaging of PLL neuromast distribution in a Tg(Brn3c:mGFP) embryo treated with VPA at 5 dpf.
{kind=link}
Figure S3. Time dependence of HDAC inhibitor effects on PLL neuromast development.
{kind=link}
Figure S4. Histone acetylation level.
{kind=link}
Figure S5. Cleaved caspase‐3 staining.
{kind=link}
Acknowledgments
The authors thank Min Yu, Jingying Hu and Shaoyang Sun for their expert fish care, and Shanhe Yu, Jin Li and Yalin Huang for technical assistance. The authors are also grateful to Zhengyi Chen for his generous gift of the Brn3c:mGFP transgenic line.
Funding: This work was supported by grants from the Major State Basic Research Development Program of China (973 Program) (2011CB504506, 2010CB945503) and the National Natural Science Foundation of China (No. 81230019, 81070793, 81371094, 81300825), and was sponsored by the Shanghai Rising‐Star Program (12QA1400500), the Program for Changjiang Scholars and Innovative Research Team in University (IRT1010), the Specialized Research Fund for the Doctor Program of Higher Education (20120071110077), the Program of Outstanding Shanghai Academic Leader (11XD1401300), the Innovation Programme of Major Basic Research Project, the Science and Technology Commission of Shanghai Municipality (09DJ1400602) and the Program of Leading Medical Personnel in Shanghai. This study was partly supported by the Novartis‐Fudan collaboration.
References
- 1. Montgomery J, Carton G, Voigt R, Baker C, Diebel C (2000) Sensory processing of water currents by fishes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gompel N, Cubedo N, Thisse C, Thisse B, Dambly‐Chaudiere C, Ghysen A (2001) Pattern formation in the lateral line of zebrafish. Mech. Dev. 105, 69–77. [DOI] [PubMed] [Google Scholar]
- 3. Ghysen A, Dambly‐Chaudiere C (2004) Development of the zebrafish lateral line. Curr. Opin. Neurobiol. 14, 67–73. [DOI] [PubMed] [Google Scholar]
- 4. Aman A, Piotrowski T (2009) Multiple signaling interactions coordinate collective cell migration of the posterior lateral line primordium. Cell Adh. Migr. 3, 365–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aman A, Piotrowski T (2008) Wnt/beta‐catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev. Cell 15, 749–761. [DOI] [PubMed] [Google Scholar]
- 6. Lecaudey V, Cakan‐Akdogan G, Norton WH, Gilmour D (2008) Dynamic Fgf signaling couples morphogenesis and migration in the zebrafish lateral line primordium. Development 135, 2695–2705. [DOI] [PubMed] [Google Scholar]
- 7. Dambly‐Chaudiere C, Cubedo N, Ghysen A (2007) Control of cell migration in the development of the posterior lateral line: antagonistic interactions between the chemokine receptors CXCR4 and CXCR7/RDC1. BMC Dev. Biol. 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. David NB, Sapede D, Saint‐Etienne L, Thisse C, Thisse B, Dambly‐Chaudiere C, et al (2002) Molecular basis of cell migration in the fish lateral line: role of the chemokine receptor CXCR4 and of its ligand, SDF1. Proc. Natl. Acad. Sci. USA 99, 16297–16302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gamba L, Cubedo N, Lutfalla G, Ghysen A, Dambly‐Chaudiere C (2010) Lef1 controls patterning and proliferation in the posterior lateral line system of zebrafish. Dev. Dyn. 239, 3163–3171. [DOI] [PubMed] [Google Scholar]
- 10. Chen X, Lou Q, He J, Yin Z (2011) Role of zebrafish lbx2 in embryonic lateral line development. PLoS One 6, e29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jenuwein T, Allis CD (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 12. Berger SL (2002) Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12, 142–148. [DOI] [PubMed] [Google Scholar]
- 13. Kurdistani SK, Grunstein M (2003) Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 4, 276–284. [DOI] [PubMed] [Google Scholar]
- 14. Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 15. Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marks PA, Miller T, Richon VM (2003) Histone deacetylases. Curr. Opin. Pharmacol. 3, 344–351. [DOI] [PubMed] [Google Scholar]
- 17. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munster PN, Troso‐Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM (2001) The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 61, 8492–8497. [PubMed] [Google Scholar]
- 19. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S et al (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karen J, Rodriguez A, Friman T, Dencker L, Sundberg C, Scholz B (2011) Effects of the histone deacetylase inhibitor valproic acid on human pericytes in vitro. PLoS One 6, e24954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Felisbino MB, Tamashiro WM, Mello ML (2011) Chromatin remodeling, cell proliferation and cell death in valproic acid‐treated HeLa cells. PLoS One 6, e29144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marks PA, Richon VM, Rifkind RA (2000) Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 92, 1210–1216. [DOI] [PubMed] [Google Scholar]
- 23. Farooq M, Sulochana KN, Pan X, To J, Sheng D, Gong Z et al (2008) Histone deacetylase 3 (hdac3) is specifically required for liver development in zebrafish. Dev. Biol. 317, 336–353. [DOI] [PubMed] [Google Scholar]
- 24. Kim YS, Kim MJ, Koo TH, Kim JD, Koun S, Ham HJ et al (2012) Histone deacetylase is required for the activation of Wnt/beta‐catenin signaling crucial for heart valve formation in zebrafish embryos. Biochem. Biophys. Res. Commun. 423, 140–146. [DOI] [PubMed] [Google Scholar]
- 25. Yamaguchi M, Tonou‐Fujimori N, Komori A, Maeda R, Nojima Y, Li H et al (2005) Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development 132, 3027–3043. [DOI] [PubMed] [Google Scholar]
- 26. Cunliffe VT (2004) Histone deacetylase 1 is required to repress Notch target gene expression during zebrafish neurogenesis and to maintain the production of motoneurones in response to hedgehog signalling. Development 131, 2983–2995. [DOI] [PubMed] [Google Scholar]
- 27. Harrison MR, Georgiou AS, Spaink HP, Cunliffe VT (2011) The epigenetic regulator Histone Deacetylase 1 promotes transcription of a core neurogenic programme in zebrafish embryos. BMC Genomics. 12, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN (2009) Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. USA 106, 7876–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cunliffe VT, Casaccia‐Bonnefil P (2006) Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mech. Dev. 123, 24–30. [DOI] [PubMed] [Google Scholar]
- 30. Shen S, Li J, Casaccia‐Bonnefil P (2005) Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J. Cell Biol. 169, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marin‐Husstege M, Muggironi M, Liu A, Casaccia‐Bonnefil P (2002) Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J. Neurosci. 22, 10333–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Provenzano MJ, Domann FE (2007) A role for epigenetics in hearing: establishment and maintenance of auditory specific gene expression patterns. Hear. Res. 233, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slattery EL, Speck JD, Warchol ME (2009) Epigenetic influences on sensory regeneration: histone deacetylases regulate supporting cell proliferation in the avian utricle. J. Assoc. Res. Otolaryngol. 10, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 35. Xiao T, Roeser T, Staub W, Baier H (2005) A GFP‐based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development 132, 2955–2967. [DOI] [PubMed] [Google Scholar]
- 36. Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP (2001) FM1‐43 dye behaves as a permeant blocker of the hair‐cell mechanotransducer channel. J. Neurosci. 21, 7013–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murko C, Lagger S, Steiner M, Seiser C, Schoefer C, Pusch O (2010) Expression of class I histone deacetylases during chick and mouse development. Int. J. Dev. Biol. 54, 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aman A, Nguyen M, Piotrowski T (2011) Wnt/beta‐catenin dependent cell proliferation underlies segmented lateral line morphogenesis. Dev. Biol. 349, 470–482. [DOI] [PubMed] [Google Scholar]
- 39. Laguerre L, Soubiran F, Ghysen A, Konig N, Dambly‐Chaudiere C (2005) Cell proliferation in the developing lateral line system of zebrafish embryos. Dev. Dyn. 233, 466–472. [DOI] [PubMed] [Google Scholar]
- 40. Valdivia LE, Young RM, Hawkins TA, Stickney HL, Cavodeassi F, Schwarz Q et al (2011) Lef1‐dependent Wnt/beta‐catenin signalling drives the proliferative engine that maintains tissue homeostasis during lateral line development. Development 138, 3931–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552. [DOI] [PubMed] [Google Scholar]
- 42. Jin H, Liang L, Liu L, Deng W, Liu J (2013) HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J Cell Biochem. 114, 1498–1509. [DOI] [PubMed] [Google Scholar]
- 43. Baumann P, Junghanns C, Mandl‐Weber S, Strobl S, Oduncu F, Schmidmaier R (2012) The pan‐histone deacetylase inhibitor CR2408 disrupts cell cycle progression, diminishes proliferation and causes apoptosis in multiple myeloma cells. Br. J. Haematol. 156, 633–642. [DOI] [PubMed] [Google Scholar]
- 44. McGraw HF, Drerup CM, Culbertson MD, Linbo T, Raible DW, Nechiporuk AV (2011) Lef1 is required for progenitor cell identity in the zebrafish lateral line primordium. Development 138, 3921–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Head JR, Gacioch L, Pennisi M, Meyers JR (2013) Activation of canonical Wnt/beta‐catenin signaling stimulates proliferation in neuromasts in the zebrafish posterior lateral line. Dev. Dyn. 242, 832–846. [DOI] [PubMed] [Google Scholar]
- 46. Lightman EG, Harrison MR, Cunliffe VT (2011) Opposing actions of histone deacetylase 1 and Notch signalling restrict expression of erm and fgf20a to hindbrain rhombomere centres during zebrafish neurogenesis. Int. J. Dev. Biol. 55, 597–602. [DOI] [PubMed] [Google Scholar]
- 47. Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G et al (2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 21, 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dose dependence of VPA's effect on the early development of zebrafish embryos.
Figure S2. Imaging of PLL neuromast distribution in a Tg(Brn3c:mGFP) embryo treated with VPA at 5 dpf.
Figure S3. Time dependence of HDAC inhibitor effects on PLL neuromast development.
Figure S4. Histone acetylation level.
Figure S5. Cleaved caspase‐3 staining.