

Abstract
Abstract. Objectives: Both interleukin‐6 (IL‐6) and transforming growth factor‐β (TGF‐β) are crucially involved in fibrotic events that characterize interstitial lung diseases (ILD). Therefore, the aim of this study was to investigate in primary cultures of normal and fibrotic human lung fibroblasts (HLF), exposed to either IL‐6 or TGF‐β1, the effects on phosphorylation of mitogen‐activated protein kinases (MAPK) and cell growth of IL‐6 signalling inhibition, performed by the IL‐6 receptor superantagonist Sant7.Materials and methods: MAPK phosphorylation was detected by Western blotting, HLF viability and proliferation were evaluated using the trypan blue staining and the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay, respectively. Results: Sant7, at a concentration of 1 µg/mL, was capable of significantly inhibiting HLF proliferation and MAPK phosphorylation induced by cell exposure to IL‐6 (100 ng/mL) or TGF‐β1 (10 ng/mL), whose actions were more evident in fibrotic cells. Conclusions: These findings suggest that, in HLFs derived from patients with ILDs, the proliferative mechanisms activated by TGF‐β1 are at least in part mediated by an increased release of IL‐6, leading to phosphorylation‐dependent MAPK activation. Such preliminary findings may thus open new therapeutic perspectives for fibrogenic ILDs, based on inhibition of signal transduction pathways stimulated by the IL‐6 receptor.
INTRODUCTION
Interleukin‐6 (IL‐6) belongs to a cytokine family comprised of multifunctional mediators (e.g. interleukin‐11, oncostatin M, leukaemia inhibitory factor, ciliary neutrophic factor, cardiotrophin‐1) involved in several biological and pathophysiological processes, including those underlying some chronic inflammatory respiratory disorders such as interstitial lung diseases (ILD). Within the respiratory system, IL‐6 is secreted by many immune/inflammatory and structural cells, such as T and B lymphocytes, macrophages, lung fibroblasts and alveolar epithelial cells (Taga 1997). IL‐6 production is increased in patients with sarcoidosis or systemic sclerosis with pulmonary involvement (Crestani et al. 1994; Shahar et al. 1996). Thus, this cytokine appears to be actively implicated in profibrogenic mechanisms responsible for lung fibrosis. In particular, IL‐6 is a powerful mitogenic agent for human lung fibroblasts (HLF) obtained from patients with idiopathic pulmonary fibrosis (IPF) (Moodley et al. 2003a).
Interleukin‐6, like the other members of the IL‐6‐type cytokine family, exerts its biological actions by activation of target genes that regulate cell differentiation, death, survival and proliferation. All physiological and pathophysiological effects of IL‐6 are mediated by its interaction with a plasma membrane receptor system comprising of an 80‐kDa ligand‐binding α‐chain glycoprotein (gp80 or IL‐6Rα), and a 130‐kDa signal‐transducing β‐chain called gp 130. In particular, IL‐6 binds to the gp80 subunit thus forming a complex that leads to recruitment and homodimerization of the gp130 signalling component (Kamimura et al. 2003) that then activates the Janus‐activated kinase 1 (JAK1)/signal transducer and activation of transcription 3 (STAT3) signal transduction pathway, thereby promoting translocation of STAT3 to the nucleus, where this transcription factor significantly affects gene expression. In addition to phosphorylating both gp130 and STAT3, JAK1 also phosphorylates the gp130‐associated protein SHP2 (SH2 domain‐containing tyrosine phosphatase), which subsequently interacts with the adaptor protein Grb2 (growth factor receptor‐bound protein 2) (Heinrich et al. 2003). The latter is constitutively complexed with the Ras‐activating factor Sos (son of sevenless), which thus enables the sequential, downstream activation of the Ras/Raf‐dependent cascade of mitogen‐activated protein kinases (MAPK). These include ERK (extracellular signal‐regulated kinases), JNK (c‐Jun N‐terminal kinases) and p38 subgroups, which upon phosphorylation‐dependent activation becomes able to interact with their cytoplasmic substrates and also to translocate into the nucleus, where many MAPK targets such as transcription factors and histones are located (Widmann et al. 1999; Chang & Karin 2001). The family of IL‐6‐type cytokines can stimulate ERK1/2, the MAPK subgroup mostly involved in cell survival and growth, as well as JNK and p38, which predominantly regulate stress, inflammatory and apoptotic responses.
Given the biological and pathological relevance of IL‐6 pleiotropic functions, the IL‐6 receptor is currently considered to be an important therapeutic target for IL‐6‐mediated diseases (Nishimoto & Kishimoto 2006). Among the possible molecular strategies aimed at blocking IL‐6 receptor activation, one of the most promising approaches is based on the use of the potent IL‐6 receptor antagonist called superantagonist Sant7. The strategy has been developed through molecular‐modelling guided mutagenesis experimentation, targeting key residues of human IL‐6 amino acid sequence (Savino et al. 1994, 1997). The resulting mutant binds the gp80 receptor subunit with a 65‐fold higher affinity than wild‐type IL‐6, but it is completely unable to bind and activate the gp130 signalling chain (Sporeno et al. 1996). Due to these specific features, Sant7 is capable of inhibiting all IL‐6 biological activities tested so far, both in vitro and in vivo (Tassone et al. 2000, 2005).
In the respiratory system, one of the most effective inducers of IL‐6 production is transforming growth factor‐β (TGF‐β), which is overexpressed in lung parenchymal tissue of patients with IPF and other ILDs (Khalil et al. 1996). In primary HLFs it has been demonstrated that TGF‐β1 up‐regulates IL‐6 expression at both mRNA and protein levels (Eickelberg et al. 1999). This action is dependent on TGF‐β1‐induced activation of the transcription factor AP‐1 (activating protein‐1), which thus acts as the effector molecule of TGF‐β1 signalling at the level of the IL‐6 gene promoter, that contains DNA consensus sequences for AP‐1 binding. Moreover, it has also been shown in HLFs that TGF‐β1‐induced AP‐1 activation and IL‐6 gene expression are mediated by hydrogen peroxide generation and MAPK stimulation (Junn et al. 2000). We recently reported that increase in IL‐6 secretion elicited by TGF‐β1 may play an important role in the proliferative effect exerted by this growth factor on primary HLFs, obtained from patients with IPF (Pelaia et al. 2007).
Therefore, in light of all these considerations we decided to evaluate, in primary cultures of normal and fibrotic HLFs, the effects of Sant7 on cell proliferation induced by IL‐6 and TGF‐β1. In addition, a further objective of our study was to investigate interference of Sant7 with MAPK signalling pathways activated in these cells by either IL‐6 or TGF‐β1.
MATERIALS AND METHODS
Reagents
Recombinant human IL‐6 and TGF‐β1 were purchased from PeproTech (Rocky Hill, NJ, USA). Sant7 was produced as previously described. Anti‐phospho‐p38, anti‐phospho‐ERK1/2 and anti‐phospho‐JNK monoclonal antibodies were obtained from New England Biolabs (Beverly, MA, USA); anti‐(total)‐p38, anti‐(total)‐ERK1/2 and anti‐(total)‐JNK polyclonal antibodies were provided from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Trypan blue, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT), and all reagents needed for cell culture, protein extraction and Western blotting were purchased from Sigma (St. Louis, MO, USA).
Primary cultures of normal and fibrotic human lung fibroblasts
Normal lung fibroblasts were obtained from histologically unaltered areas of surgical specimens taken from patients who underwent either lobectomy or pneumonectomy for lung cancer. Primary lines of HLFs were also derived from fibrotic lung tissue, obtained from patients with the UIP (usual interstitial pneumonia) form of IPF. Cell cultures were established by using an outgrowth from explant according to the method described by Jordana and co‐workers (Jordana et al. 1988; Vancheri et al. 2000). Lung specimens were chopped into pieces less than 1 mm3 and were washed once with phosphate‐buffered saline (PBS) and twice with RPMI containing 10% foetal calf serum (FCS), penicillin 100 U/mL, streptomycin 100 µg/mL and fungizone 25 µg/mL (supplemented RPMI) (Gibco, Paisley, UK); 8–10 pieces of washed specimens were then plated in a 100‐mm polystyrene dish (Falcon, Becton‐Dickinson, Lincoln Park, NJ, USA) and were overlaid with a coverslip held to the dish with sterile vaseline. Ten millilitres of supplemented RPMI were added and the tissue was incubated at 37 °C with 5% CO2. The medium was changed weekly. When a monolayer of fibroblast‐like cells covered the bottom of the dish, usually 5–6 weeks later, the explant tissue was removed, and the cells were then trypsinized for 10 min, re‐suspended in 10 mL of supplemented RPMI, and were plated in 100‐mm tissue culture dishes. Subsequently, cells were split 1 : 2 at confluence, usually weekly. Aliquots of cells were frozen and stored in liquid nitrogen. In all experiments, we used cell lines at a passage earlier than the 10th.
Cell stimulation and pre‐treatment with Sant7
Normal and fibrotic HLFs were incubated for different periods of time (24, 48 and 72 h) with either IL‐6 (10 and 100 ng/mL) or TGF‐β1 (10 ng/mL), in the absence or presence of pre‐treatment with Sant7 (1 µg/mL), initiated 12 h before cell exposure to IL‐6 or TGF‐β1. The solvent used to dissolve IL‐6 and TGF‐β1, alone, was used as control. After cell stimulation, viability and proliferation assays were performed, and HLFs were processed for protein extraction and immunoblotting.
Cell viability and proliferation
Cell viability was assessed by light microscopy and dye exclusion, using trypan blue; cell numbers were evaluated by direct counting, performed using a haemocytometer. Cell proliferation was investigated by MTT assay, based on the conversion by mitochondrial dehydrogenases of the substrate containing a tetrazolium ring into blue formazan, detectable spectrophotometrically. The level of blue formazan was then used as an indirect index of cell density. Briefly, after treatment with either IL‐6 or TGF‐β1, cells were exposed to MTT (5 µg/mL) for 150 min at 37 °C. The medium was then removed and cells were solubilized with acidified isopropanol and 2% sodium dodecyl sulfate (SDS). After complete solubilization, presence of blue formazan was evaluated spectrophotometrically at a reference wavelength of 650 nm. All experiments were carried out in triplicate.
Protein extraction and immunoblot analysis
Following stimulation with IL‐6 or TGF‐β1, preceded or not by pre‐treatment with Sant7, cells were lysed for Western blotting in a radioimmunoprecipitation assay buffer (150 mm NaCl, 1.5 mm MgCl2, 10 mm NaF, 10% glycerol, 4 mm ethylenediaminetetraacetic acid, 1% Triton X‐100, 0.1% SDS, 1% deoxycholate, 50 mm 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid, pH 7.4, plus PPIM, 25 mmβ‐glycerophosphate, 1 mm Na3VO4, 1 mm phenylmethylsulphonyl fluoride, 10 µg/mL leupeptin, 10 µg/mL aprotinin). Protein extracts were then separated on a 12.5% SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE), and transferred on to polyvinylidene difluoride membranes (Amersham Pharmacia, Little Chalfont, UK). Immunoblotting was performed using anti‐phospho‐p38, anti‐phospho‐ERK1/2 and anti‐phospho‐JNK monoclonal antibodies. After being ‘stripped’, membranes were re‐probed with polyclonal antibodies against total (phosphorylated and unphosphorylated) p38, ERK1/2 and JNK. Antibody binding was visualized by enhanced chemiluminescence (ECL‐Plus; Amersham Pharmacia); intensities of experimental bands were analysed by computer‐assisted densitometry and expressed as arbitrary units. These experiments were performed in triplicate.
Statistical analysis
All data are expressed as mean ± SEM. Statistical evaluation of the results was performed by anova. Differences identified by anova were pinpointed by unpaired Student's t‐test. The threshold of statistical significance was set at P < 0.05.
RESULTS
Effects of Sant7 on IL‐6‐dependent cell viability and proliferation
Cell number was not affected by the lowest concentration (10 ng/mL) of IL‐6, whereas this cytokine, when used at the concentration of 100 ng/mL, induced a significant increase (P < 0.01) in HLF numbers, that was completely prevented by Sant7. This effect was observed for 48 h in fibrotic cells (Fig. 1, panel A), but for only 24 h in normal HLFs (Fig. 1, panel B). At the concentration of 100 ng/mL, IL‐6 also enhanced the proliferation rate of our primary cultures of fibrotic HLFs, evaluated as percentage of MTT reduction and detected after 24 and 48 h of cell stimulation (Fig. 2). This effect, which at 24 h resulted in being above 20% with respect to control levels, was completely inhibited (P < 0.01) by Sant7 (1 µg/mL).
Figure 1.
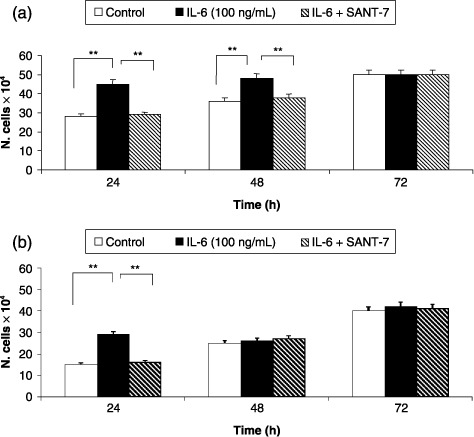
Effects of IL‐6 and Sant7 on cell viability. Exposure to IL‐6 (100 ng/mL) induced a significant increase in cell numbers, detected after 24 and 48 h in fibrotic HLFs (a), and only after 24 h in normal cells (b). At both these times, such an effect was inhibited by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Figure 2.
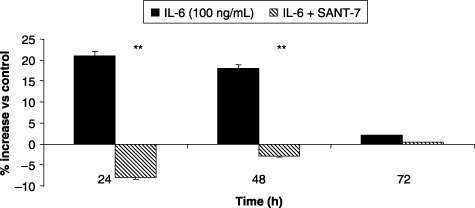
Effects of IL‐6 and Sant7 on cell proliferation. Exposure of primary cultures of fibrotic HLFs to IL‐6 (100 ng/mL) induced a relevant increase in cell proliferation, evaluated by MTT assay, that was detected after 24 and 48 h. At both these times the effect was inhibited by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Effects of Sant7 on IL‐6‐induced MAPK phosphorylation
Sant7 prevented the increase in MAPK phosphorylation elicited by IL‐6 (100 ng/mL). In particular, IL‐6‐induced ERK1/2 phosphorylation resulted in being much greater and lasting for at least 48 h in fibrotic cells (Fig. 3, panel A), when compared to normal HLFs, where this effect was detected only after 24 h (Fig. 3, panel B). After 24 h of cell stimulation with IL‐6, JNK phosphorylation was also more marked in fibrotic cells (Fig. 4, panel A) with respect to normal HLFs, where a second peak of JNK phosphorylation was observed after 72 h (Fig. 4, panel B). IL‐6‐induced p38 phosphorylation was detected by 24 h in both fibrotic (Fig. 5, panel A) and normal HLFs (Fig. 5, panel B), the latter showing a lower increase in phosphorylated p38 levels.
Figure 3.
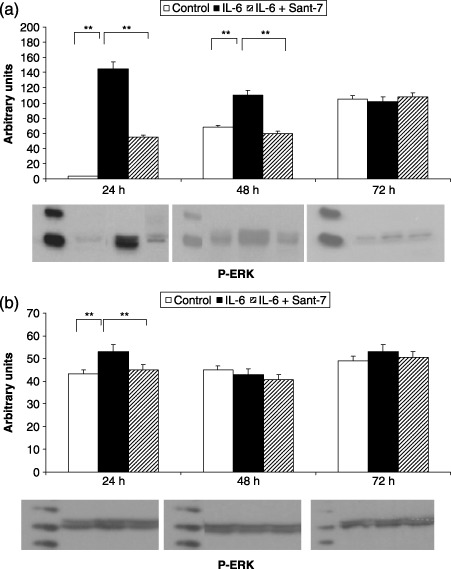
Effects of IL‐6 and Sant7 on ERK1/2 phosphorylation (p‐ERK). Exposure to IL‐6 (100 ng/mL) induced a significant increase in ERK1/2 phosphorylation, detected after 24 and 48 h in fibrotic HLFs (a), and only after 24 h in normal cells (b). At both these times, the effect was inhibited by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Figure 4.
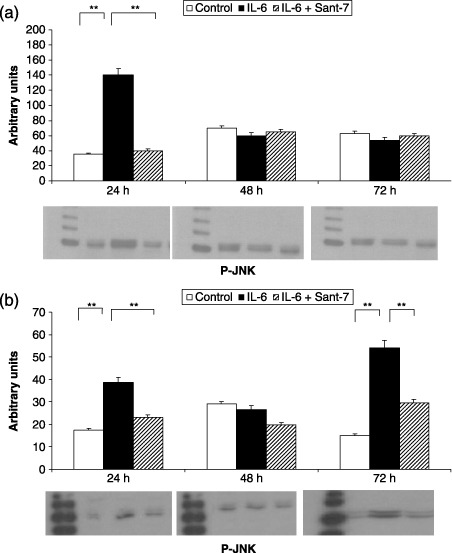
Effects of IL‐6 and Sant7 on JNK phosphorylation (p‐JNK). Exposure to IL‐6 (100 ng/mL) induced a significant increase in JNK phosphorylation, detected after 24 h in fibrotic HLFs (a), and after 24 and 72 h in normal cells (b). This effect was inhibited by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Figure 5.
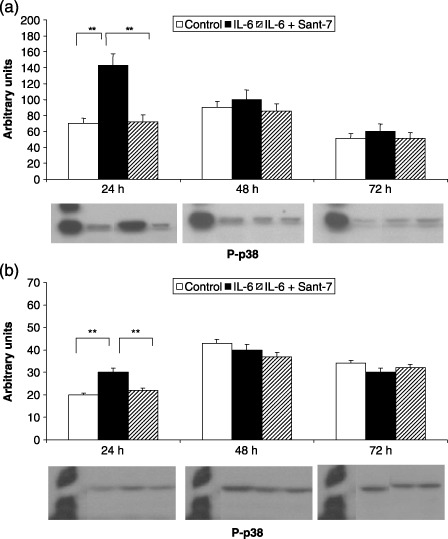
Effects of IL‐6 and Sant7 on p38 phosphorylation (p‐p38). Exposure of primary cultures of fibrotic HLFs to IL‐6 (100 ng/mL) induced a significant increase in p38 phosphorylation, detected after 24 h in both fibrotic (a) and normal (b) HLFs. This effect was inhibited by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Effects of Sant7 on TGF‐β1‐dependent HLF viability and proliferation
We have already shown in HLFs that TGF‐β is able to induce proliferation and MAPK phosphorylation, as well as IL‐6 secretion into the cell culture supernatant. Thus, in order to test whether action of TGF‐β is indeed mediated, at least in part, by IL‐6, we evaluated the effects of pre‐treatment with the IL‐6 receptor superantagonist Sant7 on TGF‐β‐induced cell growth and MAPK activation.
After 24 and 48 h, TGF‐β1 elicited a significant increase (P < 0.01) in viability of fibrotic HLFs, as detected by cell counts expressed as total numbers (Fig. 6, panel A). This was evident only at 24 h in normal lung fibroblasts (Fig. 6, panel B). Total cell numbers were significantly decreased by pre‐treatment with Sant7 at 24 (P < 0.01), but not at 48 h. TGF‐β1 also increased proliferation of our primary cultures of fibrotic HLFs, evaluated as percentage of MTT reduction, detected after 24 and 48 h at rates of almost 25% and 20%, respectively (Fig. 7). This effect was significantly inhibited (P < 0.01) by pre‐treatment with Sant7 only at 24 h.
Figure 6.
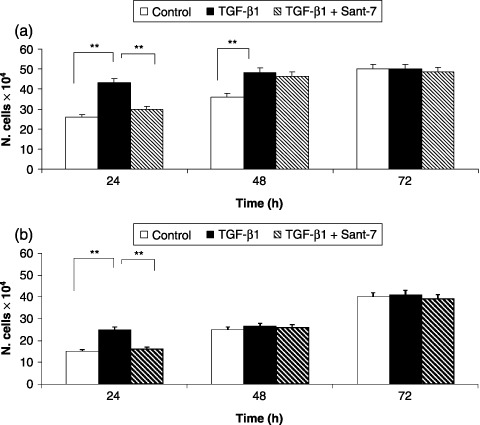
Effects of TGF‐β1 and Sant7 on cell viability. Exposure to TGF‐β1 (10 ng/mL) induced a significant increase in cell numbers, detected after 24 and 48 h in fibrotic HLFs (a), and only after 24 h in normal cells (b). This effect was inhibited at 24 h by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Figure 7.
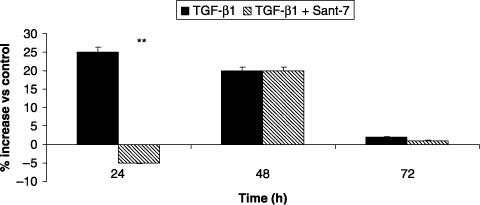
Effects of TGF‐β1 and Sant7 on cell proliferation. Exposure of primary cultures of fibrotic HLFs to TGF‐β1 (10 ng/mL) induced a relevant increase in cell proliferation, evaluated by MTT assay, that was detected after 24 and 48 h. This effect was inhibited at 24 h by pre‐treatment with Sant7 (1 µg/mL). **P < 0.01.
Effects of Sant7 on TGF‐β1‐induced MAPK phosphorylation
Evaluation of the results obtained in three independent sets of experiments showed that exposure of normal and fibrotic HLFs to TGF‐β1 (10 ng/mL) induced significant increases, with respect to control levels, in amounts of phosphorylated MAPKs. In particular, ERK1/2 phosphorylation increased after 24 h (P < 0.01) and 48 h (P < 0.05) of cell stimulation only in fibrotic (Fig. 8, panel A), but not in normal HLFs (Fig. 8, panel B). JNK phosphorylation raised (P < 0.01) after 24 and 48 h in fibrotic cells (Fig. 9, panel A) and, although to lower levels, at all three times (24, 48 and 72 h) in normal HLFs (Fig. 9, panel B). The p38 phosphorylation increased (P < 0.05) after 24 and 72 h in both fibrotic (Fig. 10, panel A) and normal HLFs (Fig. 10, panel B). TGF‐β1‐induced phosphorylation of these three MAPK subgroups was significantly inhibited by Sant7 (P < 0.05), but only at the 24th hour (8, 9, 10).
Figure 8.
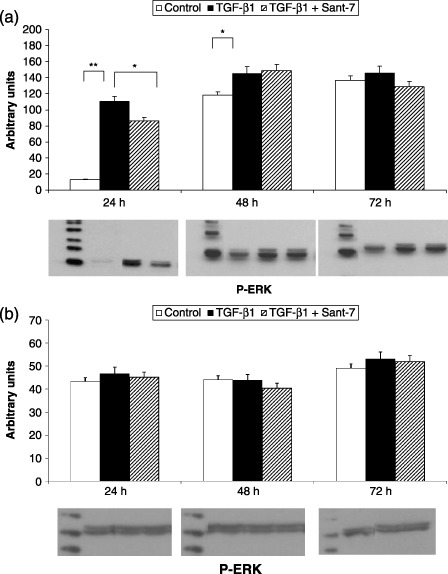
Effects of TGF‐β1 and Sant7 on ERK1/2 phosphorylation (p‐ERK). Exposure to TGF‐β1 (10 ng/mL) induced a significant increase in ERK1/2 phosphorylation, detected after 24 and 48 h in fibrotic (a), but not in normal HLFs (b). This effect was inhibited at 24 h by pre‐treatment with Sant7 (1 µg/mL). *P < 0.05; **P < 0.01.
Figure 9.
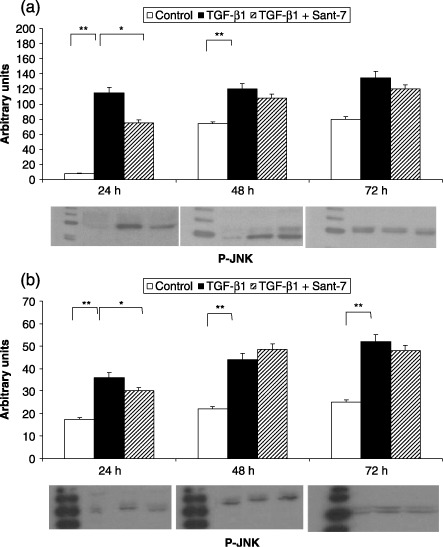
Effects of TGF‐β1 and Sant7 on JNK phosphorylation (p‐JNK). Exposure to TGF‐β1 (10 ng/mL) induced a significant increase in JNK phosphorylation, detected after 24 and 48 h in fibrotic HLFs (a), and also after 72 h in normal cells (b). This effect was inhibited at 24 h by pre‐treatment with Sant7 (1 µg/mL). *P < 0.05; **P < 0.01.
Figure 10.
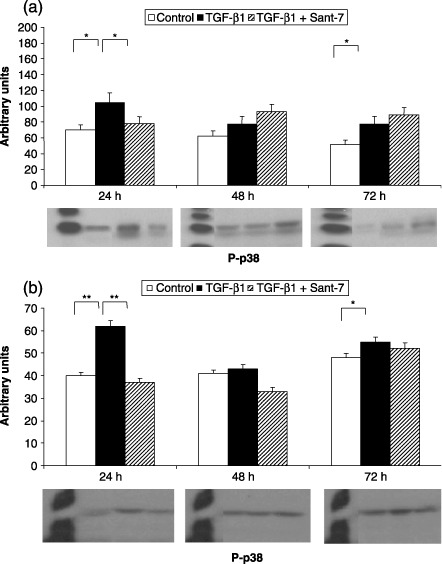
Effects of TGF‐β1 and Sant7 on p38 phosphorylation (p‐p38). Exposure to TGF‐β1 (10 ng/mL) induced a significant increase in p38 phosphorylation, detected after 24 and 72 h in both fibrotic (a) and normal (b) HLFs. This effect was inhibited at 24 h by pre‐treatment with Sant7 (1 µg/mL). *P < 0.05; **P < 0.01.
Since the monoclonal antibodies (anti‐phospho‐ERK1/2, anti‐phospho‐JNK and anti‐phospho‐p38) used in this study specifically recognize the phosphorylated, active forms of MAPKs, the remarkable induction of MAPK phosphorylation can be considered as a reliable indicator of highly efficient activation elicited by TGF‐β1. The latter exerted its effects uniquely on phosphorylation‐dependent activation of MAPKs, without affecting their total expression, as demonstrated by unchanged binding patterns of anti‐(total) MAPK polyclonal antibodies (data not shown).
DISCUSSION
This study has shown that the IL‐6 antagonist Sant7 was able to prevent, in primary cultures of human lung fibroblasts, the proliferative effects of both IL‐6 and TGF‐β1, which resulted in being more prominent in fibrotic cells when compared to normal HLFs. Moreover, Sant7 inhibited IL‐6 and TGF‐β1‐induced increases in MAPK phosphorylation, which were also higher in fibrotic HLFs. Our findings thus suggest that, especially in lung tissue derived from patients with IPF, fibrogenic mechanisms activated by TGF‐β1 are at least in part mediated by increased release of IL‐6, whose biological actions related to MAPK signalling and cell proliferation were effectively blocked by such a receptor superantagonist. We hence infer that in HLFs phosphorylation‐dependent MAPK activation is crucially involved in coupling IL‐6 receptor stimulation with IL‐6‐dependent cell viability and proliferation. The effects of Sant7 were mostly evident after 24 h of cell stimulation, when IL‐6 and TGF‐β1 elicited, with respect to control levels, the highest increases in MAPK phosphorylation. In particular, the proliferative action of IL‐6 was paralleled by a concomitant increase in phosphorylated ERK1/2, which is indeed the MAPK pathway usually associated with cell proliferation and protection from apoptosis (Xia et al. 1995). Furthermore, ERK1/2 phosphorylation was stimulated by TGF‐β1 only in fibrotic, but not in normal HLFs. It is thus possible that the ERK1/2 MAPK subgroup markedly contributes to implementation of the biological circuit, sequentially coordinated by TGF‐β1 and IL‐6, leading to proliferation of HLFs, especially in patients with IPF. In this regard, an autocrine loop between TGF‐β1 and IL‐6, mediated by IL‐6‐induced ERK activation, has also been shown to be crucial in promoting pancreatic fibrosis (Aoki et al. 2006). ERK1/2 seems to be involved in lung fibroblast survival also by mediating IL‐6 production stimulated by another fibrogenic factor such as the CC chemokine ligand 2 (CCL2) (Liu et al. 2007).
In lung fibrosis, a pivotal role is played by the known mitogenic action of IL‐6 (Fries et al. 1994), whose proliferative effects are mediated by activation of its receptors expressed on HLF surfaces. By acting at a key receptor level, Sant7 is thus able to prevent the cascade of molecular events linking IL‐6 receptor stimulation to intracellular signalling operated by MAPKs, which are very important for HLF viability and growth. In this regard, it has already been demonstrated, in lung fibroblasts obtained from IPF patients, that IL‐6 was capable of eliciting a powerful proliferative effect, via ERK1/2 activation, down‐regulation of the cyclin‐dependent kinase (cdk) inhibitor p27kip1, and consequent induction of cyclins D1 and E1 that are responsible for progression through the cell cycle (Moodley et al. 2003a). In particular, following IL‐6 exposure, the enhanced levels of ERK phosphorylation resulted in being sustained in fibrotic HLFs, lasting at least 12 h, whereas STAT3 phosphorylation was only transient. In contrast, in normal pulmonary fibroblasts, phosphorylated ERK was only transiently expressed, while STAT3 phosphorylation was detected for at least 6 h (Moodley et al. 2003a). Therefore, because cell membrane expression of gp130 does not seem to differ between normal and IPF–HLFs, fibrotic lung fibroblasts are probably characterized by relevant abnormalities in cytokine production and signalling (Knight et al. 2003), the latter possibly consisting of a tendency to marked and prolonged activation of IL‐6 receptor and/or MAPKs. In such a regard, our present findings also confirm that the effects of both IL‐6 and TGF‐β1 on cell viability and ERK1/2 phosphorylation were much more evident in fibrotic HLFs. The aberrant fibroproliferative response to IL‐6 might in part depend on genetic mechanisms affecting whatever component of the signal transduction network operating in HLFs, including IL‐6 interactions with its receptor and the downstream intracellular pathways. For instance, studies performed in IPF subjects have shown that some IL‐6 gene polymorphisms are associated with impaired carbon monoxide lung transfer typical of such patients (Pantelidis et al. 2001). Based on all these reports, we believe that strong antagonism of the IL‐6 receptor, such as that provided by Sant7, could be very useful to interfere with cell signalling at an upstream membrane level in hyperproliferative HLFs. Altered IL‐6 signalling in IPF–HLFs may also be implicated in the resistance of such cells to apoptosis (Moodley et al. 2003b), thereby further contributing to the profibrotic action of this cytokine. Indeed, in normal HLFs, IL‐6 enhanced Fas‐dependent apoptosis as well as the expression of the pro‐apoptotic protein Bax, whereas in fibrotic lung fibroblasts the same cytokine inhibited apoptosis and evoked high expression of the anti‐apoptotic protein Bcl‐2; these latter effects were mediated by ERK activation (Moodley et al. 2003b). It can thus be speculated that disruption of both IL‐6 receptor stimulation and subsequent MAPK phosphorylation, which we achieved through pre‐treatment of fibrotic HLFs with Sant7, may also contribute to restore the apoptotic behaviour of these cells.
An interesting aspect of the current study concerns the ability of Sant7 to inhibit not only IL‐6 action, but also TGF‐β1‐induced cell viability and proliferation. This effect can be explained by the well‐known positive action exerted by TGF‐β1 on IL‐6 release from lung fibroblasts (Eickelberg et al. 1999; Junn et al. 2000), also confirmed by our recently published data (Pelaia et al. 2007). However, the inhibitory effects of Sant7 were detectable only after 24 h of cell exposure to TGF‐β1, whereas such a growth factor continued to increase cell viability and proliferation at least until the 48th h in fibrotic HLFs. These results suggest that MAPK‐dependent mitogenesis caused by TGF‐β1 in fibrotic HLFs is initially due to IL‐6, and thereafter mediated by some other effector molecules. Indeed, we have shown in the same cells that TGF‐β1 can induce secretion of both IL‐6 and IL‐11, the latter being also able to promote HLF growth via MAPK signalling (Pelaia et al. 2007). Therefore, if IL‐11 is responsible for late effects of TGF‐β1, these cannot be antagonized by Sant7. The latter is unable to interfere with IL‐11 activity because it interacts selectively with the ligand‐binding subunit of the IL‐6 receptor (gp80), while it does not bind the common transducing chain (gp130) of both IL‐6 and IL‐11 receptors (Ren‐Xiao et al. 1997). The results of our present study indicate that Sant7 may significantly contribute to inhibit, at least for a certain period of time, the molecular mechanisms underlying TGF‐β‐dependent proliferation of pulmonary fibroblasts. Current knowledge suggests that the very important role played by TGF‐β in lung fibrogenesis is predominantly attributable to the Smad signalling system (Gauldie et al. 2006). Smad pathways might also be involved in mediating TGF‐β1‐induced synthesis of IL‐6. In our primary cultures of fibrotic HLFs, for example, MAPK inhibitors did not suppress the stimulatory effect of TGF‐β1 on IL‐6 release (Pelaia et al. 2007), which could therefore be regulated by Smad activation. On the other hand, Smad proteins and MAPK enzymes probably interact in HLFs through dynamic cross‐talk circuits that coordinate, integrate and amplify cellular responses to TGF‐β. IL‐6 production may thereby represent a downstream effect resulting from convergence of multiple signal transducing networks activated by several fibrogenic agents, including chemical and physical stimuli. In this regard, excessive synthesis of TGF‐β1 and IL‐6 has been found to be implicated in development of pulmonary fibrosis induced by either bleomycin or ionizing radiation (Barthelemy‐Brichant et al. 2004; Cavarra et al. 2004; Tabata et al. 2006). Sant7 then provides a strategic tool to effectively interfere with profibrotic pathways triggered by IL‐6 autocrine and paracrine effects on fibrotic HLFs.
With regard to lung fibrosis, another aspect that is worth being investigated refers to potential interactions between Sant7 and corticosteroids. The latter are widely used to attempt to limit, with uncertain outcomes, pulmonary fibrogenesis. For instance, blockade of IL‐6 signalling by Sant7 has been shown to significantly potentiate the inhibitory action of dexamethasone on myeloma cell growth (Tassone et al. 2000, 2005). Thus, a rational basis may exist for a therapeutic approach consisting of such a drug combination (Sant7 and corticosteroids), eventually aimed at preventing or treating lung fibrosis. Sant7 and glucocorticoids could synergize, by interfering in fibrotic HLFs, with different but complementary molecular mechanisms. In particular, Sant7 can impair IL‐6 receptor stimulation and subsequent MAPK activation, the latter also being modulated by corticosteroids via induction of a powerful endogenous inhibitor of MAPK phosphorylation, namely MAPK phosphatase‐1 (MKP‐1) (Barnes 2006). Furthermore, we have shown that glucocorticosteroids are able to prevent TGF‐β1‐mediated HLF proliferation by inhibiting MAPK phosphorylation (Pelaia et al. 2007). Because proliferative effects exerted by TGF‐β1 on fibrotic HLFs appear to be at least in part due to IL‐6, it is thereby conceivable that in these cells Sant7 might effectively enhance antiproliferative action of glucocorticoids.
In conclusion, taken together our results strongly suggest that IL‐6 receptor superantagonist Sant7 is capable of inhibiting cell proliferation and MAPK phosphorylation induced by cell exposure to either IL‐6 or TGF‐β1, especially in fibrotic HLFs. Such preliminary findings may thus open new therapeutic perspectives for fibrogenic ILDs, based on modulation of signal transduction pathways activated by IL‐6. In this regard, the IL‐6 receptor is already considered as a suitable molecular target for treatment of chronic inflammatory disorders like rheumatoid arthritis and Crohn's disease (Nishimoto & Kishimoto 2006). Therefore, the present study could contribute to extending the development of anti‐IL‐6 therapies to the respiratory system.
Both authors (Luca Gallelli, Daniela Falcone) contributed equally to the work presented in this article.
REFERENCES
- Aoki H, Ohnishi H, Hama K, Shinozaki S, Kita H, Yamamoto H, Osawa H, Sato K, Tamada K, Sugano K (2006) Existence of autocrine loop between interleukin‐6 and transforming growth factor‐β1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 99, 221–228. [DOI] [PubMed] [Google Scholar]
- Barnes PJ (2006) Corticosteroid effects on cell signalling. Eur. Respir. J. 27, 413–426. [DOI] [PubMed] [Google Scholar]
- Barthelemy‐Brichant N, Bosquée L, Cataldo D, Corhay JL, Gustin M, Seidel L, Thiry A, Ghaye B, Nizet M, Albert A, Deneufbourg JM, Bartsch P, Nusgens B (2004) Increased IL‐6 and TGF‐β1 concentrations in bronchoalveolar lavage fluid associated with thoracic radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 58, 758–767. [DOI] [PubMed] [Google Scholar]
- Cavarra E, Carraro F, Fineschi S, Naldini A, Bartalesi B, Pucci A, Lungarella G (2004) Early response to bleomycin is characterized by different cytokine and cytokine receptor profiles in lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 287, L1186–L1192. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40. [DOI] [PubMed] [Google Scholar]
- Crestani B, Seta N, De Bandt M, Soler P, Rolland C, Dehoux M, Boutien A, Dombret MC, Palazzo E, Kahn MF (1994) Interleukin‐6 secretion by monocytes and alveolar macrophages in systemic sclerosis with lung involvement. Am. J. Respir. Crit. Care Med. 149, 1260–1265. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Roth M (1999) Transforming growth factor‐β1 induces interleukin‐6 expression via activating protein‐1 consisting of JunD homodimers in primary human lung fibroblasts. J. Biol. Chem. 274, 12933–12938. [DOI] [PubMed] [Google Scholar]
- Fries KM, Felch ME, Phipps RP (1994) Interleukin‐6 is an autocrine growth factor for murine lung fibroblast subsets. Am. J. Respir. Cell. Mol. Biol. 11, 552–560. [DOI] [PubMed] [Google Scholar]
- Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D (2006) Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc. Am. Thorac. Soc. 3, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller‐Newen G, Schaper F (2003) Principles of interleukin (IL)‐6‐type signalling and its regulation. Biochem. J. 374, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordana M, Schulman J, McSharry C, Gauldie J (1988) Heterogenous proliferative characteristics of human adult lung fibroblasts from control and fibrotic tissue. Am. Rev. Respir. Dis. 137, 579–584. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee KN, Ju HR, Han SH, Im JY, Kang HS, Lee TH, Bae YS, Ha KS, Lee ZW, Rhee SG, Choi I (2000) Requirement of hydrogen peroxide generation in TGF‐β1 signal transduction in human lung fibroblast cells: involvement of hydrogen peroxide and Ca2+ in TGF‐β1‐induced IL‐6 expression. J. Immunol. 165, 2190–2197. [DOI] [PubMed] [Google Scholar]
- Kamimura D, Ishihara K, Hirano T (2003) IL‐6 signal transduction and its physiological roles: the signal orchestration model. Rev. Physiol. Biochem. Pharmacol. 149, 1–38. [DOI] [PubMed] [Google Scholar]
- Khalil N, O’Connor RN, Flanders KC, Unruh H (1996) TGF‐β1, but not TGF‐β2 or TGF‐β3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am. J. Respir. Cell. Mol. Biol. 14, 131–138. [DOI] [PubMed] [Google Scholar]
- Knight DA, Ernst M, Anderson GP, Moodley YP, Mutsaers SE (2003) The role of gp130/IL‐6 cytokines in the development of pulmonary fibrosis: critical determinants of disease susceptibility and progression? Pharmacol. Ther. 99, 327–338. [DOI] [PubMed] [Google Scholar]
- Liu X, Das AM, Seldeman J, Griswold D, Afuh CN, Kobayashi T, Abe S, Fang Q, Hashimoto M, Kim H, Wang X, Shen L, Kawasaki S, Rennard SI (2007) The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL‐6. Am. J. Respir. Cell. Mol. Biol. 37, 121–128. [DOI] [PubMed] [Google Scholar]
- Moodley YP, Scaffidi AK, Misso NL, Keerthisingam C, McAnulty RJ, Laurent GJ, Mutsaers SE, Thompson PJ, Knight DA (2003a) Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin‐6/gp 130‐mediated cell signaling and proliferation. Am. J. Pathol. 163, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley YP, Misso NL, Scaffidi AK, Fogel‐Petrovic M, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA (2003b) Inverse effects of interleukin‐6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell. Mol. Biol. 29, 490–498. [DOI] [PubMed] [Google Scholar]
- Nishimoto N, Kishimoto T (2006) Interleukin 6: from bench to bedside. Nat. Clin. Pract. Rheumatol. 2, 619–626. [DOI] [PubMed] [Google Scholar]
- Pantelidis P, Fanning GC, Wells AU, Welsh KI, Du Bois RM (2001) Analysis of tumor necrosis factor‐alpha, lymphotoxin‐alpha, tumor necrosis factor receptor II, and interleukin‐6 polymorphisms in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 163, 1432–1436. [DOI] [PubMed] [Google Scholar]
- Pelaia G, Gallelli L, D’Agostino B, Vatrella A, Cuda G, Fratto D, Renda T, Galderisi U, Piegari E, Crimi N, Rossi F, Caputi M, Costanzo FS, Vancheri C, Maselli R, Marsico SA (2007) Effects of TGF‐β and glucocorticoids on MAP kinase phosphorylation, IL‐6/IL‐11 secretion and cell proliferation in primary cultures of human lung fibroblasts. J. Cell. Physiol. 210, 489–497. [DOI] [PubMed] [Google Scholar]
- Ren‐Xiao S, Ciliberto G, Savino R, Zong‐Jiang G, Klein B (1997) Interleukin‐6 receptor antagonists inhibit interleukin‐11 biological activity. Eur. Cytokine Netw. 8, 51–56. [PubMed] [Google Scholar]
- Savino R, Lahm A, Salvati AL, Ciapponi L, Sporeno E, Altamura S, Paonessa G, Toniatti C, Ciliberto G (1994) Generation of interleukin‐6 receptor antagonists by molecular‐modeling guided mutagenesis of residues important for gp 130 activation. EMBO J. 13, 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino R, Demartis A, Ciapponi L, Sporeno E, Toniatti C, Bernassola F, Melino G, Klein B, Ciliberto G (1997) The receptor super‐antagonist Sant7: a potent and safe inhibitor of IL‐6 on human myeloma cells. Oncol. Rep. 4, 485–492. [DOI] [PubMed] [Google Scholar]
- Shahar I, Fireman E, Topilsky M, Grief J, Kivity S, Spirer Z, Ben Efraim S (1996) Effect of IL‐6 on alveolar fibroblast proliferation in interstitial lung diseases. Clin. Immunol. Immunopathol. 79, 244–251. [DOI] [PubMed] [Google Scholar]
- Sporeno E, Savino R, Ciapponi L, Paonessa G, Cabibbo A, Lahm A, Pulkki K, Sun RX, Toniatti C, Klein B, Ciliberto G (1996) Human IL‐6 receptor super‐antagonists with high potency and wide spectrum on multiple myeloma cells. Blood 87, 4510–4519. [PubMed] [Google Scholar]
- Tabata C, Kubo H, Tabata R, Wada M, Sakuma K, Ichikawa M, Fujita S, Mio T, Mishima M (2006) All‐trans retinoic acid modulates radiation‐induced proliferation of lung fibroblasts via IL‐6/IL‐6R system. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L597–L606. [DOI] [PubMed] [Google Scholar]
- Taga T (1997) gp130 and the interleukin‐6 family of cytokines. Annu. Rev. Immunol. 15, 797–819. [DOI] [PubMed] [Google Scholar]
- Tassone P, Forciniti S, Galea E, Savino R, Turco MC, Iacopino P, Tagliaferri P, Morrone G, Ciliberto G, Venuta S (2000) Synergistic induction of growth arrest and apoptosis of human myeloma cells by the IL‐6 super‐antagonist Sant 7 and dexamethasone. Cell Death Differ. 7, 327–328. [DOI] [PubMed] [Google Scholar]
- Tassone P, Neri P, Burger R, Savino R, Shammas M, Catley L, Podar K, Chauhan D, Masciari S, Gozzini A, Tagliaferri P, Venuta S, Munshi NC, Anderson KC (2005) Combination therapy with interleukin‐6 receptor superantagonist Sant7 and dexamethasone induces antitumor effects in a novel SCID‐hu in vivo model of human multiple myeloma. Clin. Cancer Res. 11, 4251–4258. [DOI] [PubMed] [Google Scholar]
- Vancheri C, Sortino MA, Tomaselli V, Mastruzzo C, Condorelli F, Bellistri G, Pistorio MP, Canonico PL, Crimi N (2000) Different expression of TNF‐α receptors and prostaglandin E2 production in normal and fibrotic lung fibroblasts: potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell. Mol. Biol. 22, 628–634. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen‐activated protein kinase: conservation of a three kinase module from yeast to human. Physiol. Rev. 79, 143–180. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK‐p38 MAP kinases on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]