

Abstract
Checkpoints are mechanisms that regulate progression through the cell cycle insuring that each step takes place only once and in the right sequence. Mutations of checkpoint proteins are frequent in all types of cancer as defects in cell cycle control can lead to genetic instability. This review will focus on three major areas of cell cycle transition control, with particular attention to the alterations found in human cancer. These areas include the G1/S transition, where most cancer‐related defects occur, the G2/M checkpoint and its activation in response to DNA damage, and the spindle checkpoint.
Abbreviations:
- BRCA1
AT
ATM
ATR
APC
INK4
HDAC
ORC
HPV
ARF
AT
APC
Breast Cancer susceptibility gene 1
Ataxia Telengiectasia
AT Mutated
AT Related
Anaphase Promoting Complex
Inhibitors of Cdk4
Histone Deacetylase
Origin of Replication Complex
Human Papillomavirus
Alternative Reading Frame
Ataxia Telengiectasia
Anaphase Promoting Complex
THE CELL CYCLE ENGINE
The cell cycle is driven by kinase complexes formed by a catalytic subunit, the cdk (cyclin dependent kinase) and a regulatory subunit, the cyclin (cyc), which confers specificity. Such kinases have spikes of activity that vary with orderly periodicity, which is essential to ensure that cells replicate their DNA only once per cell cycle, and that chromosomes are segregated equally to daughter cells and at the right moment. The activity of cyc‐cdk complexes is regulated at several levels: (1) Formation of the complex depends on the availability of the cyclin subunit, which is regulated by periodic synthesis and proteolysis during the cell cycle. (2) Phosphorylation. In order to be active, Cyc/cdk complexes need to be phosphorylated on Thr 161 of the cdk subunit by a large kinase complex named CAK (cdk activating complex) ( Draetta 1997). On the contrary, inhibitory phosphorylation by the protein kinases wee1 and myt1 keeps the complexes inactive. Protein phosphatases of the cdc25 family remove such inhibitory phosphates, resulting in cyc/cdk activation. Regulation of cyc/cdks by a phosphorylation/dephosphorylation cycle allows the accumulation of inactive kinase complexes independently of the presence of their activator, and permits their rapid activation when it is required. (3) Inhibitory molecules. Studies on the regulation of cdks activity has lead to the identification of inhibitory molecules, named CKIs (cdk inhibitors). CKIs are divided into two groups, the INK4 and the Cip/Kip families, depending on their specificity ( Sherr & Roberts 1999). The former includes p16INK4a ( Serrano, Hannon & Beach 1993) p15INK4b ( Hannon & Beach 1994), p18INK4c ( Guan et al. 1994 ), and p19INK4d ( Chan et al. 1995 ; Hirai & Sherr 1996), which bind and inhibit cdk4‐and cdk6‐associated activity. The Cip/Kip family includes p21CIP1 ( Dulic et al. 1993 ; El‐Deiry et al. 1993 ; Gu, Turck & Morgan 1993; Harper et al. 1993 ) p27KIP1 ( Polyak et al. 1994 ; Toyoshima 1994), and p57KIP2 ( Lee, Reynisdottir & Massague 1995; Matsuoka et al. 1995 ). These molecules have a wider specificity, being able to inhibit cyc D‐dependent kinases as well as cyc E and cyc A in complex with cdk2. Paradoxically, at lower concentrations p21 and p27 can also facilitate the formation of the cycD/cdk4 kinase complex, that retains its ability to phosphorylate the Retinoblastoma (Rb) protein ( Labaer et al. 1997 ; Cheng et al. 1999 ; Parry et al. 1999 ).
THE (G0‐)G1‐S TRANSITION
Although in most tissues of the body cells are maintained in a quiescent state, called G0, they can be driven to re‐enter the cell cycle upon mitogenic induction. One of the key characteristics of cancer cells is independence from growth stimuli. This typically occurs due to modification of the growth signal, for example by autocrine stimulation, due to modification or overexpression of growth factor receptors or to mutations of components of the intracellular pathway carrying the signal, most notably Ras ( Hanahan & Weinberg 2000). Once such independence is established, cells can re‐enter the cell cycle regardless of positive or negative external stimuli.
In quiescent or serum starved cells cycD is unstable and it is maintained at low levels by active proteolysis. Addition of growth factors activate cell surface receptors and initiates a signalling cascade that induces cycD accumulation ( Cheng et al. 1998 , and references therein). The consequent rise in cycD/cdk4 activity results in Rb phosphorylation and release of E2F inhibition, which in turn elicits S phase initiation (see Fig. 1). Once cells are committed to divide, they do not have any further requirement for external stimuli, but growth factor deprivation results in cycD degradation, resetting the dependency on extracellular signals before the cell can re‐enter the cell cycle. In short, cycD acts as a growth sensor and provides a link between mitogenic stimuli and the cell cycle machinery ( Sherr 1996).
Figure 1.
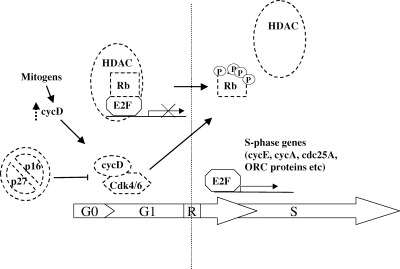
The G1/S transition in mammalian cells. CycD is a sensor of mitogenic stimuli linking the cell cycle with the external environment. Growth factor‐induced cycD accumulation results in the formation of active cycD/cdk4, which overcomes inhibition by p16 and phosphorylates Rb. This crucial event marks the transition through the restriction point (R) and induces the release of E2F transcriptional repression mediated by the HDAC protein, with the consequent transactivation of genes necessary for S phase initiation and progression. The targets of mutations found in human cancer are hatched in the Figure; alterations include Rb mutations or deletions, HDAC mutations, cycD overexpression, p16 mutations, cdk4 mutations, increased degradation of p27.
Transition from quiescence to proliferation involves changes in several other cell cycle regulators. In quiescent cells p27 protein level is high, the protein level of cycD is low and its activity is inhibited by p16. CycD accumulation and its binding to cdk4 serves to change the balance between cyc/cdk complexes and CKIs as well as to initiate Rb phosphorylation. P21 and p27 bind the nascent cycD/cdk4 complexes and help their proper folding; at the same time, both inhibitors are released from the cycE/cdk2 complex, resulting in its activation ( Sherr & Roberts 1999). The cycE/cdk2 kinase has two roles: firstly it co‐operates with cdk4/cycD to phosphorylate Rb, secondly it phosphorylates its inhibitor p27, inducing its proteasome‐dependent degradation ( Montagnoli et al. 1999 ).
The G1/S transition is regulated by the phosphorylation state of the tumour suppressor Rb ( Ewen 1994; Weinberg 1995). In its hypophosphorylated state Rb binds and inhibits the E2F family of transcription factors, and this inhibition is released upon Rb phosphorylation. Recent studies have elucidated the mechanism of E2F inhibition, showing that Rb binds the histone deacetylase protein (HDAC) and tethers it on E2F responsive promoters, resulting in active repression ( Brehm & Miska 1998; Luo, Postigo & Dean 1998; Magnahi‐Jaulin & Groisman 1998; Zhang, Postigo & Dean 1999). Rb phosphorylation leads to the disruption of the complex with HDAC and the subsequent release of E2F, thus preventing further E2F inhibition ( Harbour & Luo 1999). The E2F family of transcription factors includes six members which share common domains and the need to dimerize with DP proteins to achieve high affinity binding to E2F‐responsive promoters and to Rb family members. E2Fs can stimulate or repress transcription and their activity is regulated at the level of transcription, subcellular localization and degradation ( Dyson 1998; Helin 1998). The increase in E2F transcriptional activity following its release from Rb results in the transactivation of genes which products are essential for S phase initiation and progression, including cycA, cycE, cdc25A, ORC proteins, cdc6, thymidylate synthase (see Fig. 1).
The complex regulatory mechanism of G1/S transition is a major target of alterations in cancer. Such alterations involve gene deletion or overexpression, or point mutations that impair gene function, with the common result of altering the balance of Rb phosphorylation/dephosphorylation, in turn determining the proliferative state of a cell ( Hall & Peters 1996). Point mutations or deletions of Rb are found in a large variety of human cancers, particularly retinoblastomas or sarcomas. Rb function is also found to be impaired by its interaction with proteins from tumour viruses, such as HPV16 E7 and SV40 large T‐Antigen, which displace Rb interaction with HDAC. HDAC‐Rb complex formation is also inhibited by HDAC point mutations found in human cancer ( Brehm et al. 1998 ; Luo et al. 1998 ; Magnahi‐Jaulin et al. 1998 ). Escape from Rb‐mediated cell cycle regulation by cycD overexpression is another mechanism exploited by cancer cells ( Lammie et al. 1991 ; Jiang et al. 1992 ; Bianchi et al. 1993 ; Jiang et al. 1993 ; Bartkova et al. 1994 ; Gillett et al. 1994 ), leading to the idea that cycD can act as an oncogene
The INK4a locus encoding the p16 inhibitor is found to be deleted or altered in familial melanomas and a variety of other cancers ( Kamb et al. 1994 ). In agreement, INK4a knockout mice are tumour prone, suggesting that p16 could act as a tumour suppressor. A more accurate mapping of the INK4a locus has shown that it also encodes another protein, called p19ARF, which has a role in regulating p53 stability and which is frequently found to be mutated in cancer ( Roussel 1999; Sharpless & DePinho 1999). Mice lacking p19ARF are tumour prone like the INK4a ‐/‐ mice, suggesting that p19ARF acts as a tumour suppressor, and that the phenotype of the p16/p19 ‐/‐ mice is mainly due to p19 ablation. However point mutations in familial melanomas are found in the p16 gene ( Hussussian et al. 1994 ; Kamb et al. 1994 ), as well as in cdk4, with the effect of affecting its binding to p16 ( Wölfel et al. 1995 ; Zuo et al. 1996 ), strongly suggesting that p16 also has an important role in preventing melanoma susceptibility.
Intracellular abundance of the p27 protein is tightly regulated: p27 levels are high in quiescent cells and drop dramatically as cells enter S phase ( Carrano et al. 1999 ; Montagnoli et al. 1999 ), and references therein). Levels of p27 are found to be abnormally low in colon carcinomas and other cancer types, an event that often correlates with tumour aggressiveness and poor prognosis ( Pagano et al. 1995 ; Loda et al. 1997 ; Piva et al. 1999 ). Interestingly p27 downregulation does not depend on gene deletion, but on increased proteolysis, showing how precise regulation of the ubiquitin‐proteasome pathway is essential in the regulation of cellular proliferation (for a review see King et al. (1996) ; Pagano (1997)).
THE G2/M TRANSITION
Entry into mitosis is induced by increased activity of the cdc2/cycB kinase complex, also known as MPF (mitosis promoting factor). The MPF is regulated at several levels (reviewed in Pines (1999)): cdc2/cycB complex formation depends on the availability of cyclin B, which starts to be produced in late S phase, it accumulates until mitosis and it is rapidly degraded as cells enter the next G1 phase, thereby preventing a second round of division ( Gallant & Nigg 1992). The subcellular localization of cycB is also regulated ( Fig. 2): cycB contains an aminoacidic sequence that was first called CRS (cytoplasmic retention signal; Pines & Hunter 1994) which is responsible for its cytoplasmic localization during interphase. This sequence was later shown to be a nuclear exclusion signal, demonstrating that cyc B is actively exported from the nucleus until the beginning of prophase ( Gallant & Nigg 1992; Heald, McLoughlin & McKeon 1993; Hagting et al. 1998 ; Jin, Hardy & Morgan 1998; Toyoshima et al. 1998 ). A third level is regulation of MPF activity ( Fig. 2): the cdc2/cycB complex starts to form at the beginning of S phase and accumulates in parallel with cyc B protein level, but it is maintained in an inactive form until G2 due to phosphorylation on Thr14 and Tyr15 of cdc2 by the wee1 and myt1 protein kinases. Wee1 is a nuclear protein, and it is believed to protect the nucleus from prematurely activated MPF ( Heald et al. 1993 ), whereas myt1 is localized in the Golgi complex ( Liu et al. 1997 ), and it contributes to the maintenance of cdc2 phosphorylation during interphase.
Figure 2.
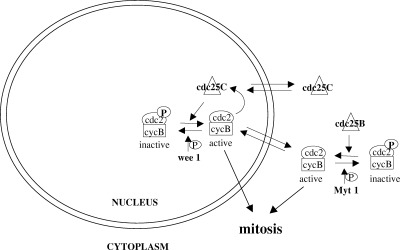
Regulation of the G2/M transition. Entry into mitosis depends on the activation of the cdc2/cycB complex and its subcellular localization. Cyc B is cytoplasmic until G2, when the rate of import exceeds nuclear export. The wee1 and myt1 protein kinases phosphorylate and inactivate the nuclear and cytoplasmic pool of cdc2/cycB, respectively, preventing premature mitotic entry. Accumulation and activation of cdc25B in G2 brings about the initial activation of cdc2/cycB. Cdc25C shuttles between the nucleus and the cytoplasm throughout interphase, with a net cytoplasmic localization. In G2 it accumulates in the nucleus and mediates further activation of cdc2/cycB, which in turn activates it, forming an autocatalytic loop that results in entry into mitosis.
Cdc2 dephosphorylation is a rate‐limiting step for entry into mitosis ( Krek & Nigg 1991), and it depends on the activity of the dual specificity protein phosphatases cdc25B and cdc25C (reviewed in Draetta & Eckstein (1997)). Cdc25B protein levels and activity increase in G2, mediating the activation of the cytoplasmic pool of cdc2/cycB and microtubule nucleation ( Gabrielli et al. 1996 ; Lammer et al. 1998 ). Cdc25B protein level is regulated by proteolysis ( Baldin et al. 1997 ) and the importance of this regulation is demonstrated by the fact that overexpression of cdc25B results in premature mitotic entry ( Karlsson et al. 1999 ). Protein levels of cdc25C are constant throughout the cell cycle, while its activity increases in G2, likely via cdc25B‐dependent activation of the cdc2/cycB complex. Cdc25C activation and its entry into the nucleus determine the activation of nuclear MPF which, in turn, further activates cdc25C forming an autocatalytic loop, and resulting in the irreversible entry into mitosis (see Fig. 2; Izumi, Walker & Maller 1992; Hoffmann et al. 1993 ; Izumi & Maller 1995; Gabrielli et al. 1997 ). Reports on the subcellular localization of cdc25C are contrasting, but it is increasingly clear that it is cytosolic during interphase and nuclear in G2/M, and that it is actively exported from the nucleus due to the presence of a nuclear exclusion signal ( Kumagai & Dunphy 1999; Seki et al. 1992 ; Nishijima et al. 1997 ; Yang et al. 1999 ), and it is kept in the cytoplasm through interaction with 14‐3‐3 family proteins ( Furnari, Rhind & Russell 1997; Peng et al. 1997 ; Sanchez et al. 1997 ). Regulation of cdc25C activity appears to be complex, since it is negatively regulated by the isomerase Pin1 ( Crenshaw et al. 1998 ; Shen et al. 1998 ; Winkler et al. 2000 ) and activated by members of the polo‐like family of protein kinases ( Kumagai & Dunphy 1996), as well as being phosphorylated by the c‐TAK1 ( Peng et al. 1998 ) and by the Chk1 and Chk2/Cds1 protein kinases ( Furnari et al. 1997 ; Sanchez et al. 1997 ).
Cellular DNA is continuously damaged, during the normal processes of cell division or as a result of external stimuli such as UV, γ‐radiation or exposure to genotoxic chemicals, and the inactivation of genes involved in the response to DNA damage have been linked to cancer predisposition. UV irradiation causes the formation of dimers between adjacent thymine bases, which are repaired by the nucleotide excision repair pathway. Mutation of proteins participating in the repair process can cause xeroderma pigmentosum, a skin disease with a high frequency of skin cancer. Defects in the repair of double strand breaks can lead to chromosomal instability, which has been linked to carcinogenesis. The DNA damage checkpoint acts at three different stages of the cell cycle, inducing G1 arrest, a block of DNA replication or a G2 delay, depending on the type of damage and cell cycle stage when the damage was detected (reviewed in Paulovich, Toczyski & Hartwell (1997)).
The G2 block ensuing after DNA damage relies on the inhibition of cdc2 dephosphorylation ( Barth et al. 1996 ; Jin, Gu & Morgan 1996; Rhind, Furnari & Russell 1997). Such inhibition is achieved by two different mechanisms ( Fig. 3). Firstly, DNA‐damage induced phosphorylation of cdc25C by the Chk1/Chk2 protein kinases (see below) stimulates its interaction with 1433 proteins, which appear to sequester it in the cytosol preventing the untimely activation of the nuclear pool of cdc2/cycB ( Furnari et al. 1999 ; Kumagai & Dunphy 1999; Lopez‐Girona et al. 1999 ; Yang et al. 1999 ). Secondly cycB is actively excluded from the nucleus ( Hagting et al. 1998 ; Jin et al. 1998 ; Toyoshima et al. 1998 ), and this depends on its increased binding to 14‐3‐3 σ, one of the transcriptional targets of p53 ( Hermeking et al. 1997 ; Chan et al. 1999 ).
Figure 3.
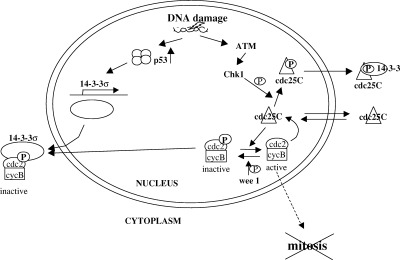
Regulation of the G2/M transition. DNA damage inhibits G2/M progression via two mechanisms. Upon DNA damage the p53 protein is stabilized and activated as a transcription factor. One of its targets is the 14‐3‐3 σ gene, which mediates the sequestration of cdc2/cycB in the cytoplasm. DNA damage also induces the activation of the Chk1/Chk2 protein kinases through an ATM‐dependent mechanism. Chk1/Chk2‐dependent phosphorylation of Cdc25C induces its binding to 14‐3‐3 proteins which inactivate it by sequestering it in the cytoplasm.
The Chk1 and Cds1/Chk2 protein kinases were first identified in yeast, and were later found to be conserved in mammalian cells. Chk1 and Chk2 are activated upon DNA damage in an ATM‐dependent manner ( Brown et al. 1999 ; Sanchez et al. 1997 ; Chaturvedi et al. 1999 ), and appear to be an essential component of the DNA damage checkpoint: phosphorylation of the cdc25C protein phosphatase is pivotal for G2 arrest (see above), Chk1/Chk2‐dependent phosphorylation of p53 on Ser20 contributes to its stabilization following DNA damage ( Chehab et al. 2000 ; Hirao et al. 2000 ; Shieh et al. 2000 ), and BRCA1 phosphorylation by Chk2 is important for its proper localization after DNA damage and its function in DNA repair ( Cortez et al. 1999 ; Lee et al. 2000 ). Notably germline mutations of the BRCA1 gene are found in about 50% of patients with familial breast cancer and ovarian cancer ( Venkitaraman 1999; Deng & Scott 2000).
The signalling pathway leading to G2 arrest after DNA damage is frequently altered or mutated in human cancer. Ionizing radiations induce the activation of protein kinases whose catalytic domains share a high degree of homology with the PI3‐kinase superfamily, including DNA‐PK, ATM and ATR in mammalian cells ( Smith et al. 1999 ). DNA‐PK is part of a multisubunit protein complex containing the kinase subunit and a DNA binding subunit named Ku, which is necessary for DNA repair and was recently shown to have a role in the prevention of chromosomal aberrations ( Difilippantonio et al. 2000 ). Mutations of the ATM protein cause Ataxia Telengiectasia, a genetic disorder characterized by hypersensitivity to ionizing radiation and cancer predisposition ( Rotman & Shiloh 1999). All three kinases have been proposed to mediate p53 phosphorylation following DNA damage, although the exact function of this modification remains to be defined ( Savitsky et al. 1995 ; Siliciano et al. 1997 ; Canman et al. 1998 ). Downstream of ATM action is the Chk2 kinase, that was recently found to be mutated in cells from Li‐Fraumeni syndrome patients, suggesting a possible role for Chk2 as a tumour suppressor ( Bell et al. 1999 ).
Among the genes involved in DNA damage response, the best known is the tumour suppressor p53, which is inactivated in 50% of all human cancers ( Hollstein, Vogelstein & Harris 1991). The importance of p53 as a tumour suppressor is underlined by the fact that mice containing a homozygous deletion for p53 are highly prone to tumours ( Donehower et al. 1992 ). In humans, individuals with the LiFraumeni syndrome, carrying a germ‐line deletion of one p53 allele, are highly susceptible to cancer development ( Malkin et al. 1990 ; Srivastava et al. 1990 ). P53 inactivation is due to either point mutations resulting in defective DNA binding capacity, or due to interaction with viral proteins, such as Large T antigen from SV40 or HPV16 E6. P53 has normally a very short half‐life in cycling cells, but it becomes stabilized after DNA damage. Studies on p53 degradation have shown that the mdm2 protein (which is also a p53 transcriptional target) stimulates p53 degradation and it has been shown to act as a p53 ubiquitin ligase in vitro. The p19ARF protein counteracts mdm2 action, sequestering it in the nucleoli and preventing its interaction with p53 ( Sherr & Weber 2000). P53 phosphorylation has been shown to displace the interaction between p53 and mdm2, resulting in p53 stabilization. Phosphorylation and acetylation of p53 on several sites also activate it, inducing the formation of active tetrameres and the ensuing transactivation of target genes. The effects of p53 activation and its targets are diverse and the object of very abundant literature (for a recent review, see Giaccia & Kastan 1998). Depending on cell type and context, p53 can induce G1 arrest (through induction of the p21CIP1 protein; El‐Deiry et al. 1993 ; Harper et al. 1993 ), or apoptosis ( Yonish‐Rouach et al. 1991 ; Clarke et al. 1993 ; Lowe et al. 1993 ; Ryan et al. 1993 ). P53 is also involved in DNA repair, directly through the association with DNA repair proteins ( Wang et al. 1995 ), or indirectly via the transactivation of p53R2, a protein similar to the R2 subunit of ribonucleotide reductase ( Tanaka et al. 2000 ). The role of p53 in G2 arrest is less clear, although it appears to mediate cycB downregulation and disruption of the cdc2/cycB complex via induction of GADD45 ( Taylor et al. 1999 ; Wang et al. 1999 ; Badie et al. 2000 ), as well cdc2/cycB sequestration by 14‐3‐3 σ induction ( Hermeking et al. 1997 ). P53 is also a mediator of the spindle checkpoint ( Cross et al. 1995 ).
THE SPINDLE CHECKPOINT
Activation of the cdc2/cycB complex results in microtubule nucleation from the centrosomes and spindle formation. The spindle is a highly organized and regulated structure which is responsible for chromosomal separation during cell division.
During DNA replication the newly formed sister chromatids become attached through a protein complex called cohesin, more tightly at the kinetochores than along chromosome arms, and this is indispensable for proper chromosomes segregation ( Nasmyth 1999). The spindle checkpoint detects both microtubule attachment on kinetochores and their tension ( Rieder et al. 1994 ; Li & Nicklas 1995), inhibiting metaphase‐anaphase transition until any defects are repaired (see Fig. 4). Components of the spindle checkpoint regulate the activity of the multisubunit ubiquitin ligase known as APC (anaphase promoting complex). This large protein complex has the double task of inducing the degradation of cohesin, allowing chromatid separation, and of mitotic cyclins, necessary for exit from mitosis. Regulatory subunits of APC determine its specificity: association with the p55CDC/cdc20 protein mediates cohesin cleavage, while Cdh1 is involved in cyclin destruction ( King et al. 1996 ).
Figure 4.
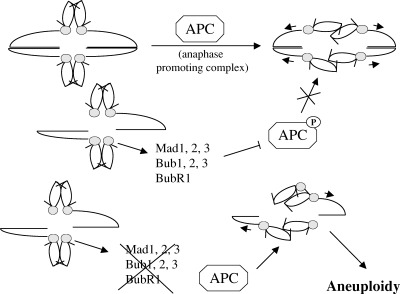
The spindle checkpoint. Proper chromosome alignment in metaphase and microtubule attachment to the kinetochores induces the activation of APC, which mediates the degradation of proteins acting as a ‘molecular glue’ to hold together sister chromatids, thus allowing sister chromosomes separation. Defects in microtubule attachment result in activation of the Mad and Bub proteins, that phosphorylate the regulatory subunit of APC inhibiting its activity, with the result of preventing metaphase‐anaphase transition. Loss of function of these sensor proteins results in chromosome segregation even in conditions of a defective spindle, leading to aneuploidy.
The spindle checkpoint is essential for the maintenance of genomic integrity and its components are fairly conserved throughout evolution. In yeast three BUB genes (budding uninhibited by benomyl) and three MAD genes (mitotic arrest deficient) have been identified as essential for proper chromosome segregation. In mammalian cells protein kinases with a high homology to MAD2 and the BUB proteins have been identified (for a review see Amon (1999)). Perturbations of the spindle lead to the activation of MAD proteins, which bind and inhibit the cdc20 subunit of APC, resulting in cell cycle arrest in metaphase ( Fang & Kirschner 1998; Kallio et al. 1998 ). The kinases hBub1 and hBub1R have an essential role in the control of mitotic progression, and their inactivation leads to premature mitotic exit with an unequal distribution of the genetic material ( Chan et al. 1999 ; see Fig. 4).
It is not clear if genomic instability is a causative event contributing to carcinogenesis or if it is a consequence of cellular transformation. However colorectal cancers have been shown to frequently undergo chromosome segregation errors (every 102 cell divisions), giving rise to aneuploidy and loss of heterozygosity at many loci ( Lengauer, Kinzler & Vogelstein 1997). Cells derived from this type of cancer exit mitosis prematurely after spindle disruption, an event possibly ascribed to mutations of the hBub1 and hBub1R kinases ( Cahill et al. 1998 ). Although not a definite proof (mutations were found only in two out of 20 carcinomas), this finding is an important indication that chromosomal instability and aneuploidy could be involved in carcinogenesis, and furthermore provides a link between the spindle checkpoint and cellular transformation.
CONCLUSION
Genetic analysis of human cancers has revealed that proteins involved in the G1/S checkpoint are inactivated in the majority of cases, and that alterations of the DNA damage checkpoint are likely to be responsible for resistance of tumour cells to chemotherapic agents or irradiation. In contrast, alterations of the G2/M checkpoint are found more rarely.
Our present understanding of G1/S progression is much greater than our knowledge of the remaining cell cycle transitions. Efforts are currently underway to close this gap: a better understanding of the regulation of mitosis might help to elucidate the connection between cell cycle regulation and the maintenance of genetic stability.
Acknowledgements
I wish to thank M. Pearson and D. Prickett for critical reading and scientific discussions. M. M. is recipient of an FIRC fellowship
REFERENCES
- Amon A (1999). The spindle checkpoint. Curr. Op Gen Dev 9,69. [DOI] [PubMed] [Google Scholar]
- Badie C, Bourhis J, Sobczak‐Thepot J, Haddada H, Chiron M, Janicot F, Türsz T, Vassal G (2000). p53‐dependent G2 arrest is associated with a decrease in cyclin A2 and B1 levels in human carcinoma cell line. Br J Cancer 82,642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldin V, Cans C, Knibiehler M, Ducommun B (1997). Phosphorylation of human CDC25B phosphatase by CDK1‐cyclin A triggers its proteasome‐dependent degradation. J. Biol. Chem 272,32731. [DOI] [PubMed] [Google Scholar]
- Barth H, Hoffmann I, Klein S, Kaszkin M, Richards J, Kinzel V (1996). Role of cdc25‐C phosphatase in the immediate G2 delay induced by the exogenous factors epidermal growth factor and phorbolester. J. Cell Physiol. 168,589. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Strauss M, Bartek J (1994). The PRAD‐1/cyclin D1 oncogene product accumulates aberrantly in a subset of colorectal carcinomas. Int J. Cancer 58,568. [DOI] [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE et al. (1999). Heterozygous germ line hChk2 mutations in LiFraumeni syndrome. Science 286,2528. [DOI] [PubMed] [Google Scholar]
- Bianchi AB, Fischer SM, Robles AI, Rinchik EM, Conti CJ (1993). Overexpression of cyclin D1 in mouse skin carcinogenesis. Oncogene 8,1127. [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ et al. (1998). Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391,597. [DOI] [PubMed] [Google Scholar]
- Brown AL, Lee CH, Schwarz JK, Mitiku N, Piwnica‐Worms H, Chung JH (1999). A human Cds1‐related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc. Natl. Acad. Sci. U S A 96,3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J et al. (1998). Mutations of mitotic checkpoint genes in human cancers. Nature 392,300. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA et al. (1998). Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of p53. Science 281,1677. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M (1999). SKP2 is required for ubiquitin‐mediated degradation of the CDK inhibitor p27. Nature Cell Biol. 1,193. [DOI] [PubMed] [Google Scholar]
- Chan TA, Hermeking H, Lengauer C, Kinzler KW, Volgeistein B (1999). 14‐3‐3σ is required to prevent mitotic catastrophe after DNA damage. Nature 401,616. [DOI] [PubMed] [Google Scholar]
- Chan GKT, Jablonski SA, Sudakin V, Hittle JC, Yen TJ (1999). Human BubR1 is a mitotic checkpoint kinase that monitors CENP‐E functions at kinetocores and binds the cyclosome/APC. J. Cell Biol. 146,941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Zhang J, Cheng L, Shapiro DN, Winoto A (1995). Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol Cell Biol. 15,2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi P, Eng WK, Zhu Y et al. (1999). Mammalian Chk2 is a downstream effector of the ATM‐dependent DNA damage checkpoint pathway. Oncogene 18,4047. [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000). Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilising p53. Genes Dev 14,278. [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA et al. (1999). The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin d‐dependent kinases in murine fibroblasts. EMBO J. 18,1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF (1998). Assembly of cyclin D‐dependent kinase and titration of p27Kip1 regulated by mitogen‐activated protein kinase kinase (MEK1). Proc. Natl. Acad. Sci. U S A 95,1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ et al. (1993). Thymocyte apoptosis induced by p53‐dependent and independent pathways [see comments]. Nature 362,849. [DOI] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Quin J, Elledge SJ (1999). Requirement of ATM‐dependent phosphorylation of Brca1 in the DNA damage response to double‐strand breaks. Science 286,1162. [DOI] [PubMed] [Google Scholar]
- Crenshaw DG, Yang J, Means AR, Kornbluth S (1998). The mitotic peptidyl‐prolyl isomerase, Pin1, interacts with Cdc25 and Plx1. Embo J. 17,1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SM, Sanchez CA, Morgan CA et al. (1995). A p53‐dependent mouse spindle checkpoint. Science 267,1353. [DOI] [PubMed] [Google Scholar]
- Deng CX & Scott F (2000). Role of the tumor suppressor gene Brca1 in genetic stability and mammary gland tumor formation. Oncogene 19,1059. [DOI] [PubMed] [Google Scholar]
- Difilippantonio MJ, Zhu J, Chen HT et al. (2000). DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404,510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL et al. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356,215. [DOI] [PubMed] [Google Scholar]
- Draetta GF (1997). Will the real Cdk‐activating kinase please stand up. Curr. Biol. 7,50. [DOI] [PubMed] [Google Scholar]
- Draetta G & Eckstein J (1997). Cdc25 protein phosphatases in cell proliferation. Biochim. Biophys. Acta 1332,M53. [DOI] [PubMed] [Google Scholar]
- Dulic V, Drullinger L, Lees E, Reed S, Stein G (1993). Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E‐Cdk2 and Cyclin D1‐Cdk2 complexes. Proc. Natl. Acad. Sci. USA 90,11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N (1998). The regulation of E2F by pRB‐family proteins. Genes Dev. 12,2245. [DOI] [PubMed] [Google Scholar]
- El‐Deiry WS, Tokino T, Velculescu VE et al. (1993). WAF1, a Potential Mediator of p53 Tumor Suppression. Cell 75,817. [DOI] [PubMed] [Google Scholar]
- Ewen M (1994). The cell cycle and the retinoblastoma protein family. Cancer Metastasis Reviews 13,45. [DOI] [PubMed] [Google Scholar]
- Fang G & Kirschner MH (1998). The checkpoint protein MAD2 and the mitotic regulator cdc20 form a ternary complex with the anaphase‐promoting complex to control anaphase initiation. Genes Dev 12,1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Blasina A, Boddy MN, McGowan CH, Russell P (1999). Cdc25 Inhibited In Vivo and In Vitro by Checkpoint Kinases Cds1 and Chk1. Mol Biol. Cell 10,833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P (1997). Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase [see comments]. Science 277,1495. [DOI] [PubMed] [Google Scholar]
- Gabrielli BG, Clark JM, McCormack AK, Ellem KA (1997). Hyperphosphorylation of the N‐terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J. Biol. Chem 272,28607. [DOI] [PubMed] [Google Scholar]
- Gabrielli BG, De Souza CPC, Tonks ID, Clark JM, Hayward NK, Ellem KAO (1996). Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 109,1081. [DOI] [PubMed] [Google Scholar]
- Gallant P & Nigg E (1992). Cyclin B2 undergoes cell cycle dependent nuclear translocation and, when expressed as a non destructable mutant, causes mitotic arrest in HeLa cells. J. Cell Biol. 117,213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia AJ & Kastan MB (1998). The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 12,2973. [DOI] [PubMed] [Google Scholar]
- Gillett C, Fantl V, Smith R et al. (1994). Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 54,1812. [PubMed] [Google Scholar]
- Gu Y, Turck C, Morgan D (1993). Inhibition of Cdk2 activity in vivo by an associated 20K regulatory subunit. Nature 366,707. [DOI] [PubMed] [Google Scholar]
- Guan K, Jenkins C, Nichols M et al. (1994). Growth suppression by p18, a p16Ink4/Mts1‐ and Mts2‐related Cdk6 inhibitor, correlates with wild‐type pRb function. Genes & Dev. 2939. [DOI] [PubMed]
- Hagting A, Karlsson C, Clute P, Jackman M, Pines J (1998). MPF localisation is controlled by nuclear export. EMBO J. 17,4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M & Peters G (1996). Genetic alterations of cyclins, cyclin‐dependent kinases, and cdk inhibitors in human cancer. Adv. Cancer Res. 68,67. [DOI] [PubMed] [Google Scholar]
- Hanahan D & Weinberg RA (2000). The hallmarks of cancer. Cell 100,57. [DOI] [PubMed] [Google Scholar]
- Hannon GJ & Beach D (1994). p15INK4B is a potential effector of TGF‐b‐induced cell cycle arrest. Nature 371,257. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC (1999). Cdk phosphorylation triggers sequential interactions that progressively block Rb function as cells move through G1. Cell 98,859. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993). The p21 Cdk‐Interacting Protein Cip1 Is a Potent Inhibitor of G1 Cyclin‐Dependent Kinases. Cell 75,805. [DOI] [PubMed] [Google Scholar]
- Heald R, McLoughlin M, McKeon F (1993). Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74,463. [DOI] [PubMed] [Google Scholar]
- Helin K (1998). Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet Dev 8,28. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K et al. (1997). 14‐3‐3 sigma is a p53‐regulated inhibitor of G2/M progression. Mol Cell 1,3. [DOI] [PubMed] [Google Scholar]
- Hirai H & Sherr CJ (1996). Interaction of D‐type cyclins with a novel myb‐like transcription factor, DMP1. Mol Cell Biol. 16,6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A, Kong Y‐Y, Matsuoka S et al. (2000). DNA damage‐induced activation of p53 by the checkpoint kinase Chk2. Science 287,1824. [DOI] [PubMed] [Google Scholar]
- Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G (1993). Phosphorylation and activation of human cdc25‐C by cdc2‐cyclin B and its involvement in the self‐amplification of MPF at mitosis. EMBO J. 12,53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein M, Vogelstein B, Harris CC (1991). p53 mutations in human cancer. Science 253,49. [DOI] [PubMed] [Google Scholar]
- Hussussian CJ, Struewing JP, Goldstein AM et al. (1994). Germline p16 mutations in familial melanoma. Nature Genet 8,15. [DOI] [PubMed] [Google Scholar]
- Izumi T & Maller JL (1995). Phosphorylation and activation of the Xenopus Cdc25 phosphatase in the absence of Cdc2 and Cdk2 kinase activity. Mol Biol. Cell 6,215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Walker D, Maller J (1992). Periodic changes in phosphorylation of the Xenopus cdc25 phosphatase regulates its activity. Mol. Biol. Of Cell 3,927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Kahan S, Tomita N, Zhang Y, Lu S, Weinstein B (1992). Amplification and expression of the human cyclin D gene in esophageal cancer. Cancer Res. 52,2980. [PubMed] [Google Scholar]
- Jiang W, Zhang Y, Kahn SM et al. (1993). Altered expression of the cyclin D1 and retinoblastoma genes in human esophageal cancer. Proc. Natl. Acad. Sci. USA 90,9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Gu Y, Morgan DO (1996). Role of inhibitory cdc2 phosphorylation in radiation‐induced G2 arrest in human cells. J. Cell Biol. 134,936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Hardy S, Morgan DO (1998). Nuclear localisation of byblin B1 controls mitotic entry after DNA damage. J. Cell. Biol. 141,875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio MJ, Weinstein J, Daum JR, Burke DJ, Gorbsky GJ (1998). Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase promoting complex, and is involved in regulating anaphase onset and late mitotic events. J. Cell. Biol. 141,1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A, Shattuck‐Eidens D, Eeles R et al. (1994). Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nature Genet 8,22. [DOI] [PubMed] [Google Scholar]
- Karlsson C, Katich S, Hagting A, Hoffmann I, Pines J (1999). Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell Biol. 146,573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW (1996). How proteolysis drives the cell cycle. Science 274,1652. [DOI] [PubMed] [Google Scholar]
- Krek W & Nigg E (1991). Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10,3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A & Dunphy WG (1996). Purification and molecular cloning of Plx1, a Cdc25‐regulatory kinase from Xenopus egg extracts. Science 273,1377. [DOI] [PubMed] [Google Scholar]
- Kumagai A & Dunphy WG (1999). Binding of 14‐3‐3 proteins and nuclear export control the intracellular localisation of the mitotic inducer Cdc25C. Genes Dev 13,1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labaer J, Garrett MD, Stevenson LF et al. (1997). New functional activities for the p21 family of CDK inhibitors. Genes Dev 11,847. [DOI] [PubMed] [Google Scholar]
- Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I (1998). The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell Sci. 111,2445. [DOI] [PubMed] [Google Scholar]
- Lammie GA, Fantl V, Smith R et al. (1991). D11S287, a putative oncogene on chromosome 11q13, is amplified and expressed in squamous cell and mammary carcinomas and linked to BCL‐1. A truncated cyclin D1 gene encodes a stable mRNA in a human breast cancer cell line. Oncogene 6,439. [PubMed] [Google Scholar]
- Lee J‐SL, Collins KM, Brown AL, Lee C‐H, Chung JH (2000). hCds1‐mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404,201. [DOI] [PubMed] [Google Scholar]
- Lee M, Reynisdottir I, Massague J (1995). Cloning of p57KIP2, cyclin dependent kinase inhibitor with unique domain structure and tissue distribution . Genes & Dev. 9,639. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B (1997). Genetic instability in colorectal cancers. Nature 386,623. [DOI] [PubMed] [Google Scholar]
- Li X & Nicklas RB (1995). Mitotic forces control a cell‐cycle checkpoint. Nature 373,630. [DOI] [PubMed] [Google Scholar]
- Liu F, Stanton JJ, Wu Z, Piwnica‐Worms H (1997). The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol. 17,571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loda M, Cukor B, Tam SW et al. (1997). Increased proteasome‐dependent degradation of the cyclin‐dependent kinase inhibitor p27 in aggressive colorectal carcinomas [see comments]. Nat Med. 3,231. [DOI] [PubMed] [Google Scholar]
- Lopez‐Girona A, Furnari B, Mondesert O, Russell P (1999). Nuclear localization of Cdc25 is regulated by DNA damage and a 1433 protein [see comments]. Nature 397,172. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993). p53 is required for radiation‐induced apoptosis in mouse thymocytes [see comments]. Nature 362,847. [DOI] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC (1998). Rb interacts with histone deacetylase to repress transcription. Cell 92,463. [DOI] [PubMed] [Google Scholar]
- Magnahi‐Jaulin L, Groisman R, Naguibneva I et al. (1998). Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391,601. [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC et al. (1990). Germ Line p53 Mutations in a Familial Syndrome fo Breast Cancer, Sarcomas, and Other Neoplasms. Science 250,1233. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Edwards M, Bai C et al. (1995). p57KIP2, a struturally distinct member of the p21CIP1 Cdk inhibitor family is a candidate tumor suppressor gene. Genes &. Dev. 9,650. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Fiore F, Eytan E et al. (1999). Ubiquitination of p27 is regulated by Cdk‐dependent phosphorylation and trimeric complex formation. Genes Dev 13,1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K (1999). Separating sister chromatids. Trends Biochem. Sci. 24,98. [DOI] [PubMed] [Google Scholar]
- Nishijima H, Nishitani H, Seki T, Nishimoto T (1997). A dual‐specificity phosphatase Cdc25B is an unstable protein and triggers p34 (cdc2) /cyclin B activation in hamster BHK21 cells arrested with hydroxyurea. J. Cell Biol. 138,1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M (1997). Cell cycle regulation by the ubiquitin pathway. Faseb J. 11,1067. [DOI] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM et al. (1995). Role of the ubiquitin‐proteasome pathway in regulating abundance of the cyclin‐dependent kinase inhibitor p27 [see comments]. Science 269,682. [DOI] [PubMed] [Google Scholar]
- Parry DA, Mahony D, Wills K, Lees E (1999). CyclinD‐CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol. Cell. Biol. 19,1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Toczyski DP, Hartwell LH (1997). When checkpoints fail. Cell 88,315. [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Ogg S, Thoma RS, Byrnes MJ (1998). 3rd, Wu, Z, Stephenson, MT and Piwnica‐Worms, H C‐TAK1 protein kinase phosphorylates human Cdc25C on serine 216 and promotes 14‐3‐3 protein binding. Cell Growth Differ 9,197. [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H (1997). Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216 [see comments]. Science 277,1501. [DOI] [PubMed] [Google Scholar]
- Pines J (1999). Four‐dimensional control of the cell cycle. Nat Cell Biol. 1,E73. [DOI] [PubMed] [Google Scholar]
- Pines J & Hunter T (1994). The differential localisation of human cyclin A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 13,3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piva R, Cancelli I, Cavalla P et al. (1999). Proteasome‐dependent degradation of p27/kip1 in gliomas. J. Neuropathol Exp Neurol. 58,691. [DOI] [PubMed] [Google Scholar]
- Polyak K, Lee M, Erdjement‐Bromage H, et al. (1994). Cloning of p27kip1, a cyclin‐dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 79,59. [DOI] [PubMed] [Google Scholar]
- Rhind N, Furnari B, Russell P (1997). Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev 11,504. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G (1994). Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell. Biol. 127,1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman G & Shiloh Y (1999). ATM: a mediator of multiple responses to genotoxic stress. Oncogene 18,6135. [DOI] [PubMed] [Google Scholar]
- Roussel MF (1999). The INK4 family of cell cycle inhibitors in cancer. Oncogene 18,5311. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Danish R, Gottlieb CA, Clarke MF (1993). Cell cycle analysis of p53‐induced cell death in murine erythroleukemia cells. Mol Cell Biol. 13,711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS et al. (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25 [see comments]. Science 277,1497. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar‐Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L (1995). Tagle DA, Smith S, Uziel T, Sfez S. and et al. A single ataxia telangiectasia gene with a product similar to PI‐3 kinase [see comments]. Science 268,1749. [DOI] [PubMed]
- Seki T, Yamashita K, Nishitani H, Russell P, Nishimoto T (1992). Chromosome condensation caused by loss of RCC1 function requires the cdc25C protein that is located in the cytoplasm. Mol. Biol. Of Cell 3,1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D (1993). A new regulatory motif in cell‐cycle control causing specific inhibition of cyclin D/CDK4 [see comments]. Nature 366,704. [DOI] [PubMed] [Google Scholar]
- Sharpless NE & Depinho RA (1999). The INK4A/ARF locus and its two gene products. Curr. Op Genet Dev 9,22. [DOI] [PubMed] [Google Scholar]
- Shen M, Stukenberg PT, Kirschner MW, Lu KP (1998). The essential mitotic peptidyl‐prolyl isomerase Pin1 binds and regulates mitosis‐specific phosphoproteins. Genes Dev 12,706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ (1996). Cancer cell cycles. Science 274,1672. [DOI] [PubMed] [Google Scholar]
- Sherr CJ & Roberts JM (1999). CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 13,1501. [DOI] [PubMed] [Google Scholar]
- Sherr CJ & Weber JD (2000). The ARF/p53 pathway. Curr. Opin. Genet Dev 10,94. [DOI] [PubMed] [Google Scholar]
- Shieh S‐Y, Ahn J, Tamai K, Taya Y, Prives C (2000). The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage‐inducible sites. Genes Dev 14,289. [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB (1997). DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev 11,3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GC, Divecha N, Lakin ND, Jackson SP (1999). The DNA‐dependent protein kinase and related proteins. Biochem. Soc. Symp 64,91. [PubMed] [Google Scholar]
- Srivastava S, Zou Z, Pirollo K, Blattner W, Chang EH (1990). Germ‐line transmission of a mutated p53 gene in a cancer‐prone gamily with LiFraumeni syndrome. Nature 348,747. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Arakawa H, Yamaguchi T et al. (2000). A ribonucleotide reductase gene involved in a p53‐dependent cell‐cycle checkpoint for DNA damage. Nature 404,42. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Deprimo SE, Argawal A, Argawal ML, Schontal AH, Katula KS, Stark GR (1999). Mechanisms of G2 arrest in response to over expression of p53. Mol Biol Cell 10,3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima & Hunter T (1994). p27, a novel inhibitor of G1‐cyclin‐cdk protein kinase activity, is related to p21. Cell 78,67. [DOI] [PubMed] [Google Scholar]
- Toyoshima F, Moriguchi T, Wada A, Fukuda M, Nishida E (1998). Nuclear export of cyclin B1 and its possible role in the DNA damage‐induced G2 checkpoint. EMBO J. 17,2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR (1999). Breast cancer genes and DNA repair. Science 286,1100. [DOI] [PubMed] [Google Scholar]
- Wang XW, Yeh H, Schaeffer L et al. (1995). p53 modulation of TFIIH‐associated nucleotide excision repair activity. Nat Genet 10,188. [DOI] [PubMed] [Google Scholar]
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Conner PM, Fornace AJ jr, Harris CC (1999). GADD4S induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 96,3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995). The retinoblastoma protein and cell cycle control. Cell 81,323. [DOI] [PubMed] [Google Scholar]
- Winkler KE, Swenson KI, Kornbluth S, Means AR (2000). Requirement of the Prolyl isomerase Pin 1 for the replication checkpoint. Science 287,1644. [DOI] [PubMed] [Google Scholar]
- Wöfel T, Hauer M, Schneider J et al. (1995). Meyer zum Büschenfelde, K and Beach, D A p16INK4a‐Insensitive CDK4 Mutant Targeted by Cytolytic T Lymphocytes in a Human Melanoma . Science 269,1281. [DOI] [PubMed] [Google Scholar]
- Yang J, Winkler K, Yoshida M, Kornbluth S (1999). Maintenance of G2 arrest in the Xenopus oocyte: a role for 14‐3‐3‐mediated inhibition of Cdc25 nuclear import. Embo J. 18,2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonish‐Rouach E, Resnitzky D, Lotem J, Sacks L, Kimchi A, Dren M (1991). Wild‐type p53 induces apoptosis of myeloid leukemia cells that is inhibited by IL‐6. Nature 345. [DOI] [PubMed]
- Zhang HS, Postigo AA, Dean DC (1999). Active transcriptional repression by the Rb‐E2F complex mediates G1 arrest triggered by p16INK4a, TGFb, and contact inhibition . Cell 97,53. [DOI] [PubMed] [Google Scholar]
- Zuo L, Weger J, Yang B et al. (1996). Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 12,97. [DOI] [PubMed] [Google Scholar]