

Abstract
Objectives: Translational research using adult stem cells derived from various tissues has been highlighted in cell‐based therapy. However, there are many limitations to using conventional culture systems of adult stem cells for clinically applicability, including limited combinations of cytokines and use of nutrients derived from animals. Here, we have investigated the effects of placental extract (PE) for culture of placenta‐derived stem cells (PDSCs) as well as their potential for hepatogenic differentiation.
Materials and methods: Placental extract, extracted using water‐soluble methods, was used as a supplement for culture of PDSCs. Cell viability was determined using the MTT assay, and cytokine assay was performed using Luminex assay kit. Gene expression, indocyanine green (ICG) up‐take, PAS (Periodic Acid‐Schiff) staining and urea production were also analysed.
Results: The placental extract contained several types of cytokine and chemokine essential for maintenance and differentiation of stem cells. Expression of stemness markers in PDSCs cultured with PE is no different from that of PDSCs cultured with foetal bovine serum (FBS). After hepatogenic differentiation, expression patterns for hepatocyte‐specific markers in PDSCs cultured with PE were consistent and potential for hepatogenic differentiation of PDSCs cultured with PE was similar to that of PDSCs cultured with FBS, as shown by PAS staining and urea production assays.
Conclusions: Our findings revealed that placental extract could be used as a new component for culture of adult stem cells, as well as for development of human‐based medium, in translational research for regenerative medicine.
Introduction
Adult stem cells, including mesenchymal stem cells (MSCs) derived from various tissues, are attractive sources for use in cell therapy in the field of regenerative medicine (1). Among adult stem cells, bone marrow‐derived MSCs (BM‐MSCs) are multi‐potent cell populations with immunomodulatory functions. When cultured using appropriate conditions for differentiation into target cells (that include osteocytes, chondrocytes, adipocytes, neuronal cells, cardiomyocytes, hepatocytes and others), they have potential for multi‐lineage differentiation (2, 3). However, in terms of efficacy and safety, a number of difficulties is associated with their isolation and large‐scale culture for transplantation.
Otherwise, placenta‐derived stem cells (PDSCs), which are primitive cell sources derived from foetal origins, have multipotential differentiation capacities for various cell types. In addition, no ethical problems or immune rejection problems are associated with use of PDSCs. Other advantages include ease of accessibility and abundant cell numbers (4, 5, 6). Thus, many scientists have demonstrated the possibility of cell‐based therapy using PDSCs.
At present, the problem of obtaining multipotential cells that can be safely expanded and manufactured for use as clinical‐grade stem cells with multilineage differentiation potential, is a major obstacle to successful cell therapy (7, 8, 9). One important point is that there are limitations to production of clinically applicable adult stem cells using conventional culture systems.; these include limited combinations of cytokines and sources of nutrients, such as foetal bovine serum and animal‐derived feeder cells (9, 10). Furthermore, mechanisms that govern differentiation and interactions within cytokine mixtures are still controversial. For this reason, new sources derived from human materials must be sought for recently required clinical applications.
In general, the placenta can synthesize/derive various nutrients and metabolites, which are supplied to the foetus from the mother during pregnancy (11). There are many types of cytokine that are essential for foetal growth and development. A number of these plus hormones and growth factors, such as hepatocyte growth factor (HGF), epidermal growth factor (EGF), transforming growth factor‐α, ‐β (TGF‐α, ‐β) and their receptors have also recently been identified in human placenta (12, 13, 14). Thus, the potential of placental extracts for hepatic disorders has been conceived (15). Although placental extracts have broad use in clinical practice, activities of various cytokines in them are often destroyed by processing or use of organic solvents or high temperatures. This can limit their application in biological as well as in clinical research.
The aim of this study was to develop effective extraction methods for high quality proteins from placentas and to evaluate the effects of placental extract for culture and hepatogenic differentiation of PDSCs and to devise new culture parameters that affect clinical grades for cell therapy.
Materials and methods
Isolation and culture of placenta‐derived stem cells
Full‐term human placentas were collected after obtaining written informed consent from donors. PDSCs were harvested from their chorionic plates after Caesarean section, using mechanical and enzymatic digestion methods. Briefly, cells scraped from the entire chorioamniotic membranes (approximately 10 g) were washed in phosphate‐buffered saline then treated with 0.5% collagenase IV (Sigma, St Louis, MO, USA) for 20 min at 37 °C. Cells were then enzymatically digested twice for approximately 20 min each, at 37 °C and then prepared as single‐cell suspensions. These samples were cultured in Dulbecco’s modified Eagle’s medium nutrient mixture F‐12 (DMEM/F12; Invitrogen, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS; Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin, 25 μg/ml FGF‐4 (Invitrogen) and 1 μg/ml heparin (Sigma‐Aldrich Co.). Cultures were maintained at 37 °C in a humidified atmosphere with 5% CO2. Three to 5 days after initiating incubation, small digested residues were removed and culture was continued. When cells were more than 80% confluent, they were recovered using 0.25% trypsin/EDTA and split at 1:3 dilution. Using the same conditions, the cells’ culture was continued. Cell viability was determined using the trypan blue exclusion test and numbers were counted using a haemocytometer.
Preparation of placental extract proteins
Red blood cells were removed from approximately 20 g of placental villus tissue by washing in buffer (50 mm Tris–HCl, 150 mm NaCl, 150 mm sucrose, pH 7.2) for 30 min at 4 °C. Washed tissue was placed in a sterile dish and chopped using sterile scissors and using pestle and mortar, chopped tissue was ground up. Samples were then snap‐frozen in liquid nitrogen for additional grinding, thawed and resuspended in 30 ml of homogenizing buffer (DMEM high glucose, 0.32 m sucrose, 2 mm EDTA). This tissue then was sonicated on ice (amplitude 60, pulse 4, repeat 4). After centrifugation (170 g for 15 min at 4 °C), total protein content of the placental extract was measured using a BCATM protein assay kit (Pierce, Rockford, IL, USA). Placental extract proteins from 10 individual placentas were measured and mixed according to the above protocols for minimized batch to batch variation. Extracts were filtered through a 0.22 μm filter and either used fresh, or frozen in liquid nitrogen and stored at −80 °C for future use. Three methods were used to obtain placental extracts, with or without heating. First, naïve protein was obtained without heat [naïve type, placental extract protein (PEP)‐A]; second, placental extract was heated for 30 min at 56 °C (pasteurized type, PEP‐B) and third, placental extract was autoclaved for 15 min at 121 °C (autoclaved type, PEP‐C).
Multiplex supernatant cytokine assay (Luminex)
Fifty‐microlitre samples were combined with coated beads using the MILLIPLEXTM MAP kit (Millipore Corp, St Charles, MO, USA). Commercial kits were run in individual plates, with buffers and standards according to previous reports (16). Incubations and washes were performed using 1.2‐μm filter membrane, 96‐well microtiter plates (Millipore, Bedford, MA, USA).
After the final wash, beads from the plates were resuspended in a 125 μl cuvette of the Luminex apparatus. An acquisition gate was set between 7500 and 13 500 for a doublet discriminator; sample volume 75 μl and 100 events/region were acquired. To obtain concentration values, raw data (mean fluorescence intensity) from all bead combinations tested were analysed using Master Plex QT3.0 quantification software (MiraiBio Inc., Alameda, CA, USA).
MTT assay
Cell viability was assessed on the 96‐multiwell culture plates. HTR‐8 SV/neo trophoblast cell lines and PDSCs were seeded at concentrations of 4 × 103 cells/well. After 3 h, media in wells were exchanged with media that either had no FBS or included serum replacement (SR). Increasing concentrations of placental protein extract (0.5, 1, 2.5 and 5 μg) were added for 24, 48 and 72 h, at which times, cell viability was estimated by the MTT assay according to the manufacturer’s instructions. Briefly, 20 μl of MTT (5 mg/ml in PBS) was added to each well and incubated for 4 h. Media were discarded and 100 μl of dimethyl sulphoxide was added. Plates were gently mixed on a gyrating shaker at room temperature for 30 min. Optical density was measured at 562 nm. Results were expressed as percentages of viable cells compared to controls (cells without test placental protein extract). All results are expressed as percent‐ages of untreated control values of three independent experiments.
Culture of placenta‐derived stem cells using placenta extract proteins
Passages 8 through 10 placenta‐derived cells, including frozen and thawed samples, were plated on 35‐mm‐diameter cell culture dishes at a density of 5 × 103 cells/cm2 in our culture medium. This consisted of Dulbecco’s modified Eagle’s medium nutrient mixture F‐12 (DMEM/F12; Invitrogen) supplemented with 10% FBS and 23.8 μg/ml (PEP‐A, PEP‐B, PEP‐C), 20% SR, 100 U/ml penicillin and 100 μg/ml streptomycin, 25 μg/ml FGF‐4 and 1 μg/ml heparin. Cells were cultured for 10 days. Total viable cell numbers were determined using the trypan blue exclusion technique and a haemocytometer.
Hepatogenic differentiation of placental‐derived stem cells using placental extract proteins
Differentiation potential of PDSCs into hepatocytes by induction, using two separate steps, was assessed (17). When the cells cultured in each mixture, including serum and PEPs, achieved 80% confluence, they were dissociated by trypsin and plated at a density of 5 × 103 cells/cm2 on culture dishes for further differentiation protocols.
Cells were pre‐cultured in 60% DMEM/F12 and 40% MCDB201 supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, 20 ng/ml EGF and 10 ng/ml bFGF to stop cell proliferation prior to induction of differentiation towards the hepatic phenotype. A two‐step differentiation protocol was then performed. Step‐1 differentiation medium, consisting of 60% DMEM/F12 and 40% MCBD201, was supplemented with 20 ng/ml HGF and 10 ng/ml bFGF for 7 days. Step‐2 differentiation medium, consisting of 60% DMEM/F12 and 40% MCDB201, was supplemented with 20 ng/ml oncostatin M, 1 μm dexa and 50 mg/ml ITS for 7 days. All chemicals were purchased from Sigma.
RT‐PCR
Approximately 105–106 placenta‐derived stem cells were lysed in 1 ml of TRIzol reagent (Invitrogen, Carlsbad, CA, USA); total RNA was extracted with 200 μl of chloroform and precipitated with 500 μl of 80% (v/v) isopropanol. After removal of the supernatant, the RNA pellet was washed in 75% (v/v) ethanol, air‐dried and dissolved in 0.1% (v/v) diethyl pyrocarbonate‐treated water. RNA concentration was determined by measurement of absorbance at 260 nm using a spectrophotometer. The reverse transcription reaction was performed with 2 μg pure total RNA using SuperScriptTM III reverse transcriptase (Invitrogen). Synthesized cDNA was amplified using PCR. Primer sequences used are given in Table 1. Amplification conditions included the following: 5 min at 95 °C, followed by 24–31 cycles at 94 °C for 30 s, 48–60 °C for 1 min and at 72 °C for 1 min. PCR products were visualized after electrophoresis on a 2% (w/v) agarose gel containing 0.5 μg/ml ethidium bromide and then photographed.
Table 1.
Sequence of primers used for RT‐PCR and length of fragments
| Genes | Sequence | T m (°C) | Size (bp) |
|---|---|---|---|
| OCT4 | F: 5′‐ACA CTC GGA CCA CGT CTT TC‐3′ | 54 | 300 |
| R: 5′‐CGT TCT CTT TGG AAA GGT GTT C‐3′ | |||
| Nanog | F: 5′‐TTC TTG ACT GGG ACC TTG TC‐3′ | 54 | 300 |
| R: 5′‐GCT TGC CTT GCT TTG AAG CA‐3′ | |||
| Sox2 | F: 5′‐GGG CAG CGT GTA CTT ATC CT‐3′ | 52 | 200 |
| R: 5′‐AGA ACC CCA AGA TGC ACA AC‐3′ | |||
| NF‐68 | F: 5′‐GAG TGA AAT GGC ACG ATA CCT A‐3′ | 58 | 500 |
| R: 5′‐TTT CCT CTC CTT CTT CTT CAC CTT C‐3′ | |||
| Cardiac | F: 5′‐GGA GTT ATG GTG GGT ATG GGT C‐3′ | 58 | 500 |
| R: 5′‐AGT GGT GAC AAA GGA GTA GCC A‐3′ | |||
| hAFP | F: 5′‐AGC TTG GTG GAT GAA AC‐3′ | 50 | 200 |
| R: 5′‐TCC AAC AGG CCT GAG AAA TC‐3′ | |||
| HLA‐G | F: 5′‐GCG GCT ACT ACA ACC AGA GC‐3′ | 58 | 550 |
| R: 5′‐GCA CAT GGC ACG TGT ATC TC‐3′ | |||
| TERT | F: 5′‐GAG CTG ACG TGG AAG ATG AG‐3′ | 55 | 300 |
| R: 5′‐CTT CAA GTG CTG TCT GAT TCC AAT G‐3′ | |||
| β‐actin | F: 5′‐TCC TTC TGC ATC CTG TCA GCA‐3′ | 58 | 300 |
| R: 5′‐CAG GAG ATG GCC ACT GCC GCA‐3′ | |||
| HNF1α | F: 5′‐AC ACC ACT CTG GCA GCC ACA CT | 58 | 114 |
| R: 5′‐CGG TGG GTA CAT TGG TGA CAG AAC | |||
| HNF4α | F: 5′‐CTG CTC GGA GCC ACC AAG AGA TCC ATG‐3′ | 55 | 370 |
| R: 5′‐ATC ATC TGC CAC GTG ATG CTC TGC A‐3′ | |||
| CXCR4 | F: 5′‐ACG TCA GTG AGG CAG ATG‐3′ | 58 | 202 |
| R: 5′‐GAT GAC TGT GGT CTT GAG‐3′ | |||
| Albumin | F: 5′‐CCC CAA GTG TCA ACT CCA AC‐3′ | 54 | 450 |
| R: 5′‐CTC CTT ATC GTC AGC CTT GC‐3′ | |||
| TAT | F: 5′‐AAC GAT GTG GAG TTC ACG G‐3′ | 59 | 288 |
| R: 5′‐GAC ACA TCC TCA GGA GAA TGG‐3′ | |||
| 28s rRNA | F: 5′‐TTG AAA ATC CGG GGG AGA G‐3′ | 52 | 100 |
| R: 5′‐ACA TTG TTC CAA CAT GCC AG‐3′ |
Indocyanine green up‐take assay in vitro and in vivo
Indocyanine green (ICG) uptake was used to follow hepatogenic differentiation of the PDSCs in vitro, according to previous reports (18). ICG solution (0.25 mg/ml; Dongindang Pharm. Co. Ltd., Gyeonggi‐do, Korea) was applied to dishes with final differentiated PDSCs and incubated at 37 °C for 30 min. These were rinsed three times in PBS and dishes were refilled with DMEM containing 10% FBS. ICG uptake by the cells was then measured using an inverted‐objective microscope.
Urea assay
Undifferentiated and differentiated PDSCs were cultured for 24 h in supplemented media. Supernatants were collected and analysed for urea production according to the manufacturer’s guidelines. Urea concentrations were determined by colorimetric assay (Quantichrom Urea assay kit; Bioassay Systems, Hayward, CA, USA). Fresh culture media and HepG2 cells were used as negative and positive controls respectively. All results are expressed as mean ± SD of three independent experiments.
Results
Cytokines in placental extract proteins variedby preparation
Proteins were extracted from placentas using water‐soluble extraction methods, filtered through a 0.22 μm pore filter, and then sterilized using different processes, including filtration (PEP‐A), pasteurization (PEP‐B) and autoclaving (PEP‐C). In general, manufacturing processes for proteins used as cell culture nutrients require several steps (for example, protein degradation, hydrolysis using strong acid, lysis by breakdown enzymes, concentrating procedures and others). Many components derived from various sources are denatured during these steps. Thus, we sought to determine whether or not placental proteins extracted by water‐soluble methods as the only essential step, could preserve functionality of molecules for maintenance and expansion of cultures of the HTR‐8 SV/neo cell line and PDSCs. Quantity and quality of placental proteins extracted from placental tissues were analysed through distribution of Ponceau S staining and expression of β‐actin, in samples of serial diluted placental extracts loaded for western blot analysis as internal control, respectively.
For analysis of changes in cytokine profiles by different processing methods, we simultaneously analysed the following 36 human cytokines and chemokines: EGF, eotaxin, FGF‐2, Flt‐3 ligand, fractalkine, GM‐CSF, IFN‐2, IFN‐γ, IL‐1α, IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, IL‐9, IL‐10, IL‐12(p40), IL‐12(p70), IL‐13, IL‐15, IL‐17, IP‐10, MCP‐1, MIP‐1α, MIP‐1β, PDGF‐AA, PDGF‐AB/BB, RANTES, sCD40L, TGF‐β, TNF‐α, TNF‐β and VEGF. Concentrations of various cytokines showed different patterns for each of the placental extracts (Table 2). Generally, concentrations of cytokines in PEP‐A were higher than those of PEP‐B and PEP‐C. Among these, cell proliferation related cytokines, such as FGF‐2 and PDGFs, were found at high levels in PEP‐A. Others, including fractakine, GM‐CSF, IFNα2a, IL‐12(p40) and sCD40L were only detected in PEP‐A. However, IFN‐γ was not detected in any of the preparations. These findings suggest that various cytokines were affected by processing, including the sterilization steps. PEP‐A, prepared by a simple, water‐soluble extraction method, could contain an adequate amount of useful components from the placenta.
Table 2.
Analysis of cytokine components in placental extract proteins
| No. | Analyte name | PEP‐A (pg/ml) | PEP‐B (pg/ml) | PEP‐C (pg/ml) |
|---|---|---|---|---|
| 1 | EGF | 7.67 | 5.68 | 4.21 |
| 2 | Eotaxin | 78.17 | 39.08 | 15.9 |
| 3 | FGF‐2 | 3452.41 | 2447.93 | 408.12 |
| 4 | Flt‐3Ligand | 386.98 | 368.41 | 355.09 |
| 5 | Fractakine | 33.51 | 0.00 | 0.00 |
| 6 | GM‐CSF | 55.30 | 0.00 | 0.00 |
| 7 | IFNα2a | 59.03 | 0.00 | 0.00 |
| 8 | IFN‐γ | 0.00 | 0.00 | 0.00 |
| 9 | IL‐10 | 31.72 | 27.75 | 24.18 |
| 10 | IL‐12(p40) | 203.82 | 0.00 | 0.00 |
| 11 | IL‐12(p70) | 3.66 | 2.89 | 1,57 |
| 12 | IL‐13 | 4.58 | 4.28 | 4.01 |
| 13 | IL‐15 | 8.54 | 7.89 | 7.34 |
| 14 | IL‐17 | 3.02 | 2.72 | 2.55 |
| 15 | IL‐1α | 19.91 | 40.17 | 12.77 |
| 16 | IL‐1β | 22.68 | 13.75 | 8.15 |
| 17 | IL‐2 | 45.16 | 40.68 | 39.17 |
| 18 | IL‐3 | 0.00 | 0.00 | 0.00 |
| 19 | IL‐4 | 2.67 | 2.65 | 2.59 |
| 20 | IL‐5 | 1.86 | 1.83 | 1.82 |
| 21 | IL‐6 | 149.28 | 138.69 | 129.71 |
| 22 | IL‐7 | 5.70 | 4.22 | 3.11 |
| 23 | IL‐8 | 10.20 | 7.54 | 5.43 |
| 24 | IL‐9 | 3.77 | 2.99 | 1.87 |
| 25 | IP‐10 | 790.49 | 138.35 | 112.01 |
| 26 | MCP‐1 | 202.20 | 133.33 | 98.71 |
| 27 | MIP‐1α | 144.18 | 85.85 | 79.12 |
| 28 | MIP‐1β | 166.50 | 51.44 | 25.15 |
| 29 | PDGF‐AA | 959.89 | 795.00 | 325.13 |
| 30 | PDGF‐BB | 262.14 | 275.54 | 258.19 |
| 31 | RANTES | >3,200 | 183.43 | 26.98 |
| 32 | sCD40L | 154.31 | 0.00 | 0.00 |
| 33 | TNF‐α | 10.07 | 4.01 | 3.51 |
| 34 | TNF‐β | 38.38 | 38.38 | 38.38 |
| 35 | TNG‐α | 10.07 | 4.01 | 12.97 |
| 36 | VEGF | 40.95 | 36.85 | 2.39 |
Cell viability of the HTR‐8 SV/neo trophoblast cell line using placental extract
For analysis of optimum concentration for each type of placental extract that did not show increased cytotoxicity, HTR‐8 SV/neo trophoblast cell line was cultured in medium with varying concentrations of each placental extract protein (PEP‐A, PEP‐B, PEP‐C) that was used as replacement of FBS for 24, 48 and 72 h. A representative experiment is shown in Data S1. As shown in Fig. 1, viability of HTR‐8 SV/neo trophoblast cells in medium without FBS (Fig. 1a) was not observed; otherwise, viability of HTR‐8 trophoblast cells with 0.5 μg concentrations of PEP‐A (Fig. 1c) was approximately 70% of that in conventional culture using FBS (Fig. 1b). In addition, viability of cells treated with PEP‐A was consistent at different times. Cell viability in PEP‐B and PEP‐C groups decreased (Fig. 1d,e). In particular, morphology of HTR‐8 cells cultured in medium with 0.5 μg of PEP‐A showed them to be very healthy compared to cells from the other groups, including PEP‐B and PEP‐C, and even in of the control group cultured with FBS (Fig. 1).
Figure 1.
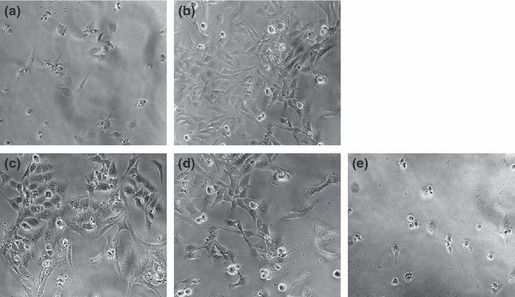
Growth features of cultured HTR‐8 SV/neo trophoblast cell line shown for various culture nutrients, in absence (a) or presence (b) of FBS and placental extract proteins (PEPs) including PEP‐A (c), PEP‐B (d) and PEP‐C (e) at 72 h. Cell viabilities of HTR‐8 SV/neo trophoblast cells with 0.5 μg concentrations of PEP‐A and PEP‐B were approximately 70% of those in conventional culture using FBS. But, growth of HTR‐8 SV/neo trophoblast cells using PEP‐C is similar to that in absence of FBS (×100 original magnification).
Cell growth and characterization of PDSCs culturedwith placental extract
In addition, we examined the question of whether or not application of placental extract could be used for maintenance and expansion of PDSCs. Optimal concentrations of placental extract proteins without cytotoxicity in the culture system were similar to those seen for HTR‐8 SV/neo cells. These results suggest that placental extract proteins, such as PEP‐A and PEP‐B could be used for culture of PDSCs (Data S2). To evaluate effects of placental extract for PDSC culture, we compared differences of growth rate and morphology of cells, using conventional medium containing FBS and the new medium containing each placental extract, for 5 days and for 10 days (Fig. 2). After culture in the different components, including FBS, PEPs, and 20% SR (used as nutrient source for culture of embryonic stem cells), most PDSCs were consistently attached to their culture vessels and maintained for 5 and 10 days, with the exception of cultures containing PEP‐C and 20% SR. In particular, morphology of PDSCs grown in media containing PEP‐A and PEP‐B appeared to be healthier than those in conventional medium with FBS (Fig. 2a). Proliferation rates of PDSCs using media containing PEP‐A and PEP‐B were slower compared to those with conventional medium containing FBS (Fig. 2b) and followed general growth curves of cells. However, PDSCs in media containing PEP‐C and 20% SR showed no proliferation changes.
Figure 2.
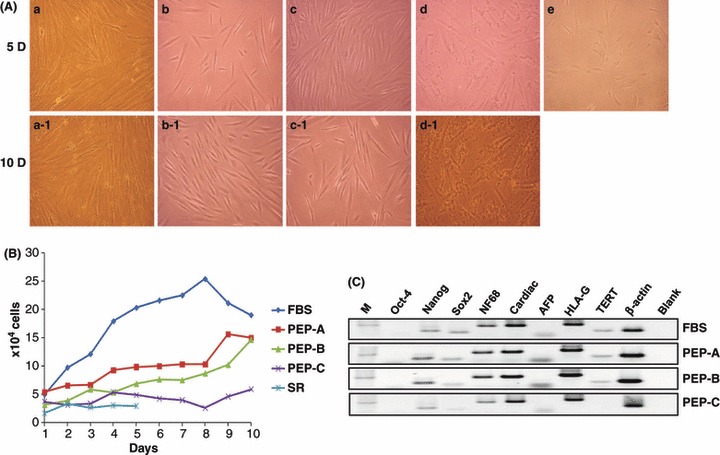
Culture and characterization of PDSCs using placental extract proteins. (A) Morphological features of the cultured PDSCs shown for various PEPs depending on incubation time from 5 days (5D) to 10 days (10D). Serum replacement (SR, e) was used as negative control to FBS (a, a‐1). Morphology of PDSCs grown in media containing PEP‐A (b, b‐1) and PEP‐B (c, c‐1) appeared healthier than those in conventional medium with FBS. PDSCs in media containing PEP‐C (d, d‐1) induced cell death; otherwise, PDSCs showed no growth in 20% SR (e). (×100 original magnification); (B) Growth rate of PDSCs for culture. Growth rates of PDSCs using media containing PEP‐A and PEP‐B were lower compared to those in conventional medium containing FBS and followed general growth curve of cells; (C) Expression of genes indicating stemness of PDSCs cultured by PEPs. Gene expression patterns of PDSCs cultured with PEP‐A were similar to those of PDSCs cultured with FBS. M: marker.
Next, we evaluated the question whether or not type of medium could alter gene expression associated with the original characteristics of PDSCs. No changes were observed in patterns using media containing FBS or PEP‐A. Expression of some genes in PDSCs cultured with media containing PEP‐B or PEP‐C showed a tendency to be lower (Fig. 2c). From these results, it is clear that medium with PEP‐A could be used as an alternative protein source, although growth rate was slower than for cells cultured using medium with FBS.
Hepatogenic differentiation of PDSCs cultured with placental extract
We have demonstrated a potential for differentiation of PDSCs into hepatocytes by induction using two separate steps. During progression towards hepatogenesis, morphology of PDSCs changed from spindle shaped to polygonal, which is a characteristic hepatocyte‐like morphology. In addition, expression of hepatogenesis‐related genes was increased. Therefore, we examined the question of whether or not gene expression patterns in hepatogenic differentiated PDSCs would be altered when cultured in different media, containing FBS and PEPs. When cultured with both FBS and PEPs, expression of hepatocyte nuclear factor 1α (HNF1α), HNF4 α, α‐fetoprotein (AFP), CXCR4, albumin and TAT was up‐regulated in hepatogeneic differentiated PDSCs compared to naïve PDSCs (Fig. 3). There were no differences in expression of hepatogenesis‐related genes for each type of medium.
Figure 3.
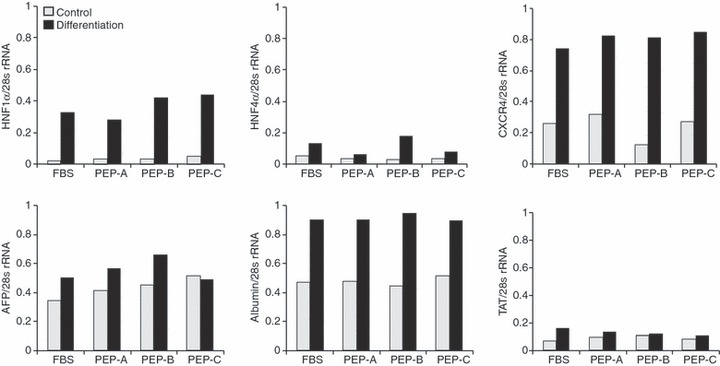
Gene expression of cultured PDSCS by placental extract proteins according to hepatogenic differentiation. Several gene expressions related to hepatogenic differentiation in PDSCs depend on PEPs before (grey bar) and after (black bar) inducing hepatogenic differentiation. Gene expressions were analysed semi‐quantitatively using 28s rRNA, which was used as an internal control.
As alteration of gene expression and effects of placental extract are considered to represent a progressive gain of hepatic function, we analysed the cells using ICG uptake assays. ICG is a non‐toxic organic anion used clinically to test hepatic function. For analysis of glycogen storage, we also stained hepatogenically differentiated PDSCs using PAS staining solution. As shown in Fig. 4a, ICG uptake was observed in hepatogenically differentiated PDSC cultures, regardless of protein composition, including FBS, PEP‐A, PEP‐B and PEP‐C (Fig. 4a). However, there were differences in efficiency of glycogen storage of hepatogenically differentiated PDSCs, depending on the protein source (Fig. 4b). Although urea production in hepatogenically differentiated PDSCs cultured with FBS was higher than for PDSCs cultured with PEPs, the order of urea production was PEP‐A, PEP‐B and PEP‐C (Fig. 4c). From these results, we concluded that PEP‐A and PEP‐B, but not PEP‐C, could be used as alternative protein sources for expansion of PDSCs as well as for inducing their hepatogenic differentiation of.
Figure 4.
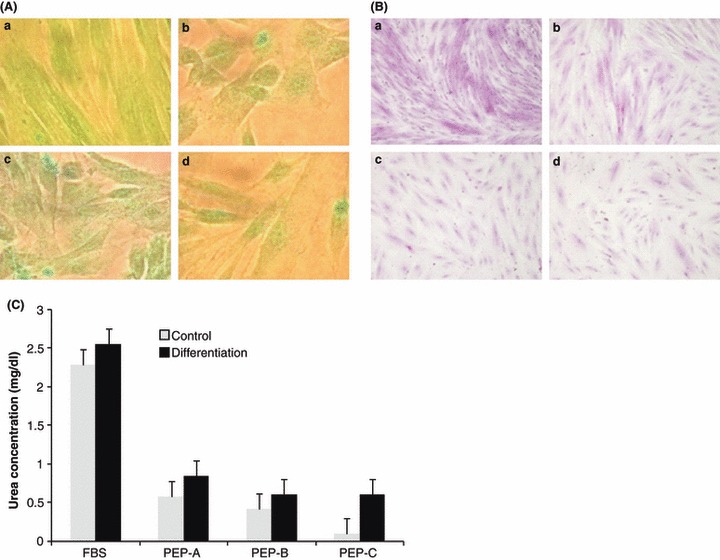
Potential of hepatogenic differentiation of cultured PDSCs by placental extract proteins. (A) ICG up‐taken PDSCs were observed to be green in colour in hepatogenic induced PDSCs after differentiation in cultured PDSCs by FBS (a), PEP‐A (b), PEP‐B (c) and PEP‐C (d). (B) After hepatogenic differentiation, glycogen deposition in cultured PDSCs by FBS (a), PEP‐A (b), PEP‐B (c), and PEP‐C (d) stained by PAS. (C) Urea production of cultured PDSCs by placental extract depended on hepatogenic differentiation (×200 original magnification).
Discussion
In the present study we have demonstrated for the first time, feasibility of placental proteins extracted using water‐soluble methods for culture and hepatogenic differentiation of placenta‐derived stem cells for clinical grade use.
Cell‐based therapy using human stem cells in regenerative medicine is an emerging field. However, there are many obstacles to overcome before cell therapy can be used in clinical trials. In this regard, source and number of stem cells obtained, proliferation and differentiation potentials of stem cells and culture and expansion of human stem cells to be transplanted into a target organ, are essential factors for cell therapy (3, 8, 9, 19). In particular, safety of stem cells for transplantation has been a critical issue because of uses of animal sources, such as FBS or foetal calf serum, for the cells’ expansion (20, 21).
Conventional culture methods using animal serum for cell therapy could involve several risks, including immune inflammatory rejection and pathogenic transmission as a result of contamination of proteins derived from animals. These cannot be effectively eliminated through processing after culture for clinical use of stem cells (22). For these reasons, many scientists have attempted to develop new culture methods capable of maintaining the ‘stemness’ of stem cells, using serum‐free medium.
Although various supplements and several types of cytokine have been used in these approaches, serum‐free culture systems do not allow for expansion or maintenance of differential potential of stem cells for clinical grade use (9, 23). Other possibilities include use of human serum or pooled human platelet lysate for their culture (9, 24, 25). However, these have several limitations, including high cost and limited quality. However, we have been able to demonstrate that expansion and differentiation of placenta‐derived stem cells is feasible using placental extract proteins in FBS‐free medium. Several types of cytokine in PEPs were extracted using water‐soluble methods (Table 2). As cytokines derived from placentas can be easily denatured using traditional methods, such as temperature and organic solvents, we compared effects of placental extract, including PEP‐A, PEP‐B and PEP‐C, which varied over heat treatments during sterilization for culture. Effects of PEP‐A and PEP‐B were shown to be similar, whereas PEP‐C was not. These findings indicate that high temperature can induce protein denaturation of PEs and that finally, it can reduce the effect of PE during PDSCs culture. Moreover, we have concluded that various cytokines in placental extract are sufficient for maintenance of PDSCs with FBS‐free medium, at least from a small amount of PEP‐A (approximately 2.5 μg/ml). Also, PDSCs, which originate from mesodemal lineages, grown in medium with PEP‐A, maintained their proliferative capacity and sustained their ability for hepatogenic differentiation, as well as for osteogenic differentiation (Data S3). However, using Multiplex cytokine assay, we observed that cytokines in placental extract were unstable, even when stored at low temperature (for example, 4 °C, data not shown).
These results suggest that water‐soluble extraction methods described here are a valuable source for the future of PDSC expansion and hepatogenic differentiation, which may aid in progression to clinical‐grade cell therapy. However, more work will be needed for further optimization of concentration of PEP‐A and for characterization of properties of PEP‐A obtained by water‐soluble extraction for use in stem cell therapy. Taken together, use of PEPs is a promising approach that allows circumvention of use of FBS, not only for expansion but also for differentiation of PDSCs for clinical applications.
Supporting information
Data S1. Comparison of cell viability in cultured HTR‐8 SV/neo trophoblast cell line by placental extract proteins.
Data S2. Comparison of cell viability in cultured PDSCs by placental extract proteins.
Data S3. Osteogenic differentiation of cultured PDSCs by placental extract proteins. Mineralization pattern of PDSCs after osteogenic differentiation was determined by the von‐Kossa staining technique. Black arrows indicate strong positive reaction (×100 original magnification).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
We especially thank Dr Charles H. Graham (Queen’s University, Canada) for HTR‐8 SV/neo cell line. This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MEST) (KRF‐2008‐313‐E00247).
Kyung‐Sun Shin and Hyun‐Jung Lee equally contributed to the present work.
References
- 1. Porada CD, Zanjani ED, Almeida‐Porad G (2006) Adult mesenchymal stem cells: a pluripotent population with multiple applications. Curr. Stem Cell Res. Ther. 1, 365–369. [DOI] [PubMed] [Google Scholar]
- 2. Delorme B, Charbord P (2007) Culture and characterization of human bone marrow mesenchymal stem cells. Methods Mol. Med. 140, 67–81. [DOI] [PubMed] [Google Scholar]
- 3. Kassem M (2004) Mesenchymal stem cells: biological characteristics and potential clinical applications. Cloning Stem Cells 6, 369–374. [DOI] [PubMed] [Google Scholar]
- 4. Yen BL, Huang HI, Chien CC, Jui HY, Ko BS, Yao M et al. (2005) Isolation of multipotent cells from human term placenta. Stem Cells 23, 3–9. [DOI] [PubMed] [Google Scholar]
- 5. Parolini O, Alviano F, Bagnara GP, Bilic G, Buhring HJ, Evangelista M et al. (2008) Concise review: isolation and characterization of cells from human term placenta: outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 26, 300–311. [DOI] [PubMed] [Google Scholar]
- 6. Ilancheran S, Moodley Y, Manuelpillai U (2009) Human fetal membranes: a source of stem cells for tissue regeneration and repair? Placenta 30, 2–10. [DOI] [PubMed] [Google Scholar]
- 7. Lepperdinger G, Brunauer R, Jamnig A, Laschober G, Kassem M (2008) Controversial issue: is it safe to employ mesenchymal stem cells in cell‐based therapies? Exp. Gerontol. 43, 1018–1023. [DOI] [PubMed] [Google Scholar]
- 8. Tateishi K, Ando W, Higuchi C, Hart DA, Hashimoto J, Nakata K et al. (2008) Comparison of human serum with fetal bovine serum for expansion and differentiation of human synovial MSC: potential feasibility for clinical applications. Cell Transplant. 17, 549–557. [DOI] [PubMed] [Google Scholar]
- 9. Sotiropoulou PA, Perez SA, Papamichail M (2007) Clinical grade expansion of human bone marrow mesenchymal stem cells. Methods Mol. Biol. 407, 245–263. [DOI] [PubMed] [Google Scholar]
- 10. Chavez SL, Meneses JJ, Nguyen HN, Kim SK, Pera RA (2008) Characterization of six new human embryonic stem cell lines (HSF7, ‐8,‐9, ‐10, ‐12, and ‐13) derived under minimal‐animal component conditions. Stem Cells Dev. 17, 535–546. [DOI] [PubMed] [Google Scholar]
- 11. Miller RK, Faber W, Asai M, D’Gregorio RP, Ng WW, Shah Y et al. (1993) The role of the human placenta in embryonic nutrition. Impact of environmental and social factors. Ann. N Y Acad. Sci. 678, 92–107. [DOI] [PubMed] [Google Scholar]
- 12. Chen CF, Kurachi H, Fujita Y, Terakawa N, Miyake A, Tanizawa O (1988) Changes in epidermal growth factor receptor and its messenger ribonucleic acid levels in human placenta and isolated trophoblast cells during pregnancy. J. Clin. Endocrinol. Metab. 67, 1171–1177. [DOI] [PubMed] [Google Scholar]
- 13. Wolf HK, Zarnegar R, Oliver L, Michalopoulos GK (1991) Hepatocyte growth factor in human placenta and trophoblastic disease. Am. J. Pathol. 138, 1035–1043. [PMC free article] [PubMed] [Google Scholar]
- 14. Filla MS, Zhang CX, Kaul KL (1993) A potential transforming growth factor alpha/epidermal growth factor receptor autocrine circuit in placental cytotrophoblasts. Cell Growth Differ. 4, 387–393. [PubMed] [Google Scholar]
- 15. Liu KX, Kato Y, Kaku T, Sugiyama Y (1998) Human placental extract stimulates liver regeneration in rats. Biol. Pharm. Bull. 21, 44–49. [DOI] [PubMed] [Google Scholar]
- 16. Giavedoni LD (2005) Simultaneous detection of multiple cytokines and chemokines from nonhuman primates using luminex technology. J. Immunol. Methods 301, 89–101. [DOI] [PubMed] [Google Scholar]
- 17. Talens‐Visconti R, Bonora A, Jover R, Mirabet V, Carbonell F, Castell JV et al. (2006) Hepatogenic differentiation of human mesenchymal stem cells from adipose tissue in comparison with bone marrow mesenchymal stem cells. World J. Gastroenterol. 12, 5834–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamada T, Yoshikawa M, Kanda S, Kato Y, Nakajima Y, Ishizaka S et al. (2002) In vitro differentiation of embryonic stem cells into hepatocyte‐like cells identified by cellular uptake of indocyanine green. Stem Cells 20, 146–154. [DOI] [PubMed] [Google Scholar]
- 19. Klimanskaya I, Rosenthal N, Lanza R (2008) Derive and conquer: sourcing and differentiating stem cells for therapeutic applications. Nat. Rev. Drug Discov. 7, 131–142. [DOI] [PubMed] [Google Scholar]
- 20. Doucet C, Ernou I, Zhang Y, Llense JR, Begot L, Holy X et al. (2005) Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell‐based therapy applications. J. Cell. Physiol. 205, 228–236. [DOI] [PubMed] [Google Scholar]
- 21. Schallmoser K, Rohde E, Reinisch A, Bartmann C, Thaler D, Drexler C et al. (2008) Rapid large‐scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng. Part C Methods 14, 185–196. [DOI] [PubMed] [Google Scholar]
- 22. Unger C, Skottman H, Blomberg P, Dilber MS, Hovatta O (2008) Good manufacturing practice and clinical‐grade human embryonic stem cell lines. Hum. Mol. Genet. 17, R48–R53. [DOI] [PubMed] [Google Scholar]
- 23. Genbacev O, Krtolica A, Zdravkovic T, Brunette E, Powell S, Nath A et al. (2005) Serum‐free derivation of human embryonic stem cell lines on human placental fibroblast feeders. Fertil. Steril. 83, 1517–1529. [DOI] [PubMed] [Google Scholar]
- 24. Stute N, Holtz K, Bubenheim M, Lange C, Blake F, Zander AR (2004) Autologous serum for isolation and expansion of human mesenchymal stem cells for clinical use. Exp. Hematol. 32, 1212–1225. [DOI] [PubMed] [Google Scholar]
- 25. Shetty P, Bharucha K, Tanavde V (2007) Human umbilical cord blood serum can replace fetal bovine serum in the culture of mesenchymal stem cells. Cell Biol. Int. 31, 293–298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Comparison of cell viability in cultured HTR‐8 SV/neo trophoblast cell line by placental extract proteins.
Data S2. Comparison of cell viability in cultured PDSCs by placental extract proteins.
Data S3. Osteogenic differentiation of cultured PDSCs by placental extract proteins. Mineralization pattern of PDSCs after osteogenic differentiation was determined by the von‐Kossa staining technique. Black arrows indicate strong positive reaction (×100 original magnification).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item