

Abstract
Objectives
Phosphodiesterase 9 (PDE9) is a major isoform of phosphodiesterase hydrolysing cGMP and plays a key role in proliferation of cells, their differentiation and apoptosis, via intracellular cGMP signalling. The study described here was designed to investigate expression, activity and apoptotic effect of PDE9 on human breast cancer cell lines, MCF‐7 and MDA‐MB‐468.
Materials and methods
Activity and expression of PDE9 were examined using colorimetric cyclic nucleotide phosphodiesterase assay and real‐time RT‐PCR methods respectively; cGMP concentration was also measured. MTT viability test, annexin V‐FITC staining, Hoechst 33258 staining and caspase3 activity assay were used to detect apoptosis.
Results
Treatment of both cell lines with BAY 73‐6691 lead to reduction in PDE9 mRNA expression, PDE9 cGMP‐hydrolytic activity and elevation of the intracellular cGMP response. BAY 73‐6691 significantly reduced cell proliferation in a dose‐ and time‐dependent manner and caused marked increase in apoptosis through caspase3 activation.
Conclusion
Our results revealed that BAY 73‐6691 induced apoptosis in these breast cancer cell lines through the cGMP pathway. These data suggest that BAY 73‐6691 could be utilized as an agent in treatment of breast cancer.
Introduction
Mammalian cyclic nucleotide phosphodiesterases (PDEs) hydrolysis cyclic adenosine 3′,5′‐monophosphate (cAMP) and cyclic guanosine 3′,5′‐monophosphate (cGMP), which are omnipresent, second messengers and responsible for transducing extra‐cellular signaling pathways 1. cGMP is involved in regulation of various physiological functions including neurotransmission 3, platelet aggregation 2 and vascular smooth‐muscle modulation 4. It also plays a key role in cell proliferation, differentiation and apoptosis 5, 6.
Intracellular concentration of cGMP is determined by net balance between synthesis from guanylylcyclases and breakdown by 3′, 5′ cyclic nucleotide phosphodiesterases 7. Eleven different subfamilies of PDE isozymes (PDE1–PDE11) are known. These enzymes are differentiated in terms of substrate selectivity, thus PDEs 1, 2, 3, 10, 11 degrade both cAMP and cGMP, PDEs 4, 7 and 8 degrade merely cAMP, and PDEs 5, 6 and 9 degrade cGMP selectively. PDE9 has the highest affinity for cGMP (Km = 170 nm) 8. All PDE family members share 25–50% amino acid sequence identity within the catalytic domain, suggesting that it should be feasible to identify isoform selective PDE inhibitors 8. More than 20 splice variants of PDE9 have been identified 1 but one variant, PDE9A1, appears to be located within the nucleus 9, suggesting that different variants may have a role in metabolizing cGMP in different intracellular compartments.
Recently, it has been reported that PDE activities are elevated in tumours 10, 11 and overexpression occurs in many carcinomas 12, 13. Phosphodiestrase inhibitors have been used for treatment of non‐malignant condition such as inflammatory diseases 14, and also have been suggested for treatment of different types of cancer such as acute promyelocytic leukaemia 15, and malignant glioma 16.
The compound 1‐(2‐chlorophenyl)‐6‐[(2R)‐3, 3, 3‐trifluoro‐2‐methylpropyl]‐1,5‐dihydro‐4H‐Pyrazolo (3, 4‐d) pyrimidine‐4‐one (BAY 73‐6691, Fig. 1) has been reported to be a novel and selective PDE9 inhibitor 17, which (remarkably) has been shown to improve learning and memory in rodents 18. BAY 73‐6691 only moderately inhibits human PDE1 and human PDE11 with selectivity ratios of 25 and 47 respectively, and also has only very low potency against other human PDEs with selectivity ratios of >>73 17. It could conceivably be used for induction of apoptosis and as a new potential anti‐cancer therapy. In the present study, PDE9 expression and effects of PDE9 inhibitor, BAY 73‐6691 on cell population growth and induction of apoptosis, were investigated in ER positive and ER negative, MCF‐7 and MDA‐MB‐468, human breast cancer cell lines.
Figure 1.
Selective PDE 9 inhibitor BAY 73‐6691.
Materials and methods
Chemical reagents
Culture media (RPMI 1640) and growth supplements [trypsin/EDTA, phosphate‐buffered saline (PBS), penicillin and streptomycin and foetal bovine serum (FBS)] were obtained from Gibco (Rockville, Maryland, USA). cGMP direct immunoassay (ELISA) kit and caspase‐3 colorimetric assay kit were purchased from R&D System CO. (Minneapolis, Minnesota, USA). Annexin V‐FITC apoptosis detection kit was procured from Biovision (San Francisco, California, USA). Bisbenzimide H33258 (Hoechst 33258), 3‐(4,5‐Dimethyl thiazol‐2‐yl)‐2,5 diphenyltetrazolium bromide (MTT), DMSO (dimethyl sulfoxide) and BAY73‐6691(1‐(2‐chlorophenyl)‐6‐[(2R)‐3,3,3‐trifluoro‐2‐methylpropyl]‐1,5‐dihydro‐4H‐pyrazolo[3,4‐d]pyrimidine‐4‐one), selective PDE9 inhibitor were purchased from Sigma‐Aldrich (St Louis, MO, USA). Revert Aid M‐Mulv Reverse Transcriptase was obtained from Fermentas (Burlington, ON, Canada).
Cell culture
Human breast cancer cell lines MCF‐7 and MDA 46‐ 468 were obtained from the National Cell Bank of Iran (NCBI). Cell lines were grown adherently in RPMI‐1640 media supplemented with 10% foetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin at 37 °C in 5% CO2:95% air.
Gene expression assay
For expression profile analysis, total RNA was extracted by Trizol reagent (Sigma‐Aldrich). RNA concentration and purity were carried out using UV spectrophotometry as previously described 19. Total RNA was reverse transcribed using RevertAid‐First‐Strand cDNA Synthesis Kit with random hexamer according to the manufacturer's protocol (Fermentas Cat. No. K1631). Quantitative real‐time RT‐PCR assays of PDE9 cDNA were carried out using gene‐specific double fluorescently labelled TaqMan MGB probe (HS00610023‐mL) in a fast real‐time PCR system (ABI 7500 fast Real Time PCR System; Applied Biosystem, Foster, California, USA) according to the manufacturer's instructions. Identical PCR conditions were performed using 1 μl of cDNA, and relative expression levels of genes were normalized to TaqMan probe GAPDH (HS99999905‐m1). Relative mRNA expression level was determined using the 2−ΔΔCt analysis method.
PDE9 activity assay
After treatment, MCF‐7 and MDA‐MB‐468 cells were harvested for total protein, in lysis buffer [(50 mm Tris–HCl (pH 7–8), 150 mm NaCl, 100 mm EDTA, Triton X‐100 1%, 0.1% (w/v) sodium dodecyl sulphate (SDS) 10 ml 10X and PMSF, with protease and phosphatase inhibitors)]. Total protein was determined using the Bradford method 20. cGMP hydrolytic activity phosphodiesterase 9 was determined using a colorimetric cyclic nucleotide phosphodiesterase assay kit (Biomol Life Science, Exeter, UK) as described previously 21. Briefly, protein was purified using column and P6 DG desalting resin (BML‐KI 100) to remove excess phosphate. The basis for the assay is the cleavage of cGMP by a cyclic nucleotide phosphodiesterase, such as PDE9, followed by the release of free phosphate by a 5' nucleotidase. Phosphate released was assessed using Biomol Green reagent in a modified Malachite Green assay. Optimal time reactions were obtained. To measure PDE9‐specific cGMP hydrolytic activity, each sample per time‐dependent manner was read, with and without PDE9 inhibitor (BAY 73‐6691). Samples were incubated at 30 °C for 30 min and appropriately stopped by addition of the Biomol Green reagent. Results were measured using an automated plate reader Synergy HT (Bio Tek instrument, Winooski, Vermont, USA) at 620 nm and compared to a standard curve drawn with cGMP and 5′‐nucleotidase. Differences between pmol cGMP hydrolysed per mg total protein per minute without and with BAY 73‐6691 PDE9 inhibitor, showed PDE9‐specific cGMP‐hydrolytic activity.
Cyclic GMP assay
Cyclic guanosine 3′, 5′‐monophosphate was quantitatively determined using an immunoassay cGMP kit provided by R&D Pharmacia Biotech, according to the manufacturer's instructions, as described previously 22. In brief, cells were cultured for 24 h. At 80% confluence they were treated with PDE9 inhibitor (BAY 73‐6691) in a time‐dependent fashion. Cells were lysed by cell lysis buffer and mixtures were placed in wells of a 96‐well plate pre‐coated with goat anti‐rabbit antibody. Following washing to remove excess conjugate and unbound sample, substrate solution was added to the wells and incubated for 30 min at room temperature. Then measurement of cGMP was carried out using a microplate reader (Bio‐Rad., Hemel Hempstead, UK) set at 450 nm.
MTT viability assay
Cell viability was determined by MTT assay as described previously 23. Briefly, approximately 5000 cells/well were seeded in 96‐well plates and incubated for 24 h. On reaching 60–80% confluence, used medium was replaced by fresh medium containing appropriate concentrations of specific compound, and was incubated for 24, 48 and 72 h. Then, 20 μl MTT (5 mg/ml in PBS) was added to each well and incubated at 37 °C for 4 h, culture medium was then removed carefully and dye dissolved in 200 μl dimethyl sulphoxide (DMSO). Plates were incubated in the dark for an additional 10 min and absorbance was measured 570 nm wavelength, using a microplate reader (Tekan Sunrise Instruments, Wiesbaden, Austria). All assays were performed in triplicate.
Flow cytometry assay using annexin V/PI staining
Apoptotic cell death induced by BAY 73‐6691 was quantified by flow cytometry using an annexin‐V–FITC kit, according to the manufacturer's protocol. Briefly, cells were plated to 3 × 105/well density in six‐well plates and incubated with or without appropriate concentrations of BAY 73‐6691, for 48 h. Treated and untreated cells were harvested and washed twice in cold PBS. Cell pellets were re‐suspended in 500 μl 1× binding buffer, and 5 μl annexin V‐FITC and 5 μl of PI were added into the cell suspension, followed by gentle vortexing. Staining samples were incubated for 15 min at room temperature in the dark. Samples were analysed using a FACScalibur flow cytometer (BD Biosciences, San Jose, CA, USA) using software supplied with the instrument. This allows discrimination of living cells (unstained with either fluorochrome) from early apoptotic cells (stained with annexin V only) and late apoptotic cells (stained with both annexin V and PI).
Morphological analysis using Hoechst 33258 staining
To confirm cells undergoing apoptosis, Hoechst 33258 staining was performed as described previously 24. Briefly, Cells were grown in eight well chamber slides and allowed to adhere. After treatment with 75 μm of BAY 73‐6691 for 48 h, cells were fixed in 4% paraformaldehyde for 30 min at room temperature then washed twice in PBS. Hoechst 33258 (50 ng/ml) was added to the fixed cells, incubated for 30 min at 37 °C in dark, then washed in PBS. Cells were examined by fluorescence microscopy. Apoptotic cells were identified by their characteristic nuclear condensation, whereas nuclei of non‐apoptotic cells had normal uniform chromatin.
Measurement of caspase‐3 activity
Caspase‐3 activity was measured using Colorimetric Assay Kit (R&D systems Co., Grodig, Germany) according to the manufacturer's instructions, as described previously 19. Briefly, cells were treated with PDE9 inhibitor (BAY 73‐6691) for 24 h, then were washed in PBS and lysed in 50 μl lysis buffer for 10 min; then they were centrifuged (10 min at 10 000 g). Supernatants were incubated in 50 μl reaction buffer and 5 μl of caspase‐3 substrate, at 37 °C for 60 min. Absorbance was measured at 405 nm using a Tekan Sunrise microplate reader.
Statistical analysis
Results were expressed as mean ± SD and statistical analyses were performed by nonparametric analysis of variance between groups (ANOVA), followed by Dunnett's post‐hoc test. All experiments were repeated at least three times. Statistical analyses were performed using software package spss version 17 (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant at P < 0.05.
Results
Quantitative RT‐PCR assay
Differential expressions of PDE9 in both cell lines were investigated by qRT‐PCR using TaqMan probe and PDE9 mRNAs were detected in both MCF‐7 and MDA‐MB‐468 cell lines (Fig. 2a). Effects of PDE9 inhibitor BAY 73‐6691, on expression of the PDE9‐coding gene had been performed in a time‐dependent manner and results indicated that PDE9 expression level gradually reduced in MCF‐7 cells in the presence of PDE9 inhibitor (200 μm), over times of 30, 60, 120, 240 and 480 min compared to that of the untreated group (P < 0.05). Subsequently, a significant increase was observed in mRNA expression level at 720 min (Fig. 2b) (P < 0.05). Similar effects were also observed in MDA‐MB‐468 cells. As shown in Fig. 2b, mRNA expression level of PDE9 decreased significantly at times 30, 60, 120, 240 min (P < 0.05). However, gene expression level increased at 480 min (P < 0.05) in comparison to that of the untreated group.
Figure 2.
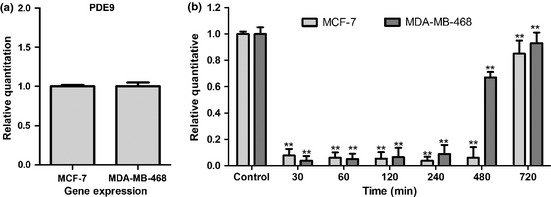
(a) PDE9 mRNA was detected on human breast cancer cell lines, MCF‐7 and MDA‐MB‐468. (b) Quantitative real‐time PCR analysis of PDE9 mRNA expression in MCF‐7 and MDA‐MB‐468 cells following BAY 73‐6691 treatment in a time‐dependent manner. Expression levels were normalized to human GAPDH mRNA. Error bars represent standard deviation.*P < 0.05; **P < 0.01 compared to untreated control groups.
PDE9 activity assay
Treatment of MCF‐7 cells with BAY 73‐6691 significantly decreased PDE9 activity at 30–240 min, but PDE9 activity increased significantly by 12–24 h (P < 0.05). Similarly, BAY 73‐6691 reduced PDE9 activity at 30–480 min in the MDA‐MB‐468 cell line, compared to that of controls (P < 0.05) (Fig. 3), whereas this activity was elevated by 24 h. Thus, PDE9 activity correlated significantly with PDE9 mRNA expression in both cell lines (data not shown).
Figure 3.
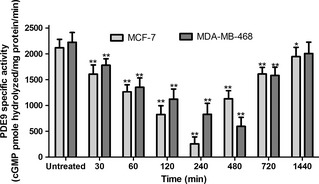
BAY 73‐6691 reduced PDE 9 activity in MCF ‐7 and MDA ‐ MB ‐468 cells. Cells were incubated to concentration of PDE9 inhibitor; BAY 73‐6691 (200 μm) in a time‐dependent manner and total protein was harvested. PDE9‐specific activity was measured as BAY 73‐6691‐inhabitable fraction of total cGMP hydrolysis, normalized to total milligrams of protein. Each point, mean ± SD of three experiments. Each experiment was repeated three times. *P < 0.05; **P < 0.01 compared to untreated control groups.
cGMP assay
Effects of PDE9 inhibition on intracellular levels of cGMP in MCF‐7 and MDA‐MB‐468 cells had been assessed. As shown in Fig. 4, BAY 73‐6691 significantly increased cGMP levels at 480 and 720 min in MCF‐7 and MDA‐MB‐468 respectively, compared to those of controls (P < 0.05). These results demonstrated that inhibition of PDE9 promoted cGMP levels, and suggests that PDE9 may be a major regulator of basal cGMP levels in these breast cancer cells.
Figure 4.
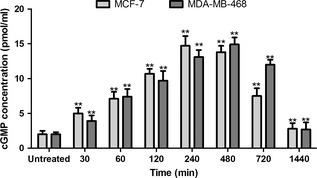
Effect of PDE 9 inhibitor ( BAY 73‐6691) on c GMP levels in MCF ‐7 and MDA ‐ MB ‐468 cells. Cells were treated with 200 μm BAY 73‐6691 over a time course. Measurement of cGMP was performed using a colorimetric competitive ELISA kit. Each point, mean ± SD of three experiments. Each experiment was repeated three times. *P < 0.05; **P < 0.01 compared to untreated control groups.
Anti‐proliferative effect of PDE9 inhibitor, BAY 73‐6691, on human breast cancer cells
Effects of selective PDE9 inhibitor (BAY 73‐6691) on the breast cancer cell lines’ viability was examined. MCF‐7 and MDA‐MB‐468 cells were treated with different concentrations of PDE9 inhibitor (10–200 μm) for 24, 48 and 72 h. BAY 73‐6691 significantly inhibited viability of both MCF‐7 (Fig. 5a) and MDA‐MB‐468 (Fig. 5b) cells in a time‐ and dose‐dependent manner (P < 0.05). The most significant inhibitory effects of BAY‐73‐6691 on MCF‐7 cells were 55.3 ± 5.5% and 46.9 ± 3.5% at 200 μm, after treatment for 48 and 72 h respectively. Similar effects were observed on MDA‐MB‐468 cells; treatment with 200 μm BAY‐73‐6691 caused reduction of 70.3 ± 3.2% and 58.8 ± 5.4% on cell viability after 48 and 72 h respectively.
Figure 5.
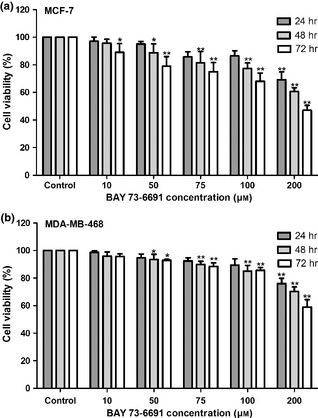
Effect of BAY 73‐6691 PDE 9 inhibitor, in inhibition of cell proliferation of the breast cancer cell lines. Cells were treated with different concentrations of BAY 73‐6691 for 24, 48 and 72 h, and proliferation was measured by MTT assay. BAY 73‐6691 reduced cell proliferation in MCF‐7(5A) and MDA‐MB‐468(5B) breast cells in a time‐ and dose‐dependent manner. Each value is presented as mean ± SD of three experiments (Each triplicate). *P < 0.05, **P < 0.01 are significant compared to untreated control groups.
BAY 73‐6691 induction of apoptosis in breast cancer cell lines
To study whether BAY 73‐6691‐induced cell population growth inhibition was related to apoptosis, effects of BAY 73‐6691 on apoptosis were evaluated. MCF‐7 and MDA‐MB‐ 468 cells were exposed to various concentrations (50–200 μm) of BAY 73‐6691 for 48 h, and analysed by flow cytometry, using annexin‐V and PI double staining. A significant increase was shown in percentage of both early (annexin‐V positive, PI negative) and late (annexin‐V positive, PI positive) apoptosis, in a dose‐dependent manner (P < 0.01) (Fig. 6). To confirm that BAY 73‐6691 had induced apoptosis in the cells, an additional apoptotic marker, Hoechst 33258 staining, was evaluated. As shown in Fig. 7, treatment with 75 μm of BAY 73‐6691 for 48 h caused increased changes in nuclear morphology such as chromatin condensation and fragmentation, in both cell lines.
Figure 6.
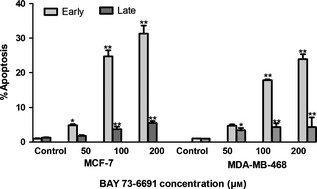
Flow cytometric evaluation of apoptosis in MCF ‐7 and MDA ‐ MB ‐468 cells, using annexin‐V and propidium iodide ( PI ) staining. After 48 h treatment, PDE9 inhibition resulted in a significant increase of early apoptotic cells and of late apoptotic cells, in a concentration‐dependent manner. Results, mean ± SD three independent experiments. *P < 0.05; **P < 0.01 compared to control.
Figure 7.
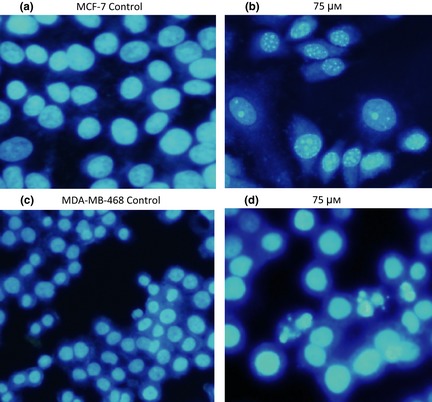
Detection of typical features of apoptotic nuclear condensation by Hoechst 33258 staining (after 48 h treatment). MCF‐7 control (a), MCF‐7 treated with 75 μm of BAY73‐6691 (7b), MDA‐MB‐468 control (7c), MDA‐MB‐468 treated with 75 μm of BAY 73‐6691 (d).
Caspase‐3 activation assay
To examine any contribution of caspases in PDE9 inhibitor ‐induced apoptosis, the role of caspase‐3 was investigated. Treatment of MDA‐MB‐468 cells with various concentrations of BAY 73‐6691 induced significant increases in activity of caspase‐3, in a dose‐dependent manner (Fig. 8) (P < 0.05). Unlike MDA‐MB‐468 cells, activity of caspase‐3 remained unchanged in MCF‐7 cells after treatment with BAY 73‐6691.
Figure 8.
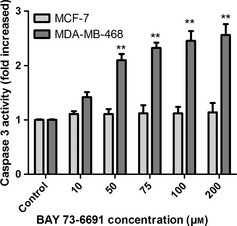
Colorimetric assay of caspase ‐3 activty after treatment with BAY73‐6691 (10‐200/xB5M). Activity of caspase‐3 increased in a concentration‐dependent manner in MDA‐MB‐468, but in MCF‐7, activity of caspase‐3 remained unchanged. *P < 0.05; **P < 0.01 significance compared to control.
Discussion
In recent studies, it has been reported that inhibition of PDE reduced cancer cell proliferation and induced apoptosis, through the cGMP pathway. Thus, selective PDE inhibitors have been considered for treatment of various human diseases such as cancer 11. BAY 73‐6691, as a selective PDE9 inhibitor, has been implicated in suppression of tumour growth both in vitro and in vivo and it has been used in clinical trials for treatment of Alzheimer's disease 17, 25. In a previous study by our group, it was demonstrated that activation of PKG by cGMP induced cell population growth inhibition and apoptosis in human breast cancer cell lines, MCF‐7 and MDA‐MB‐468 22. Biological roles of cGMP‐specific phosphodiesterase have been poorly investigated in human breast cancer cells yet in a preliminary study, Tinsley et al. showed that Sulindac sulphide (a PDE5 inhibitor), inhibited proliferation of SK‐BR‐3 and MDA‐MB‐231 breast cancer cell lines 13. However, the molecular mechanisms and apoptotic pathway of cGMP‐specific phosphodiesterase9, PDE9, have not been elucidated in breast cancer cell lines. Thus, the effect of BAY 73‐6691 on expression level and activity of PDE9, as well as the cGMP signalling pathway, has been studied in the both ER positive and ER negative human breast cancer cell lines, MCF‐7 and MDA‐MB‐46 respectively. Cell population growth inhibitory effect of BAY 73‐6691 and possible mechanisms of activity have been investigated.
cGMP‐specific PDE9 mRNAs were expressed in both cell lines and PDE9 expression level was found to gradually decrease, in a time‐dependent manner, by BAY 73‐6691 (Fig. 2b). There was significant difference between MDA‐MB‐468 and MCF‐7 cells in mRNA expression of PDE9 at 720 and 480 min after inhibitor administration, as well (Fig. 2b). Elevation was maximum after 720 min in both cell lines, although increase in PDE9 mRNA expression in MDA‐MB‐468 (ER negative breast cancer cell line) occurred earlier than in MCF‐7s (ER positive breast cancer cell line).
To verify activity of PDE9 and regulation of second messenger cGMP in human breast cancer cells, we investigated the ability of BAY 73‐6691 to modulate PDE9 activity and the cGMP signalling pathway. Our data indicated that BAY 73‐6691 significantly reduced PDE9 activity, and increased levels of intracellular cGMP, in the both cell lines. PDE9 mRNA expression significantly correlated with PDE9 activity and intracellular cGMP levels.
These results demonstrate that inhibition of PDE9 promotes cGMP accumulation, and suggest this phosphodiesterase to be a major regulator of basal cGMP levels in breast cancer cells. Recent studies have shown that inhibition of phosphodiesterases elevates intracellular cGMP 17, 26. Other studies have indicated that other PDEs are involved in the negative feedback control of cyclic nucleotides pathways 14 and our observations indicate that inhibition of PDE9 by BAY 73‐6691 leads to accumulation of intracellular cGMP and maybe, activates the cGMP signalling pathway.
The results here indicate that PDE9 activity in MCF‐7 cells is higher than in MDA‐MB‐468 by 480 min after using the inhibitor. This difference is probably due to differences in cell type. PDE activities are regulated through a variety of mechanisms including protein expression and negative feedback regulation 14. Negative feedback regulation is one of the most fundamental of biological regulatory processes and, in mammals, can occur at several levels. The most common mechanism for negative feedback control of PDEs is by mass action, that is, elevation of cAMP or cGMP increasing activity of PDE. Most studies have been focused on negative feedback regulation of PDE5 and according to them it would be expected that other PDEs within the PDE superfamily would be involved in negative feedback control of cAMP or cGMP pathways, in addition to their roles by mass action 14.
We have also demonstrated that BAY73‐6691 inhibited proliferation of these breast cancer cells through induction of apoptosis via cGMP signalling. These findings are consistent with those of previous studies using MCF‐7 and MDA‐MB‐468 breast cancer cells 22 and human colon tumour HT29 cells 27, showing that cGMP can play an important role in inhibition of cell proliferation and induction of apoptosis. Moreover, the results have shown that anti‐proliferative and apoptotic effects of BAY73‐6691 on MCF‐7s (ER positive breast cancer cell line) were higher than on MDA‐MB‐468s (ER negative human breast cancer cell line).
In addition, induction of apoptosis by treatment of the cells with various concentrations (50–200 μm) of BAY73‐6691 was observed to externalize phosphatidylserine, to activate caspase3 and to cause morphological changes. Recently, studies have shown that inhibition of selective PDEs induced apoptosis and cell cycle arrest in malignant cells; thus, PDE inhibitors may be used as novel therapeutic agents for tumour cell population growth inhibition and induction of apoptosis 11, 13, 14, 28, 29.
Activation of a caspase cascade is a critical event in induction of apoptosis in mammalian cells 30 and recently, a number of studies has demonstrated that caspase 3 plays an important role in the apoptotic response 22, 31, 32. It has been found that activation of PKG by cGMP induces cell population growth inhibition and apoptosis in breast cancer cell lines 22. We have shown that in MDA‐MB‐468 cells, apoptosis is caspase‐3 dependent. Our findings indicated that treatment of these with various concentrations of BAY 73‐6691 resulted in a significant increase in caspase‐3 activity, while activation of caspase‐3 was not observed in MCF‐7 cells. MCF‐7 cells do not express caspase‐3 protein due to presence of a 47‐bp deletion in exon 3 of the caspase‐3 gene 33.
Conclusion
This study introduces a possible mechanism for control of breast cancer cell population growth through PDE9 and shows that BAY73‐6691 can suppress MCF‐7 and MDA‐MB468 breast cancer cell proliferation, and induces apoptosis via the cGMP signalling pathway. Apoptosis induced by BAY73‐6691 was also determined by characteristic morphological changes and activation of caspase3, which indicated that the caspase pathway also, was involved in the apoptotic signalling. Thus, our results provide valuable new information concerning a novel therapeutic target for breast cancer, using a selective PDE9 inhibitor.
References
- 1. Rentero C, Monfort A, Puigdomenech P (2003) Identification and distribution of different mRNA variants produced by differential splicing in the human phosphodiesterase 9A gene. Biochem. Biophys. Res. Commun. 301, 686–692. [DOI] [PubMed] [Google Scholar]
- 2. Eigenthaler M, Lohmann SM, Walter U, Pilz RB (1999) Signal transduction by cGMP‐Dependent protein kinases and their emerging roles in the regulation of cell adhesion And gene expression. Rev. Physiol. Biochem. Pharmacol. 135, 173–209. [DOI] [PubMed] [Google Scholar]
- 3. Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM et al (1998) Functional analysis of cGMP – dependent protein kinases I and II as media – Tors of NO/cGMP effects. Naunyn‐Schmiedeberge Arch. Pharmacol. 358, 134–139. [DOI] [PubMed] [Google Scholar]
- 4. Vaandrager AB, De Jonge HR (1996) Signalling by cGMP‐dependent protein kinases. Mol. Cell. Biochem. 157, 23–30. [DOI] [PubMed] [Google Scholar]
- 5. Loweth AC, Williams GT, Scarpello JH, Morgan NG (1997) Evidence for the involvement of cGMP and protein kinase G in nitric oxide‐induced apoptosis in the pancreatic B‐cell line, HIT‐T15. FEBS Lett. 400, 285–288. [DOI] [PubMed] [Google Scholar]
- 6. Shimojo T, Hiroe M, Ishiyama S, Ito H, Nishikawa T, Marumo F (1999) Nitric oxide induces apoptotic death of cardiomyocytes via a cyclic‐GMP‐dependent pathway. Exp. Cell Res. 247, 38–47. [DOI] [PubMed] [Google Scholar]
- 7. Diederen RM, La Heij EC, Markerink‐van Ittersum M, Kijlstra A, Hendrikse F, de Vente J (2007) Selective blockade of phosphodiesterase types 2, 5 and 9 results in cyclic 3′5′ guanosine monophosphate accumulation in retinal pigment epithelium cells. Br. J. Ophthalmol. 91, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hutson PH, Finger EN, Magliaro BC, Smith SM, Converso A, Sanderson PE et al (2011) The selective phosphodiesterase 9 (PDE9) inhibitor PF‐04447943 (6‐((3S,4S)‐4‐methyl‐1‐(pyrimidin‐2‐ylmethyl)pyrrolidin‐3‐yl]‐1‐(tetrahydr o‐2H‐pyran‐4‐yl)‐1,5‐dihydro‐4H‐pyrazolo[3,4‐d]pyrimidin‐4‐one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology 61, 665–676. [DOI] [PubMed] [Google Scholar]
- 9. Wang P, Wu P, Egan RW, Billah MM (2003) Identification and characterization of a new human type 9 cGMP‐specific phosphodiesterase splice variant (PDE9A5). Differential tissue distribution and subcellular localization of PDE9A variants. Gene 314, 15–27. [DOI] [PubMed] [Google Scholar]
- 10. Marko D, Romanakis K, Zankl H, Furstenberger G, Steinbauer B, Eisenbrand G (1998) Induction of apoptosis by an inhibitor of cAMP‐specific PDE in malignant murine carcinoma cells overexpressing PDE activity in comparison to their nonmalignant counterparts. Cell Biochem. Biophys. 28, 75–101. [DOI] [PubMed] [Google Scholar]
- 11. Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F et al (2010) Targeting cancer with phosphodiesterase inhibitors. Expert Opin. Investig. Drugs 19, 117–131. [DOI] [PubMed] [Google Scholar]
- 12. Piazza GA, Thompson WJ, Pamukcu R, Alila HW, Whitehead CM, Liu L et al (2001) Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 61, 3961–3968. [PubMed] [Google Scholar]
- 13. Tinsley HN, Gary BD, Thaiparambil J, Li N, Lu W, Li Y et al (2010) Colon tumor cell growth‐inhibitory activity of sulindac sulfide and other nonsteroidal anti‐inflammatory drugs is associated with phosphodiesterase 5 inhibition. Cancer Prev. Res. (Phila) 3, 1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Francis SH, Blount MA, Corbin JD (2011) Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev. 91, 651–690. [DOI] [PubMed] [Google Scholar]
- 15. Guillemin MC, Raffoux E, Vitoux D, Kogan S, Soilihi H, Lallemand‐Breitenbach V et al (2002) In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J. Exp. Med. 196, 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen TC, Wadsten P, Su S, Rawlinson N, Hofman FM, Hill CK et al (2002) The type IV phosphodiesterase inhibitor rolipram induces expression of the cell cycle inhibitors p21(Cip1) and p27(Kip1), resulting in growth inhibition, increased differentiation, and subsequent apoptosis of malignant A‐172 glioma cells. Cancer Biol. Ther. 1, 268–276. [DOI] [PubMed] [Google Scholar]
- 17. Wunder F, Tersteegen A, Rebmann A, Erb C, Fahrig T, Hendrix M (2005) Characterization of the first potent and selective PDE9 inhibitor using a cGMP reporter cell line. Mol. Pharmacol. 68, 1775–1781. [DOI] [PubMed] [Google Scholar]
- 18. van der Staay FJ, Rutten K, Barfacker L, Devry J, Erb C, Heckroth H et al (2008) The novel selective PDE9 inhibitor BAY 73‐6691 improves learning and memory in rodents. Neuropharmacology 55, 908–918. [DOI] [PubMed] [Google Scholar]
- 19. Aghaei M, Panjehpour M, Karami‐Tehrani F, Salami S (2011) Molecular mechanisms of A3 Adenosine receptor‐induced G1 cell cycle arrest and Apoptosis in androgen‐dependent and independent prostate cancer cell line: involvement of intrinsic pathway. J. Cancer Res. Clin. Oncol. 137, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 21. Farrow KN, Groh BS, Schumacker PT, Lakshminrusimha S, Czech L, Gugino SF et al (2008) Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ. Res. 102, 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fallahian F, Karami‐Tehrani F, Salami S, Aghaei M (2011) Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor‐positive and ‐negative breast cancer cell lines. FEBS J. 278, 3360–3369. [DOI] [PubMed] [Google Scholar]
- 23. Hashemi M, Karami‐Tehrani F, Ghavami S, Maddika S, Los M (2005) Adenosine and deoxyadenosine induces apoptosis in oestrogen receptor‐positive and ‐negative human breast cancer cells via the intrinsic pathway. Cell Prolif. 38, 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghavami S, Kerkhoff C, Los M, Hashemi M, Sorg C, Karami‐Tehrani F (2004) Mechanism of apoptosis induced by S100A8/A9 in colon cancer cell lines: the role of ROS and the effect of metal ions. J. Leukoc. Biol. 76, 169–175. [DOI] [PubMed] [Google Scholar]
- 25. Hendrix M (2005) Selective inhibitors of cGMP phosphodiestrases as procognitive agents. BMC Pharmacol. 5, S5. [Google Scholar]
- 26. Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J (2009) Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology 202, 419–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu B, Vemavara L, Thompson WJ, Strada SJ (2005) Suppression of cyclic GMP‐specific phosphodiestrase 5 promotes apoptosis and inhibits growth in HT 29 cell. J. Cell. Biochem. 94, 336–350. [DOI] [PubMed] [Google Scholar]
- 28. Hirsh L, Dantes A, Suh BS, Yoshida Y, Hosokawa K, Tajima K et al (2004) Phosphodiesterase inhibitors as anti‐cancer drugs. Biochem. Pharmacol. 68, 981–988. [DOI] [PubMed] [Google Scholar]
- 29. Marko D, Pahlke G, Merz KH, Eisenbrand G (2000) Cyclic 3′,5′‐nucleotide phosphodiesterases: potential targets for anticancer therapy. Chem. Res. Toxicol. 13, 944–948. [DOI] [PubMed] [Google Scholar]
- 30. Fraser M, Chan SL, Chan SS, Fiscus RR, Tsang BK (2006) Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene 25, 2203–2212. [DOI] [PubMed] [Google Scholar]
- 31. Tai CJ, Chang SJ, Chien LY, Leung PC, Tzeng CR (2005) Adenosine triphosphate induces activation of caspase‐3 in apoptosis of human granulosa‐luteal cells. Endocr. J. 52, 327–335. [DOI] [PubMed] [Google Scholar]
- 32. Zhang YX, Yu SB, Ou‐Yang JP, Xia D, Wang M, Li JR (2005) Effect of protein kinase C alpha, caspase‐3, and survivin on apoptosis of oral cancer cells induced by staurosporine. Acta Pharmacol. Sin. 26, 1365–1372. [DOI] [PubMed] [Google Scholar]
- 33. Janicke RU, Sprengart ML, Wati MR, Porter AG (1988) Caspase‐3 required for DNA fragmentation, and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357–9360. [DOI] [PubMed] [Google Scholar]