

Abstract
Background: Mesenchymal stem cells are able to undergo adipogenic differentiation and present a possible alternative cell source for regeneration and replacement of adipose tissue. The human infrapatellar fat pad is a promising source of mesenchymal stem cells with many source advantages over from bone marrow. It is important to determine whether a potential mesenchymal stem‐cell exhibits tri‐lineage differentiation potential and is able to maintain its proliferation potential and cell‐surface characterization on expansion in tissue culture. We have previously shown that mesenchymal stem cells derived from the fat pad can undergo chondrogenic and osteogenic differentiation, and we characterized these cells at early passage. In the study described here, proliferation potential and characterization of fat pad‐derived mesenchymal stem cells were assessed at higher passages, and cells were allowed to undergo adipogenic differentiation.
Materials and methods: Infrapatellar fat pad tissue was obtained from six patients undergoing total knee replacement. Cells isolated were expanded to passage 18 and proliferation rates were measured. Passage 10 and 18 cells were characterized for cell‐surface epitopes using a range of markers. Passage 2 cells were allowed to undergo differentiation in adipogenic medium.
Results: The cells maintained their population doubling rates up to passage 18. Cells at passage 10 and passage 18 had cell‐surface epitope expression similar to other mesenchymal stem cells previously described. By staining it was revealed that they highly expressed CD13, CD29, CD44, CD90 and CD105, and did not express CD34 or CD56, they were also negative for LNGFR and STRO1. 3G5 positive cells were noted in cells from both passages. These fat pad‐derived cells had adipogenic differentiation when assessed using gene expression for peroxisome proliferator‐activated receptor γ2 and lipoprotein lipase, and oil red O staining.
Discussion: These results indicate that the cells maintained their proliferation rate, and continued expressing mesenchymal stem‐cell markers and pericyte marker 3G5 at late passages. These results also show that the cells were capable of adipogenic differentiation and thus could be a promising source for regeneration and replacement of adipose tissue in reconstructive surgery.
Introduction
There is ever‐increasing clinical need for regeneration and replacement of adipose tissue, in reconstructive surgery to replace soft tissue lost due to trauma, disease and in congenital anomalies, as well as in cosmetic surgery (1, 2).
Use of synthetic materials for example collagen, to enhance or improve soft tissue contours, invariably results in foreign body reaction (3). A patient’s own tissue is preferable to allografts and xenografts to avoid potential complications, such as pathogen transmission and immune rejection. Autologous free adipose tissue grafts have been used in many clinical settings to restore defects such as in breast reconstruction, but they have inconsistent vascularization, and result in unpredictable shrinkage up to 40–60%, and thus, poor cosmesis (4). These procedures are also associated with donor site morbidity and deformity (5), and frequently additional procedures might be necessary for tissue harvest. Adipose cell suspensions obtained through liposuction result in significant mechanical trauma to adipocytes (6), mature adipocytes obtained do not have potential to proliferate and have poor ability for volume retention when used for tissue reconstruction (1). Cell‐based repair strategies using preadipocytes have been explored, but these cells are difficult to isolate, show unpredictable variability based on age and anatomical site of origin, and exhibit limited proliferation (7, 8, 9).
A potential alternative to current treatment modalities is use of tissue engineering applications with mesenchymal stem cells (MSCs) that have been identified in many tissues, including bone marrow, adipose tissue and the pad (10, 11, 12, 13). These cells have excellent proliferative potential and have been shown to undergo adipogenic differentiation under defined culture conditions (14). A tissue engineered graft of a patient’s own MSCs would overcome shortcomings of current treatment modalities.
Adipose tissue engineering approaches have been described using bone marrow‐derived MSCs (9, 15), but initial harvest of bone marrow can be painful and has associated morbidities including risk of wound infection and sepsis (14). In addition, these cells form only 0.001–0.01% total nucleate cells in bone marrow aspirates (16). With a person’s increasing age, there is reduction in bone marrow cellularity including of MSCs (17, 18). Some studies have also shown age‐related decline in osteogenic potential of these cells (19, 20). The fat pad, however, is easily accessible with less discomfort to the patient and has a greater yield of MSCs than bone marrow (21). Fat pad‐derived MSCs also do not seem to have any age‐related decline in proliferative and differentiation potential (22). Although a biomechanical cadaveric study (23) indicated that the infrapatellar fat pad may have a biomechanical function, and may play a role in anterior knee pain syndrome, there was no clinical evidence that resecting the fat pad would cause problems. The infrapatellar fat pad is commonly resected at arthroscopy for improved surgical visualization, and in arthroplasty to prevent possible impingement of fat by the prosthesis. No adverse long‐term side effects have been noted following resection of the infrapatellar fat pad (24).
It is important to determine whether a potential mesenchymal stem‐cell source exhibits tri‐lineage differentiation potential, and is able to maintain its proliferation potential and cell‐surface characterization on expansion in culture. We have previously shown that MSCs derived from the fat pad can undergo chondrogenic and osteogenic differentiation, and we characterized these cells at early passage (22, 25). In this study, proliferation potential and characterization for fat pad‐derived MSCs were assessed at higher passages, and cells were allowed to undergo adipogenic differentiation in appropriate culture medium.
Materials and methods
Infrapatellar fat pad tissue was obtained from six patients undergoing total knee replacement for osteoarthritis, following ethical committee approval and written informed consent. Five millilitres of fat pad weighting between 4 and 5 g was dissected from each knee and cells were isolated by digestion with 0.2% (vol/vol) collagenase I (Invitrogen, Paisley, UK) for 3 h at 37 °C with constant agitation. Released cells were sieved (70‐μm mesh) and washed in basic medium consisting of Dulbecco’s modified Eagle’s medium supplemented with 20% (vol/vol) foetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Cambrex, Wokingham, UK), with l‐glutamine (2 mm). Stromal cells were separated from floating adipocytes by centrifugation at 300 g for 5 min, and were counted and plated at 100 000 cells/cm2 in monolayer culture in basic medium. Five millilitres of tissue yielded around 7.5 million mononuclear cells. Cultures were maintained at 37 °C with 5% CO2 and normal oxygen (20%).
Calculation of cell population doublings per day and proliferation rates
Growth kinetics of cell populations for different passages were calculated using an equation from a previously described method (26):
Population doublings per day = (ln (N/N o))/t
Where N is total number of cells at the end of the time period, N o is total number of cells at the beginning of the time period; t is time period in days. Cell number was determined by counting using a cytometer. Viability of cells was determined by staining with trypan blue, then assessing cells able to exclude the dye.
Cell‐surface epitope characterization and flow cytometry
Confluent passage 10 and passage 18 cells were stained using a panel of antibodies for cell‐surface epitopes previously used for passage 2 cells (27). This included antibodies to CD13 (aminopeptidase N), CD44 (hyaluronan receptor), CD90 (Thy‐1), LNGFR (low affinity nerve growth factor receptor), STRO‐1 (marker for bone marrow‐derived stem cell) and CD56 (neural cell adhesion molecule, NCAM) from BD Biosciences, Oxford, UK; CD29 (β1 integrin), CD105 (SH2 or endoglin) and CD34 (marker for haematopoetic cells) from Dako, Ely, UK; and 3G5 (marker for vascular pericytes) courtesy of Dr Ann Canfield, University of Manchester, UK. Cells were incubated for 1 hour with mouse primary antibodies (neat 3G5 and 1:100 dilution for others) followed by FITC‐conjugated anti‐mouse IgM secondary antibody (1:40 dilution) (Dako). For controls, nonspecific monoclonal mouse IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was substituted for the primary antibody. Cells were incubated in 1:100, 4′,6‐diamidino‐2‐phenylindole stain for 5 min and images were captured using an Axioplan 2 microscope with Axiocam HRc camera and AxioVision 4.3 software (all from Carl Zeiss Ltd, Welwyn Garden City, UK).
Cells were also analysed using flow cytometry. Cells in monolayer were detached using trypsin (0.05% with 5 mm EDTA), washed and incubated in mouse primary antibodies (neat 3G5 and 1:100 dilution for others) followed by FITC‐conjugated anti‐mouse IgM secondary antibody (1:40 dilution). They were then re‐washed, suspended at 1 million cells/ml and assayed in a flow cytometer (Dako cytomation cyan).
Adipogenic differentiation culture
For adipogenic differentiation, passage 2 cells were seeded at 5000 cells/cm2 density in six‐well plates in basic medium. Medium was changed every 3 days until the cells were confluent. Cells were then cultured in adipogenic inducing medium consisting of basic medium with 10 μg/ml insulin, 1 μm dexamethasone, 100 μm indomethacin and 500 μm 3‐isobutyl‐1‐methyl xanthine (IBMX) (all from Sigma, Poole, UK), for 72 h. This was followed by adipogenic maintenance medium consisting of basic medium with 10 μg/ml insulin for 24 h. The cycle was repeated four times over 16 days and cells were then cultured in adipogenic maintenance medium for one further week with medium changed once in the week (11, 14, 28). Parallel cell lines were cultured in control basic medium without adipogenic differentiation and maintenance factors.
Gene expression analysis
Three wells were used for extraction of RNA examine gene expression of peroxisome proliferator‐activated receptor γ2 (PPARγ2) gene and that of lipoprotein lipase (LPL). Quantitative real‐time gene expression analysis was performed. Total RNA was extracted using Tri Reagent (Sigma) from passage 2 cells in monolayer; cDNA was generated from 10 to 100 ng total RNA by using reverse transcription followed by poly(A) polymerase chain reaction (PCR) global amplification (29). Globally amplified cDNAs were diluted 1:1000 and 1 μl aliquot of diluted cDNA was amplified by quantitative real‐time PCR in final reaction volume of 25 μl by using MJ Research Opticon with an SYBR Green Core Kit (Eugentec, Seraing, Belgium). Gene‐specific primers were designed within 300 base pairs 3′ region of the relevant gene with use of ABI Primer Express software (Applied Biosystems, Foster City, CA, USA). Gene expression analyses were performed relative to β‐actin and calculated using the 2−ΔΔCt method (30). All primers (Invitrogen) were based on human sequences: β‐actin, 5′‐AAGCCACCCCACTTCTCTCTAA‐3′ (forward) and 5′‐AATGCTATCACCTCCCCTGTGT‐3′ (reverse); PPARγ2, 5′‐TCAGGTTTGGGCGGATGC‐3′ (forward) and 5′‐TCAGCGGGAAGGACTTTATGTATG‐3′ (reverse); LPL, 5′‐GAACCGCTGCAACAATCTGGGCTATGA‐3′ (forward) and 5′‐TGCTGCTTCTTTTGGCTCTGACTTTATTGA‐3′ (reverse).
Oil red O staining
Stock solution of 0.5% (w/v) oil red O (0.7 g in 200 ml of isopropanol) was prepared and filtered through a 0.2‐μm filter. Working solution was prepared by diluting 6 ml stock solution in 4 ml distilled water; working solution was kept for 1 h at room temperature before being filtered once more. Cells of the remaining three wells per six‐well plate, were fixed in 10% solution formaldehyde in Delbecco’s phosphate‐buffered solution (Cambrex, Wokingham, UK) for 1 h at 40 °C, washed in 60% isopropanol, then stained in 1 ml oil red O solution (in 60% isopropanol) for 10 min, followed by five rinses in distilled water. Dye retained by cells was eluted by incubation in 100% isopropanol for 15 min.
Statistical analyses
Experiments were performed separately with cells from six patients and all experiments were in triplicate. Cell expansion and gene expression data are presented as mean and standard deviation. One‐way analysis of variance (ANOVA) with Bonferroni’s correction was carried out to compare population doublings over the 18 passages. Student’s paired t‐test was used to analyse results from gene expression studies and to determine levels of significance. Statistical analyses were conducted with spss statistical software (version 11.5) (SPSS Inc., Chicago, IL, USA) and significance was set at P‐value <0.05.
Results
No effect of passage number on cell population doublings
Cells from fat pad samples were expanded to passage 18 to determine whether rate of population doublings was affected by progressive passaging. Cells from sequential passages replated at 1:3 dilution of confluence, took 7–10 days to regain confluence level. There was no significant difference between population doubling per day for any of the 18 passages (Fig. 1).
Figure 1.
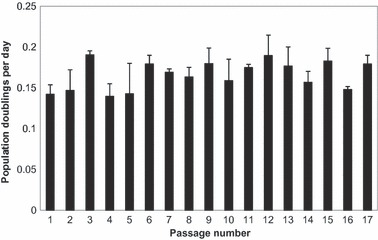
Cell population doublings per day for fat pad‐derived cells from different passages, expanding in monolayer culture, in basic medium. Data are mean ± standard deviation.
Cell‐surface staining and flow cytometry indicate maintenance of cell‐surface epitope profile in passage 10 and passage 18 cells
Cell‐surface epitope profile characterization of passage 10 and 18 fat pad‐derived MSCs was performed using the following panel of antibodies. Cells from both passages stained strongly for CD13, CD44, CD90 and CD105 (markers of MSCs), and for CD29 (marker of β1 integrin) (Fig. 2). Cells stained poorly for LNGFR and STRO1 (markers of bone marrow‐derived MSCs), CD34 (marker of haematopoetic lineage) and CD56 (marker of NCAM). Occasional cells stained positively for 3G5 (pericyte marker). No staining was observed on IgG control cells.
Figure 2.
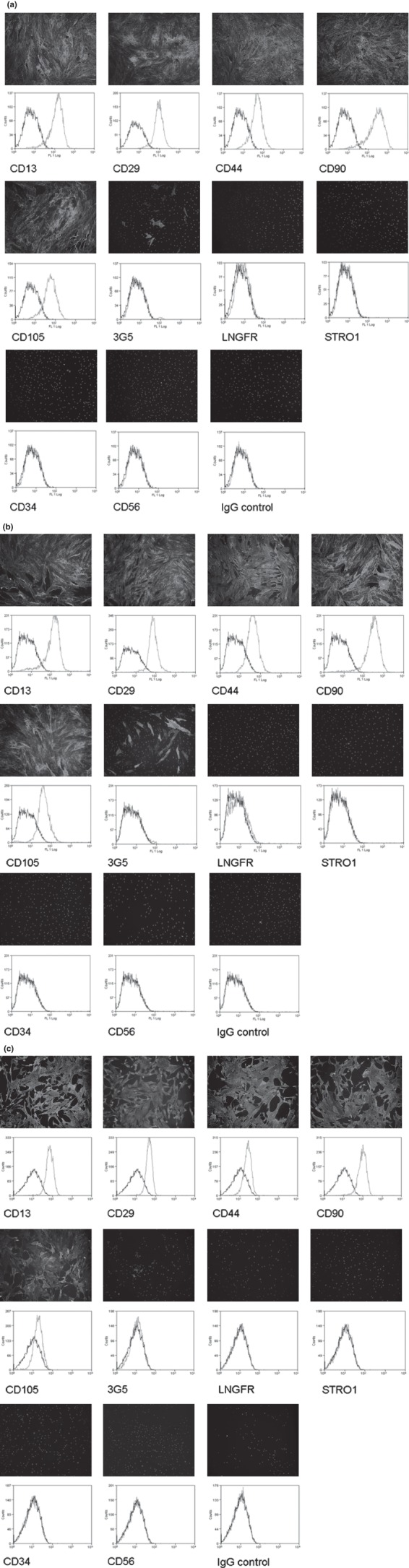
Cell‐surface epitope characterization of passage 2 (a) (21), passage 10 (b) and passage 18 (c) fat pad‐derived MSCs using a panel of antibodies. Cell‐surface staining using FITC conjugated secondary antibody (green) and DAPI (blue) shows that cells stained strongly for CD13, CD29, CD44, CD90 and CD105, and poorly for LNGFR, STRO1, CD34 and CD56. Occasional cells stained for 3G5. No staining was observed for on IgG controls. The staining pattern was confirmed by flow cytometry, and shows increase in fluorescence (green) compared to autofluorescence (black) for CD13, CD29, CD44, CD90 and CD105.
There was similar increase in fluorescence for CD13, CD29, CD44, CD90 and CD105 of cells from both passages, using flow cytometry. There was no detectable increase in fluorescence when cells were labelled with antibodies to 3G5, LNGFR, STRO1, CD34 and CD56. The fluorescence histogram for the sample containing no primary antibody had a little expected difference compared to IgG control cells, due to non‐specific binding. The fluorescence histogram of no primary control cells identified distinct peaks over a wide fluorescence range.
Fat pad‐derived cells cultured in adipogenic medium had increased expression of PPARγ2 and LPL, and increased lipid vacuole accumulation displayed by oil red O staining
Cells cultured under adipogenic conditions had 391‐fold increase in expression of PPARγ2 and more than 2 million‐fold increase in expression of LPL (P < 0.05) (Fig. 3).
Figure 3.
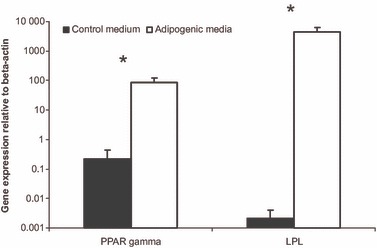
PPARγ2 and LPL relative gene expression levels are significantly greater for fat pad‐derived MSCs cultured in adipogenic medium than for cells cultured in control medium. Data are mean ± standard deviation. *P < 0.05 (Student’s paired t‐test).
Expression of adipocyte‐specific genes during adipocyte differentiation was associated with morphological changes. Vacuoles were observed in confluent fat pad‐derived cells after 3 days in adipogenic culture. Then there was gradual increase in number and size of cells and of vacuoles. By day 16, many cells had vacuoles occupying most of the cytoplasm, but no significant morphological change was observed during final adipogenic maintenance medium culture for 1 week. Oil red O staining was performed to confirm nature of observed vacuoles and confirmed that they were lipid, as shown in Fig. 4. No such vacuoles were observed in cells cultured in control medium and no staining was seen in these cells.
Figure 4.
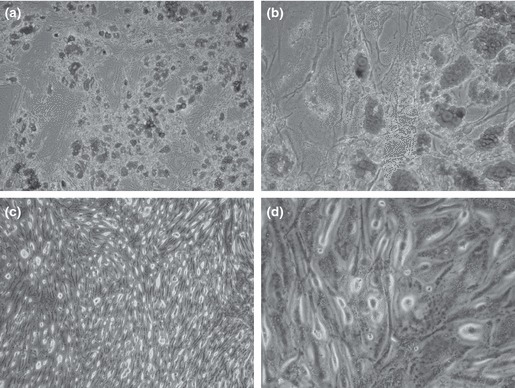
Oil red O staining for fat pad‐derived MSCs cultured in adipogenic medium at ×10 (a) and ×40 (b) magnification show significantly greater staining than cells cultured in control medium at ×10 (c) and ×40 (d) magnification.
Discussion
Ex vivo expanded MSCs may need to undergo a large number of cell divisions in monolayer culture to reach a number sufficient for clinical application. This may lead to cell senescence, and it is important to determine whether characterization is altered by passage, in monolayer culture. Full characterization of fat pad‐derived MSCs at late passage is important to achieve greater understanding of their origin and their repair potential. Cell‐surface epitope profile of synovial tissue‐derived MSCs has been shown to be stable during expansion up to passage 10 (31). Although some previous studies have looked at cell‐surface characterization of fat pad‐derived cells at early passage (12, 22, 25, 32), no previous study has observed them at late passage. To investigate effects of passage on expression of cell‐surface epitopes within the fat pad population, cell‐surface staining and flow cytometry were performed on passage 10 and passage 18 cells, employing a panel of antibodies previously used for passage 2 cells.
Passage 10 and passage 18 cells were consistently expressed CD13, CD29, CD44, CD90 and CD105, and did not express LNGFR, STRO1, CD34 or CD56, suggesting a homogenous population at this stage. Cell‐surface epitope characterization suggested that fat pad cell population had surface expression characteristics of MSCs (14, 33, 34, 35), and these were consistently maintained over passage. Fat pad‐derived MSCs have previously been shown to express CD29, CD90 and CD105 (36). Although we have not found fat pad‐derived MSCs able to express LNGFR at lower passage numbers previously and higher passages in this study, English et al. (37) found LNGFR expression of 31 ± 17% in early passage at ∼11 population doublings. Lack of CD34 expression in fat pad‐derived cells suggests that there was no contamination with haematopoetic cells during their isolation. Anti‐CD56 antibody recognizes neural (38, 39) and myogenic stem cells (40, 41), and its absence in the expression profile of fat pad‐derived cells further confirms lack of contamination by cells of other tissues during isolation and expansion. The consistent pattern of expression demonstrates that adherence to plastic, and selection through proliferation and passaging allowed selection of a homogenous MSC population.
Our gene expression results showed that the fat pad‐derived cells had the potential to undergo adipogenic differentiation when cultured in adipogenic medium. Previous studies on adipogenesis on fat pad‐derived MSCs have either looked only at staining (37) or failed to perform quantitative gene expression studies (32). Ours, thus, is the first one to observe both these parameters. Morphological changes during adipocyte differentiation were associated with expression of adipocyte‐specific genes. PPARγ2 is a central regulator of fat cell differentiation and its activation is a critical step in adipogenesis (41, 42); PPARγ2 is a key transcription factor of adipogensis in vitro and in vivo (43, 44). LPL is involved in lipid transport, storage and metabolism. Both PPARγ2 and LPL are early adipocyte markers (45). Cells cultured under adipogenic conditions had significantly greater expression of PPARγ2 and LPL. Gene expression studies were performed relative to beta‐actin rather than to glycerol‐3‐phosphate dehydrogenase (GAPDH); GAPDH is a key enzyme in biosynthesis of triglycerides and a late marker of adipogenic differentiation, making it unsuitable as a reference gene.
In fat pad‐derived cells cultured under adipogenic differentiation culture conditions, size and number of lipid vacuoles increased with time, suggesting greater degrees of maturation (9). By day 16, most cells had lipid vacuoles occupying most of the cytoplasm, probably the result of undergoing terminal differentiation (46). Oil red O staining revealed triglyceride accumulation within the cells exhibiting typical adipogenic morphology. Cells cultured in control medium failed to express genes suggestive of adipogenic differentiation and showed no staining of lipid vacuoles on oil red O staining.
To induce adipogenic differentiation, cells were cultured in adipogenic medium containing insulin, dexamethasone, indomethacin and IBMX. Insulin, dexamethasone and IBMX have been shown to be sufficient to stimulate adipogenic differentiation (11, 14, 45). Insulin is needed to generate the substrate glycerol 3‐phosphate, which is needed for biosynthesis of triglycerides (47); also, it is known to promote proliferation and differentiation of pre‐adipocytes (7). High concentrations of insulin mimic the role of insulin‐like growth factor (IGF‐1) (48) and have mitogenic effects. The anti‐inflammatory drug indomethacin is a PPARγ2 activator and was also used in our medium as it is a strong inducer of adipogenesis (49).
Adipogenesis of undifferentiated MSCs is made up of two stages: determination of adipocyte lineage, and adipogenic differentiation. Molecular pathways involved in the second stage have been studied extensively (50), but those of the first stage are not well understood (51). Our results show that serum deprivation is not necessary for adipogenic differentiation as has previously been suggested (52, 53), and supporting previous observations (54). Our results support the previous finding that adipogenic differentiation of MSCs is only possible when cells are confluent (55). Our results also support previous finding that MSCs require several cycles of hormonal stimulation to exit from the cell cycle, a feature necessary to achieve commitment to the adipogenic lineage (51).
In a patient‐matched quantitative comparison, MSCs from adipose synovium have been shown to demonstrate higher proliferative potential and colony‐forming efficiency compared to subcutaneous fat‐derived cells, both in mixed‐population and in single‐cell‐derived cultures. In addition, adipose synovium MSCs also have been shown to exhibited greater chondrogenic and osteogenic potential compared to subcutaneous fat MSCs (56). Cell expansion is an important consideration in any potential MSC‐based treatment, and fat pad‐derived cells have previously been shown to have better proliferation potential and tri‐lineage differentiation potential compared to adipose tissue, and in this study, we have shown that they also maintain their proliferation rate and cell‐surface characterization in late passages. Cell‐based adipose tissue repair strategies using fat pad‐derived MSCs will not have disadvantages of exhibiting limited proliferation as shown by preadipocytes. Harvesting MSCs from all tissue sources is associated with potential risks, and benefits of using MSCs from the fat pad need to be balanced against the small potential risk of joint sepsis. A comparative patient‐matched study is planned to determine effects of passage on proliferation and cell‐surface characterization, on MSCs from different sources. The next challenge would be to establish how these cells behave in vivo. To date, there have been no in vivo studies on fat pad‐derived MSC constructs undergoing adipogenesis, and it remains to be seen whether adipogenic potential is maintained in vivo.
Conclusion
Our results show that fat pad‐derived MSCs exhibited tri‐lineage differentiation potential and maintained their proliferation potential while continuing to express MSC markers and pericyte marker 3G5, at late passage. We believe that these cells are a promising source for regeneration and replacement of adipose tissue for reconstructive surgery.
References
- 1. Patrick CW (2001) Tissue engineering strategies for adipose tissue repair. Anat Rec. 263, 361–366. [DOI] [PubMed] [Google Scholar]
- 2. Beahm EK, Walton RL, Partick CW (2003) Progress in adipose tissue construct development. Clin. Plast. Surg. 30, 547–558. [DOI] [PubMed] [Google Scholar]
- 3. von Heimburg D, Zachariah S, Heschel I, Kühling H, Schoof H, Hafemann B et al. (2001) Human preadipocytes seeded on freeze‐dried collagen scaffolds investigated in vitro and in vivo. Biomaterials 22, 429–438. [DOI] [PubMed] [Google Scholar]
- 4. Patrick CW (2000) Adipose tissue engineering: the future of breast and soft tissue reconstruction following tumor resection. Semin. Surg. Oncol. 19, 302–311. [DOI] [PubMed] [Google Scholar]
- 5. Lee KY, Halberstadt CR, Holder WD, Mooney DJ (2000) Breast reconstruction In: Lanza RP, Langer R, Vacanti J, eds. Principles of Tissue Engineering, pp. 409–423, San Diego, CA: Academic. [Google Scholar]
- 6. Patrick CW, Chauvin PB, Robbo GL (1998) Tissue engineered adipose tissue In: Patrick CW, Mikos AG, McIntire LV, eds. Frontiers in Tissue Engineering, pp. 369–382, Oxford: Elsevier Science. [Google Scholar]
- 7. Ailhaud G (1982) Adipose cell differentiation in culture. Mol. Cell. Biochem. 49, 17–31. [DOI] [PubMed] [Google Scholar]
- 8. Sen A, Lea‐Currie YR, Sujkowska D, Franklin DM, Wilkison WO, Halvorsen YD et al. (2001) Adipogenic potential of human adipose derived stromal cells from multiple donors is heterogeneous. J. Cell. Biochem. 81, 312–319. [DOI] [PubMed] [Google Scholar]
- 9. Neubauer M, Hacker M, Bauer‐Kreisel P, Weiser B, Fischbach C, Schulz MB et al. (2005) Adipose tissue engineering based on mesenchymal stem cells and basic fibroblast growth factor in vitro. Tissue Eng. 11, 1840–1851. [DOI] [PubMed] [Google Scholar]
- 10. Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU (1998) In vitro chondrogenesis of bone marrow‐derived mesenchymal progenitor cells. Exp. Cell Res. 238, 265–272. [DOI] [PubMed] [Google Scholar]
- 11. Zuk PA, Zhu M, Mizino H, Huang J, Futrell JW, Katz AJ et al. (2001) Multilineage cells from human adipose tissue: implications for cell‐based therapies. Tissue Eng. 7, 211–228. [DOI] [PubMed] [Google Scholar]
- 12. Dragoo JL, Samimi B, Zhu M, Hame SL, Thomas BJ, Lieberman JR et al. (2003) Tissue‐engineered cartilage and bone using stem cells from human infrapatellar fat pads. J. Bone Joint Surg. 85B, 740–747. [PubMed] [Google Scholar]
- 13. Lee SY, Nakagawa T, Reddi AH (2010) Mesenchymal progenitor cells derived from synovium and infrapatellar fat pad as a source for superficial zone cartilage tissue engineering: analysis of superficial zone protein/lubricin expression. Tissue Eng. Part A 16, 317–325. [DOI] [PubMed] [Google Scholar]
- 14. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147. [DOI] [PubMed] [Google Scholar]
- 15. Choi YS, Park SN, Suh H (2005) Adipose tissue engineering using mesenchymal stem cells attached to injectable PLGA spheres. Biomaterials 26, 5855–5863. [DOI] [PubMed] [Google Scholar]
- 16. Jones EA, Kinsey SE, English A, Jones RA, Straszynski L, Meredith DM et al. (2002) Isolation and characterisation of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 46, 3349–3360. [DOI] [PubMed] [Google Scholar]
- 17. Muschler GF, Nitto H, Boehm CA, Easley KA (2001) Age and gender related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J. Orthop. Res. 19, 117–125. [DOI] [PubMed] [Google Scholar]
- 18. Nishida S, Endo N, Yamagiwa H, Tanizawa T, Takahashi HE (1999) Number of osteoprogenitor cells in human bone marrow markedly decreases after skeletal maturation. J. Bone Miner. Metab. 17, 171–177. [DOI] [PubMed] [Google Scholar]
- 19. Mueller SM, Glowacki J (2001) Age‐related decline in the osteogenic potential of human bone marrow cells cultured in three‐dimensional collagen sponges. J. Cell. Biochem. 82, 583–590. [DOI] [PubMed] [Google Scholar]
- 20. Mendes SC, Tibbe JM, Veenhof M, Bakker K, Both S, Platenburg PP et al. (2002) Bone tissue‐engineered implants using human bone marrow stromal cells: effect of culture conditions and donor age. Tissue Eng. 8, 911–920. [DOI] [PubMed] [Google Scholar]
- 21. Khan WS, Adesida AB, Hardingham TE (2007) Hypoxic conditions increase HIF2a and enhance chondrogenesis in stem cells from the infrapatellar fat pad of osteoarthritic patients. Arthritis Res. Ther. 9(3), R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan WS, Adesida AB, Tew SR, Andrew JG, Hardingham TE (2009) The epitope characterisation and the osteogenic differentiation potential of human fat pad‐derived stem cells is maintained with ageing in later life. Injury 40, 150–157. [DOI] [PubMed] [Google Scholar]
- 23. Bohnsack M, Wilharm A, Hurschler C, Rühmann O, Stukenborg‐Colsman C, Wirth CJ (2004) Biomechanical and kinematic influences of a total infrapatellar fat pad resection on the knee. Am. J. Sports Med. 32, 1873–1880. [DOI] [PubMed] [Google Scholar]
- 24. Duri ZA, Aichroth PM, Dowd G (1996) The fat pad: clinical observations. Am. J. Knee Surg. 9, 55–66. [PubMed] [Google Scholar]
- 25. Khan WS, Tew SR, Adesida AB, Hardingham TE (2008) Human infrapatellar fat pad‐derived stem cells express the pericyte marker 3G5 and show enhanced chondrogenesis after expansion in fibroblast growth factor‐2. Arthritis Res. Ther. 10, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kinner B, Gerstenfeld LC, Einhorn TA, Spector M (2002) Expression of smooth muscle actin in connective tissue cells participating in fracture healing in a murine model. Bone 30, 738–745. [DOI] [PubMed] [Google Scholar]
- 27. Khan WS, Adesida AB, Tew SR, Lowe ET, Hardingham TE (2010) Bone marrow derived mesenchymal stem cells express the pericyte marker 3G5 in culture and show enhanced chondrogenesis in hypoxic conditions. J. Orthop. Res. 28(6), 834–840. [DOI] [PubMed] [Google Scholar]
- 28. Rim JS, Mynatt RL, Gawronska‐Kozak B (2005) Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. FASEB J. 19, 1205–1207. [DOI] [PubMed] [Google Scholar]
- 29. Al Taher A, Bashein A, Nolar T, Hollingsworth M, Brady G (2000) Global cDNA amplification combined with real‐time RT‐PCR: accurate quantification of multiple human potassium channel genes at the single cell level. Yeast 17, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 31. De Bari C, Dell’Accio F, Tylzanowski P, Luyten FP (2001) Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 44, 1928–1942. [DOI] [PubMed] [Google Scholar]
- 32. Wickham MQ, Erickson GR, Gimble JM, Vail TP, Guilak F (2003) Multipotent stromal cells derived from the infrapatellar fat pad of the knee. Clin Orthop. 412, 196–212. [DOI] [PubMed] [Google Scholar]
- 33. Haynesworth SE, Baber MA, Caplan AI (1992) Cell surface antigen on human marrow derived mesenchymal cells are detected by monoclonal antibodies. Bone 13, 69–80. [DOI] [PubMed] [Google Scholar]
- 34. Baddoo M, Hill K, Wilkinson R, Gaupp D, Hughes C, Kopen GC et al. (2003) Characterisation of mesenchymal stem cells isolated from murine bone marrow by negative selection. J. Cell. Biochem. 89, 1235–1249. [DOI] [PubMed] [Google Scholar]
- 35. Shi S, Gronthos S (2003) Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J. Bone Miner. Res. 18, 696–704. [DOI] [PubMed] [Google Scholar]
- 36. Jurgens WJ, Van Dijk A, Doulabi BZ, Niessen FB, Ritt MJ, van Milligen FJ et al. (2009) Freshly isolated stromal cells from the infrapatellar fat pad are suitable for a one‐step surgical procedure to regenerate cartilage tissue. Cytotherapy 11, 1052–1064. [DOI] [PubMed] [Google Scholar]
- 37. English A, Jones EA, Corscadden D, Henshaw K, Chapman T, Emery P et al. (2007) A comparative assessment of cartilage and joint fat pad as a potential source of cells for autologous therapy development in knee osteoarthritis. Rheumatology (Oxford) 46, 1676–1683. [DOI] [PubMed] [Google Scholar]
- 38. Luthi A, Laurent JP, Figurov A, Muller D, Schachner M (1994) Hippocampal long‐term potentiation and neural cell adhesion molecules L1 and NCAM. Nature 372, 777–779. [DOI] [PubMed] [Google Scholar]
- 39. Muller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V et al. (1996) PSA‐NCAM is required for activity‐induced synaptic plasticity. Neuron 17, 413–422. [DOI] [PubMed] [Google Scholar]
- 40. Illa I, Leon‐Monzon M, Dalakas MC (1992) Regenerating and denervated human muscle fibers and satellite cells express neural cell adhesion molecule recognized by monoclonal antibodies to natural killer cells. Ann. Neurol. 31, 46–52. [DOI] [PubMed] [Google Scholar]
- 41. Sinanam ACM, Hunt NP, Lewis MP (2004) Human adult craniofacial muscle‐derived cells: neural‐cell adhesion‐molecule (NCAM; CD56)‐expressing cells appear to contain multipotential stem cells. Biotechnol. Appl. Biochem. 40, 25–34. [DOI] [PubMed] [Google Scholar]
- 42. Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA et al. (1999) Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402, 880–883. [DOI] [PubMed] [Google Scholar]
- 43. Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS et al. (1999) PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617. [DOI] [PubMed] [Google Scholar]
- 44. Spiegelman BM (1998) PPAR‐gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes 47, 507–514. [DOI] [PubMed] [Google Scholar]
- 45. Fink T, Abildtrup L, Fogd K (2004) Induction of adipocyte‐like phenotype in human mesenchymal stem cells by hypoxia. Stem Cells 22, 1346–1355. [DOI] [PubMed] [Google Scholar]
- 46. Zhang L, Baker G, Janus D, Paddon CA, Fuhrer D, Ludgate M. (2006) Biological effects of thyrotropin receptor activation on human orbital preadipocytes. Invest Ophthalmol Vis Sci. 47, 5197–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sottile V, Seuwen K (2001) A high capacity screen for adipogenic differentiation. Anal. Biochem. 293, 124–128. [DOI] [PubMed] [Google Scholar]
- 48. Qiu Z, Wei Y, Chen N, Jiang M, Wu J, Liao K. (2001) DNA synthesis and mitotic clonal expansion is not a required step for 3T3–L1 preadipocyte differentiation into adipocytes. J. Biol. Chem. 276, 11988–11995. [DOI] [PubMed] [Google Scholar]
- 49. Rosen ED, Spiegelman BM (2000) Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 16, 145–171. [DOI] [PubMed] [Google Scholar]
- 50. Gregoire FM (2001) Adipocyte differentiation: from fibroblast to endocrine cell. Exp. Biol. Med. 226, 997–1002. [DOI] [PubMed] [Google Scholar]
- 51. MacDougald OA, Mandrup S (2002) Adipogenesis: forces that tip the scales. Trends Endocrinol. Metab. 13, 5–11. [DOI] [PubMed] [Google Scholar]
- 52. Ramirez‐Zacarias JL, Castro‐Munozledo F, Kuri‐Harcuch W (1992) Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry 97, 493–497. [DOI] [PubMed] [Google Scholar]
- 53. Entenmann G, Hauner H (1996) Relationship between replication and differentiation in cultured human adipocyte precursor cells. Am. J. Physiol. 270, 1011–1016. [DOI] [PubMed] [Google Scholar]
- 54. Janderova L, McNeil M, Murrell AN, Mynatt RL, Smith SR (2003) Human mesenchymal stem cells as an in vitro model for human adipogenesis. Obes. Res. 11, 65–74. [DOI] [PubMed] [Google Scholar]
- 55. Ishino T, Hirakawa K, Takeno S, Furukido K, Sugimoto I, Yajin K (2004) Bone‐constructing cells from ethmoid bone may have multilineage differentiation potential: preliminary report. Acta Otolaryngol. Suppl. 553, 105–108. [DOI] [PubMed] [Google Scholar]
- 56. Mochizuki T, Muneta T, Sakaguchi Y, Nimura A, Yokoyama A, Koga H et al. (2006) Higher chondrogenic potential of fibrous synovium‐ and adipose synovium‐derived cells compared with subcutaneous fat‐derived cells: distinguishing properties of mesenchymal stem cells in humans. Arthritis Rheum. 54, 843–853. [DOI] [PubMed] [Google Scholar]