

Abstract
Abstract. Objectives: The aim of the current study was to investigate whether nicotine treatment would induce the proliferation of isolated rat primary pancreatic acinar cells in culture by activating mitogen‐activated protein kinase (MAPK) signalling and exocrine secretion. Materials and Methods: A nicotine dose‐ and time‐response curve was initially developed to determine the optimal dose and time used for all subsequent studies. Proliferation studies were conducted by cell counting and confirmed further by bromodeoxyuridine (BrdU) incorporation and flow cytometry assays. MAPK signalling studies were conducted by Western blot analysis. Localization of ERK1/2 signals, with or without nicotine and the MAPK inhibitor, was visualized by immunofluorescence. Results: Nicotine treatment caused dose‐dependent activation of extracellular signal‐regulated kinases (ERK1/2), the maxima occurring at 100 µm and at 3 min after treatment; the response was suppressed by the ERK1/2 inhibitor. Maximal nicotine‐induced cell proliferation occurred at 24 h, and UO126‐treatment significantly reduced this response. Exposure of cells to 100 µm nicotine for 6 min significantly enhanced both baseline and cholecystokinin‐stimulated cell function, and these effects were not affected by treatment with the inhibitor of ERK1/2 but were suppressed by mecamylamine, a nicotinic receptor antagonist. Conclusions: Our results suggest that nicotine treatment induced cell proliferation of isolated pancreatic acinar cells and that this is coupled with the activation of MAPK signalling with no effect on its function. Hence, in primary cells, the mechanism of induction and regulation of these two processes, cell proliferation and cell function, by nicotine treatment are independent of each other.
INTRODUCTION
Nicotine has been shown to behave in the manner of a growth factor for survival of human lung cancer cells (Jin et al. 2004). Thus, as various tissues and cell types have been found to be responsive to nicotine by activation of the mitogen‐activated protein kinase (MAPK) signalling pathway (Heusch & Maneckjee 1998; Tang et al. 1998; Nakayama et al. 2001; Nakayama et al. 2002), the effect of nicotine on MAPK activation of isolated pancreatic acinar cells and their cell proliferation remained unexplored. The mitogenic effect of nicotine on AR42J cells, a pancreatic tumour cell line derived from the hyperplastic nodules of male rats (Longnecker et al. 1979), has been reported (Bose et al. 2005). Cells of this line show important similarities to freshly isolated rat, primary pancreatic cells, with respect to their ability to secrete enzymes in response to hormones and cholinergic agonists (Christophe 1994). Furthermore, it has been shown that nicotine treatment can activate extracellular signal‐regulated kinases 1/2 (ERK1/2) in AR42J cells and can induce cell proliferation without affecting baseline levels and stimulated enzyme secretion (Bose et al. 2005). However, it is not known whether primary cells derived from intact rat pancreas would respond to nicotine in a similar manner.
The hypothesis for the current study was that ‘nicotine, at doses described, would induce proliferation of primary pancreatic acinar cells isolated freshly from rats’. This hypothesis was tested on the cells based on the following reported information. Pathophysiological effects of nicotine on the pancreas have been reported (1990, 2002); however, the mechanism by which it promotes this effect remained unknown. Hence, the proliferative potential of nicotine and the confirmation of the observation were sought by screening methods as described. In addition, the mechanism of the proliferative response to nicotine has been investigated further from the standpoint of MAPK activation and cell function. Although the importance of the linkage of nicotine with the development of lung cancer has been widely reported (Jin et al. 2004), its effects on pancreatic cancer development remain controversial. The current study provides important data showing that nicotine behaves in the manner of a growth factor on rat primary pancreatic acinar cells and thus may contribute to development pancreatic cancer.
The effects of nicotine on cell proliferation, its relationships to cell signalling and cell function may involve specific MAPK signalling pathways. We have elected to study the activation of ERK1/2 in this cellular system as it is generally accepted that activation of ERK1/2 is associated with cell proliferative signals, whereas the activations of c‐jun NH2‐terminal kinases 1/2 (JNK1/2) and p38 MAPKs are associated with stress‐response signalling (Xia et al. 1995; Harper & LoGrasso 2001). At present, very little is known about the signalling pathways triggered by nicotine in freshly isolated pancreatic acinar cells, although ERK1/2 activation in AR42J cells (Bose et al. 2005), gastric cancer cells (Shin et al. 2004), human lung cancer cells (Heusch & Maneckjee 1998) and p42/44 MAPK activation in hippocampal CA1 region in rats (Wang et al. 2001) have been reported.
Differential activation of MAPK by cholecystokinin (CCK) and bombesin has been reported in AR42J cells (Kiehne et al. 2002). CCK has also been found to modulate signal transduction and intracellular signalling pathways in isolated pancreatic acinar cells (Williams et al. 1978; Yule & Williams 1994; Williams 1995; Williams 2001). In addition, freshly isolated acinar cells, treated with high doses of nicotine or derived from chronically nicotine‐exposed animals, have been found to significantly reduce enzyme secretion (measured as amylase release as percentage of initial content) when subjected to CCK or cholinergic stimulus, compared to unexposed control cells (Chowdhury et al. 1989; Hosotani et al. 1989; Chowdhury et al. 1990; Chowdhury et al. 1996). The mechanism by which nicotine inhibits enzyme secretion from acinar cells is unknown. However, a pharmacological or toxic response, caused by nicotine treatment, to the function of these cells cannot be ruled out.
In the present study, we report the effects of nicotine on the activation of MAPK signalling in isolated primary pancreatic acinar cells and assess whether a correlation exists between cell proliferation, MAPK signalling and exocrine secretion, induced by nicotine. Our data suggest that nicotine can activate ERK1/2, and this event leads to the increase in proliferation of pancreatic acinar cells. Our data further show that the function of isolated pancreatic acinar cells, in response to nicotine at the studied dose and time, is an independent process from MAPK‐dependent cell proliferation.
MATERIALS AND METHODS
Isolation of pancreatic acinar cells
Adult male Sprague Dawley rats (Charlan Sprague Dawley, Indianapolis, IN, USA) were used for the study. The animals were procured through a protocol approved by the Institutional Animal Care and Use Committee, and were acclimatized for a week under controlled laboratory conditions. After an 18‐h fast, the animals were sacrificed and the pancreata were removed quickly and freed from fat and lymph nodes. The pancreatic acini were isolated by enzymatic digestion according to methods reported previously (Williams et al. 1978; Hosotani et al. 1988; Doi et al. 1995). Briefly, for each sample, Krebs–Henseleit bicarbonate buffer, pH 7.4 (KHB), containing the minimum Eagle's Medium supplement (MEM), 67 U/ml collagenase, 2 mg/ml bovine serum albumin (BSA) and 0.1 mg/ml soybean trypsin inhibitor, was injected into the pancreatic tissue interstitium. Injected pancreatic tissue was incubated at 37 °C in a shaking water bath at a frequency of 120 times/min for 40 min, followed by mechanical disruption of the tissue by gentle suction through pipettes of decreasing orifice size. Acini were then purified by filtration through 150 µm polyethylene mesh and by density gradient centrifugation with KHB containing 4% BSA. Acini were then pre‐incubated for 30 min in N‐2‐hydroxyethylpiperazine‐N′‐2‐ethanesulfonic acid (HEPES)‐buffered Ringer's solution, pH 7.4 (HR). The HR used was the same as KHB, except that it contained 10 mmol/l HEPES and 0.5% BSA. Prior to use, the buffer was gassed with 100% O2. After pre‐incubation, the acini were washed and were resuspended in fresh HR at a density of 0.3–0.4 mg/ml of acinar protein.
Primary cell culture
Purified primary acinar cells were maintained overnight in 100 mm culture dish at concentration of 1.6 × 106/10 ml, in culture media containing Ham's F‐12 nutrient medium (F12K) with 2 mm l‐glutamine, 1% antibiotic, 1.5% sodium bicarbonate and 10% foetal bovine serum albumin (FBS) at 37 °C in a 5% CO2/95% air atmosphere. On day 1, the medium was changed to a serum‐free nutrient medium. Cells were treated with 10–500 µm of nicotine (Sigma, St. Louis, MO) in 6.0 ml of serum‐free medium for periods of 30 s to 10 min. Nicotine was purchased from Sigma and was obtained in liquid form. Nicotine was dissolved initially with a few drops of ethanol and was further diluted to the required concentration with saline solution, pH adjusted 7.4 by sodium hydroxide (1 m). For control samples, medium containing the same amount of ethanol was used for dissolution of nicotine with saline, pH adjusted to 7.4 was added. Specific MAPK inhibitor, UO126 (Cell Signalling Technology, Inc., Beverly, MA) was used in the inhibitor studies. Cells were pre‐treated with UO126 (10 µm) for 30 min before adding nicotine. Cell function studies were performed with or without CCK (Bachem, Philadelphia, PA) at its maximal, previously determined stimulating dose.
Preparation of whole‐cell lysate and Western blot analysis
Procedures used for the preparation of primary cell lysate, followed by Western blot analysis, were similar to those described earlier (Bose et al. 2005).
Immunofluorescence study
Immunofluorescence studies were performed as described earlier by Sharma et al. (2003) and by us (Bose et al. 2005), except that some samples were pre‐treated with UO126, a specific ERK1/2 inhibitor prior to the addition of 100 µm nicotine. In brief, primary cells were treated with 100 µm of nicotine for 3 min or 10 µm UO126, followed by nicotine treatment for 3 min. Cells were fixed with paraformaldehyde and treated with primary antibody to phospho‐ERK1/2 for 1 h. After washing, they were treated with secondary antibody labelled with FITC. Slides were observed by fluorescence microscopy.
Cell proliferation assays
Primary cells in 10% FBS were seeded in 96‐well microplates, and were allowed to attach during overnight incubation; they were then transferred to 0.05% serum prior to the study. Nicotine at a concentration of 100 µm was added, and cell proliferation was measured over 18–40 h, utilizing a commercially available Cell Counting Kit‐8 (Dojindo Molecular Technologies, Gaithersburg, MD), which used a highly sensitive spectrophotometric assay as described by Itano et al. (2002).
Proliferation of primary cells in response to nicotine was also confirmed by 5′‐bromo‐2′‐deoxyuridine (BrdU) incorporation method using an enzyme‐linked immunosorbent assay kit procured from Roche Diagnostics Corp. (Indianapolis, IN). Briefly, primary cells were treated in a similar manner as described previously, in 96‐well microtiter plates. Four hours prior to the end of the interval of measurement, BrdU (10 µm) was added. The cells were fixed, lysed and then treated for 3 h at 25 °C with peroxidase‐conjugated, anti‐BrdU antibody supplied by the manufacturer. The cells were washed three times followed by the addition of substrate solution. Absorbance was measured at 450 nm using a microplate reader.
Cell cycle analysis for DNA content
To confirm the effect of nicotine on cell proliferation, we investigated the cell cycle phase distribution by flow cytometry. Cell cycle analysis was performed using propidium iodide (PI) staining, according to the protocol provided with cell cycle reagent by Guava Technologies (Hayward, CA). Primary cells were seeded in 12‐well culture plates (5 × 104 cells/well) and were incubated overnight in nutrient medium containing 10% FBS. The cells were then changed to 0.05% FBS and were treated with 100 µm of nicotine for 24 h. Cells were trypsinized and collected by centrifugation, washed with PBS, and were re‐suspended in PBS. They were fixed in 70% ethanol (V/V) overnight at −20 °C. After washing with PBS, cells were stained with PI reagent for 30 min in the dark. The cells were analysed for DNA content using Guava EasyCyte mini system (Guava Technologies). The cell cycle profile (percentage of cells within G0 + G1, S and G2 + M phase) was analysed using cytosoft software (Guava Technologies).
Amylase secretion from primary cells after nicotine treatment
These studies were conducted with primary cells that were washed free of media with HR buffer, pH 7.4 (2×), incubated in the same buffer with or without nicotine (50–1000 µm) for periods of 0–10 min and then washed twice with HR buffer to make the cells nicotine‐free. The cells were then dispersed in fresh HR buffer, incubated with or without CCK (10−9 M) for 30 min at 37 °C. The selection of CCK dose (10−9 M) for stimulation was based on our initial study, which showed a maximal stimulated response of amylase release in a CCK dose–response curve (Chowdhury et al. 1989). Following the incubation period, the medium was removed by centrifugation and analysed for amylase activity by the method of (Jung 1980) with Procion yellow starch as substrate (PRO Chemical & Dye, Somerset, MA). Cell pellets were washed with ice‐cold PBS, lysed with water by sonication and were centrifuged. The cell lysate was analysed for both amylase and protein content. Protein concentration was measured by the method of Bradford (1976). Amylase release was expressed as the fractional amount (%) released from total over a wide range of CCK dose.
Amylase secretion by primary cells with ERK inhibitor and nicotine receptor antagonist, mecamylamine
To study the effect of an ERK1/2 inhibitor on baseline and stimulated amylase release, primary cells were pre‐treated with UO126 (10 µm) for 30 min. After washing, the cells were incubated with 100 µm nicotine for 6 min. For studies of primary cells with mecamylamine, a nicotine receptor antagonist, the cells were pre‐treated with 500 µm mecamylamine (Sigma Aldrich, St Louis, MO) for 30 min before the addition of nicotine and were incubated further with 100 µm nicotine for an additional 6 min. At the end of the incubation period, the cells were washed with HR buffer and were incubated with either buffer or a buffer that contains 10−9 M CCK‐8, for 30 min. Amylase release in the media and protein concentration were determined as outlined in the earlier section. The method used for CCK stimulation was also similar to that described in the earlier section.
Statistics
Results were reported as mean ± standard error of the mean (SEM). The results were analysed by student's t‐test and where required, with analysis of variance (anova). Differences were considered significant at P= 0.05 or lower.
RESULTS
Activation of ERK1/2 by nicotine in primary cells
We first evaluated the nicotine‐induced activation of ERK1/2 in primary cells prior to assessing its cell proliferation effect on these cells. Western blotting analysis, employing phospho‐specific antibodies to ERK1/2, was used to accomplish these studies. Incubation of primary cells with varying doses of nicotine from 50 µm to 500 µm for 3 min produced higher phosphorylation of ERK1/2 in comparison to the control (Fig. 1). A maximal induction of up to 3‐fold increase in phosphorylated ERK1/2 occurred with a nicotine dose of 100 µm, in comparison to the control without nicotine (Fig. 1a and c), whereas incubation time needed to achieve this maximum was 3 min (Fig. 1b and d). The total ERK1/2 activity remained unchanged during all of these conditions (Fig. 1a and b). Because the highest response was observed at a nicotine dose of 100 µm and incubation time of 3 min, these conditions were employed for all subsequent ERK1/2 stimulation studies.
Figure 1.
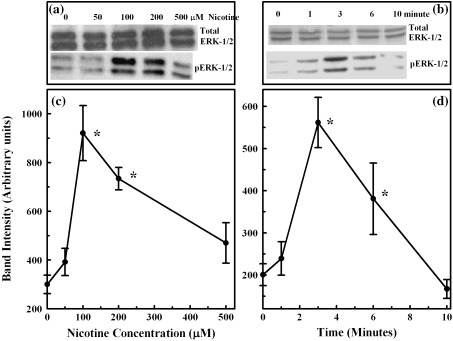
Dose‐ and time‐dependent induction of ERK1/2 in isolated pancreatic acinar cells. Cell lysates were loaded on SDS gel, separated by electrophoresis, blocked by casein and probed with antibodies to total and phospho ERK1/2. Horseradish peroxidase‐coupled anti‐IgG was used as the secondary antibody. Bands were visualized with an enhanced chemiluminescence kit and were quantified with a storm imager. Panels a and b: representative ERK1/2 bands on Western blots. Panels c and d: increased induction (band intensity in arbitrary units) of ERK1/2 as the mean ± SEM of five separate studies.
To evaluate whether the other two members of MAPK proteins, namely JNK1/2 and p38, would follow similar patterns as the ERK1/2, studies were then repeated for the following treatment with nicotine. JNK1/2 and p38 were not significantly activated after nicotine treatment (Data not shown).
ERK1/2 inhibitor studies
To evaluate the mechanism of the effect of nicotine on MAPKs and to determine the role of an upstream ERK1/2 activator, MEK1/2, a specific inhibitor of MEK1/2 activity (UO126) was used (Fig. 2). This agent blocks MEK1/2 phosphorylation and subsequent activation of ERK1/2. The inhibitor significantly blocked the increase in ERK1/2 phosphorylation induced by nicotine (Fig. 2a and c). As before, with the addition of 100 µm of nicotine, phospho‐ERK1/2 activation was significantly increased from 407 ± 29 arbitrary units to 930 ± 74 arbitrary units in 3 min (P < 0.05). Incubation with 10 µm UO126, prior to the addition of nicotine (100 µm) abolished such an increase, the value being 58 ± 13 arbitrary units, which was lower than the control (Fig. 2c). There was no effect of the inhibitor on the amount of total ERK1/2 (Fig. 2a). These studies demonstrated that nicotine activation of ERK1/2 was dependent on MEK1/2 activation.
Figure 2.
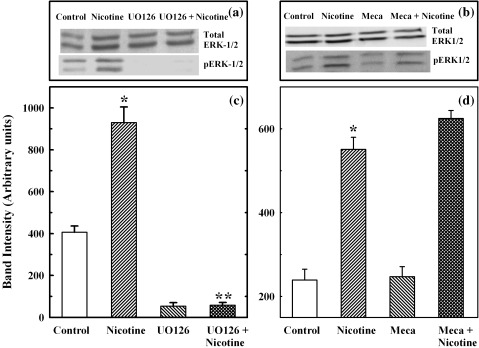
Effect of MEK1/2 inhibitor and mecamylamine on ERK1/2 induction in primary cells. Primary cells were incubated with MEK1/2 inhibitor, UO126 (10 µm) or nicotine receptor antagonist, mecamylamine (500 µm) for 30 min, after which the cells were incubated with 100 µm nicotine for 3 min. Cell extracts were prepared and used for Western blotting of total and phospho ERK1/2, as described in the legends of Figure 1. Panels a and b: representative ERK1/2 bands on Western blots. Panels c and d: induction (band intensity in arbitrary units) of phospho ERK1/2 as the mean ± SEM of five separate studies. *P < 0.05 between control and nicotine‐added samples; **P < 0.05 between nicotine‐added and UO126 + nicotine‐added samples.
MAPK activation by nicotine was not receptor‐mediated
The next study was performed to determine whether activation of ERK1/2 by nicotine was receptor‐mediated. For this study, primary cells had first been pre‐treated with a specific nicotine receptor antagonist, mecamylamine, at a concentration of 500 µm for 30 min before stimulation with 100 µm of nicotine, following which, the cell lysates were tested by Western blotting to determine the activation of phosphorylated ERK1/2. As shown in Fig. 2d, this activation of ERK1/2 by nicotine was similar in the presence or absence of mecamylamine (5.51 ± 0.29 and 6.25 ± 0.19 arbitrary units with the addition of nicotine alone, or added after mecamylamine treatment, respectively). These results indicated that the activation of ERK1/2 after nicotine treatment was not receptor‐mediated.
Proliferation of primary cells by nicotine is mediated by the activation of ERK‐MAPK signal
In order to evaluate the role of nicotine‐induced ERK1/2 activation in the population growth of primary acinar cells, we investigated their proliferation potential after treatment with nicotine by cell counting using the kit‐8 method (Fig. 3a). Cells were maintained in 0.05% serum, and their proliferation was evaluated in response to 100 µm of nicotine as described in the Materials and Methods section. Addition of 100 µm of nicotine caused an increase in cell proliferation at 24–40 h, with significant increases observed at 24 and 32 h, compared with untreated cells (Fig. 3a). Proliferation of cells in both nicotine‐treated and control cells attained a maximum at 24 h, and the proliferation measured in the control wells (by absorption), was 0.61 ± 0.04 arbitrary units, compared with the absorption noted in nicotine‐added wells of 1.34 ± 0.07 arbitrary units (P < 0.01). In both instances, proliferation of cells decreased by 40 h.
Figure 3.
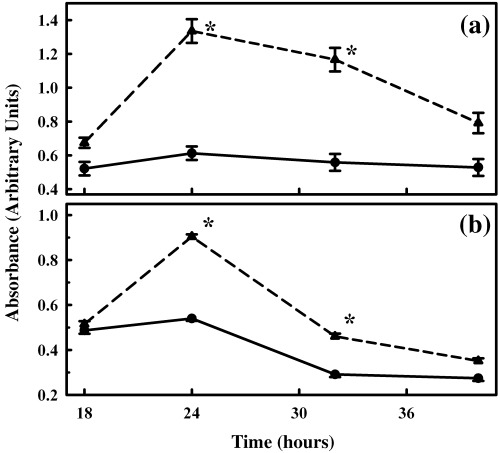
Effect of nicotine on the proliferation of primary cells. Primary cells were seeded in 96‐well microplates, allowed to attach by overnight incubation, and were transferred to 0.05% serum prior to the study. Nicotine, at a concentration of 100 µm, was added, and cell proliferation was measured for 18–40 h, using the cell counting kit‐8 from Dojindo Molecular Technologies (panel a) or BrdU incorporation method (panel b) according to the manufacturer's instructions. Data points represent mean ± SEM of five separate studies for control (circles and solid line) and nicotine added (triangles and dashed line) samples. *P < 0.05.
Proliferation of primary cells observed in the previous discussion was further confirmed by BrdU incorporation method. Maximum incorporation of BrdU also occurred at the 24‐h interval, followed by a decline in subsequent periods (Fig. 3b). At 24 h, incorporation of BrdU by nicotine‐treated primary cells was significantly higher than by control cells, the values being 0.90 ± 0.01 arbitrary units in nicotine‐treated cultures and 0.51 ± 0.01 arbitrary units for control cultures (Fig. 3b, P < 0.01).
To determine the inhibitory effect of the MEK‐ERK1/2 pathway on cell proliferation of primary cells, the specific MEK1/2 pathway inhibitor, UO126, was used. Cells were pre‐treated either with 10 µm of UO126 or with medium alone containing equivalent volume of dimethyl sulfoxide similarly diluted and without the inhibitor, for 30 min. Following this interval, 100 µm nicotine was added to both. Cell proliferation was monitored at 24 h, using the cell‐counting kit‐8 method as well as by BrdU incorporation, as described earlier. In the cell‐counting method, with the addition of UO126 alone to the primary cells, proliferation induced in these cells was 0.76 ± 0.03 arbitrary units at 24 h, which was similar to the value for control samples of 0.61 ± 0.04 arbitrary units at that interval (Table 1). With the addition of 10 µm of UO126 and 100 µm nicotine, proliferation at 24 h was 0.85 ± 0.04 arbitrary units and was not significantly different from the UO126‐alone added sample. However, it was significantly lower than the value of nicotine‐alone added samples, with a value of 1.34 ± 0.07 arbitrary units (P < 0.01). Similar inhibition was also observed by the BrdU incorporation method when UO126 was used prior to the treatment with nicotine, further confirming the data obtained from the cell‐counting method (Table 1). These studies suggested that cell proliferation induced by nicotine was ERK1/2‐dependent.
Table 1.
Proliferation of isolated pancreatic acinar cells in presence of nicotine and ERK1/2 inhibitor, UO126 at 24 h after culture initiation
| Measurement performed | Absorbance (arbitrary units) | |||
|---|---|---|---|---|
| Control | 10 µm UO126 | 100 µm nicotine | UO126 + nicotine | |
| Cell counting kit‐8 | 0.61 ± 0.04 | 0.76 ± 0.03 | 1.34 ± 0.07* | 0.85 ± 0.04** |
| BrdU method | 0.54 ± 0.01 | 0.32 ± 0.01 | 0.90 ± 0.01* | 0.32 ± 0.01** |
These studies were performed together with the observations in Fig. 3a and b. Values are mean ± SEM of five studies. *P < 0.01, between control and nicotine treated groups. **P < 0.01, between nicotine and UO126 + nicotine groups.
Confirmation of nicotine‐induced cell proliferation by flow cytometry studies
To investigate the effect of nicotine and UO126 on cell cycle phase distribution, we analysed the primary cells at 24 h after treatment with nicotine alone or UO126 treatment followed by nicotine treatment using flow cytometry. As shown in Fig. 4a, in control samples the distribution of cells in G0 + G1, S and G2 + M phases were 39.4 ± 1.4%, 52.8 ± 0.8% and 2.0 ± 0.1%, respectively. Incubation with UO126 alone increased the proportion of primary cells in G2 + M phase but cells in the other two phases were not altered significantly. This indicated an accumulation of some cells in G2 + M without any major alteration in the cell cycle. Incubation of primary cells with nicotine significantly decreased the cell number in G0 + G1 phase to 29.7 ± 1.5%, and increased the number of cells in S phase and G2 + M to 60.9 ± 1.6% and 9.4 ± 0.5%, respectively, indicating an increase in cell cycling parameters (P < 0.01). Incubation of primary cells with UO126 and nicotine caused most of them to accumulate in the G2 + M phase and indicated an elongation of the cell cycle when increase in ERK1/2 stimulation by nicotine was prevented by pre‐treatment of primary cells with UO126.
Figure 4.
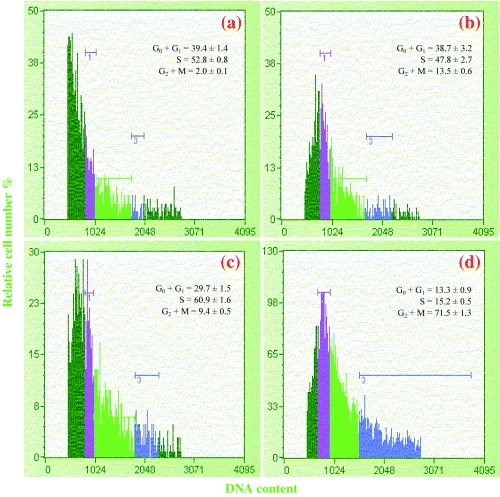
Cell cycle study after nicotine treatment of primary cells. Primary pancreatic cells were treated with nicotine for 24 h and were sorted for their DNA contents using Guava EasyCyte mini flowcytometer and ‘Guava Cell cycle kit’. Panel a represents control cells. Panel b represents cells treated with 10 µm of ERK1/2 inhibitor, UO126. Panel c represents cells treated with 100 µm nicotine alone. Panel d represents cells treated with UO126 and nicotine. In each panel, region 1 (purple) represents cells in G0 + G1 phase, region 2 (green) represents cells in S phase and region 3 (blue) represents cells in G2 + M phase. Cells on the left of region 1 (dark green) represent debris and sub‐G0 cells, whereas cells on the right of region 3 represent aggregates. Percentages in different phases of the cell cycle were calculated, excluding debris and sub‐G0 cells as well as aggregates.
Localization of phospho‐specific ERK1/2 in primary cells
An immunohistochemical technique was used for the localization of phospho‐specific ERK1/2 in primary cells after nicotine treatment. As depicted in Fig. 5, immunofluorescence staining showed a considerably higher quantity of phosphorylated ERK1/2 present in primary cells 3 min after nicotine (100 µm) treatment, as indicated by higher fluorescence intensity, corresponding to phospho‐ERK1/2 in the cytoplasm and nucleus (panel b), compared to the nicotine‐free control cells (panel a). This observation confirmed our result of activation of phospho‐ERK1/2 by nicotine, noted in the Western blots. Pre‐incubation with the ERK inhibitor abolished the higher fluorescence intensity corresponding to phospho‐ERK1/2, following treatment with nicotine (panel c), suggesting that the localization of MAPK signalling in the cell nucleus is ERK‐specific.
Figure 5.
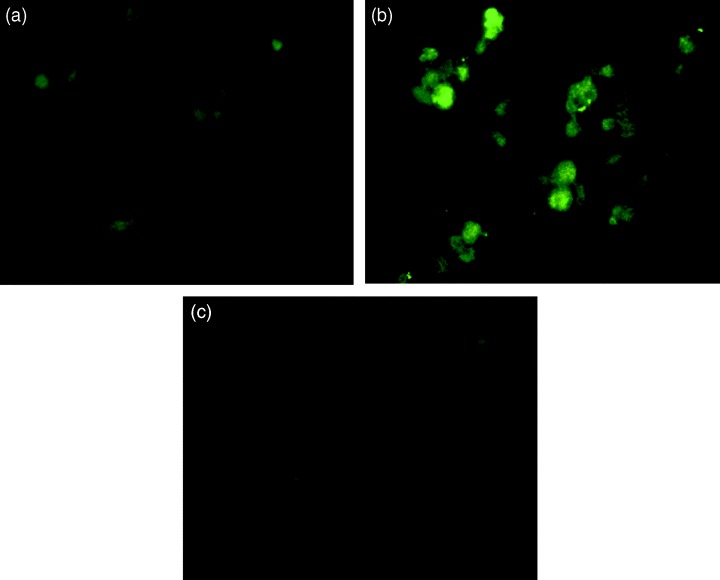
Immunolocalization of phospho‐ERK1/2 in primary cells after nicotine treatment, as indicated by immunohistochemistry. Primary cells were treated with 100 µm of nicotine for 3 min. Cells were fixed with paraformaldehyde and were covered with antibody to phospho ERK1/2 for 1 h. After washing, they were treated with secondary antibody and labelled with FITC. Slides were observed under a fluorescence microscope. Panel a: primary cells untreated control. Panel b: primary cells following treatment with nicotine for 3 min. Panel c: primary cells following ERK1/2 inhibitor and nicotine treatment. The increase in phospho ERK1/2 is clearly seen following treatment with nicotine for 3 min. This is absent in cells treated with the ERK1/2 inhibitor, UO126. No corresponding increase in fluorescence was seen after nicotine treatment when antibody to total ERK1/2 was used.
Effect of nicotine on amylase secretion in primary cells
In the next set of studies, we investigated whether the activation of ERK1/2 was related to amylase secretion. Here we established the optimum dose and time course for nicotine addition to achieve maximal release of amylase from the primary cells with or without CCK. As shown in Fig. 6a, with 100 µm of nicotine alone, amylase secretion increased in primary cells, reaching a maximum at 3 min, with an increase of 1.34‐fold over the amylase secretion noted in untreated cells. There was only a slight decrease in amylase secretion between 3 and 6 min of nicotine exposure, after which secretion decreased. In the presence of CCK, maximum amylase release achieved by nicotine in primary cells was for 6 min of incubation, at a nicotine dose of 100 µm (Fig. 6a). Hence, we selected 6 min as the incubation time for all subsequent secretory studies.
Figure 6.
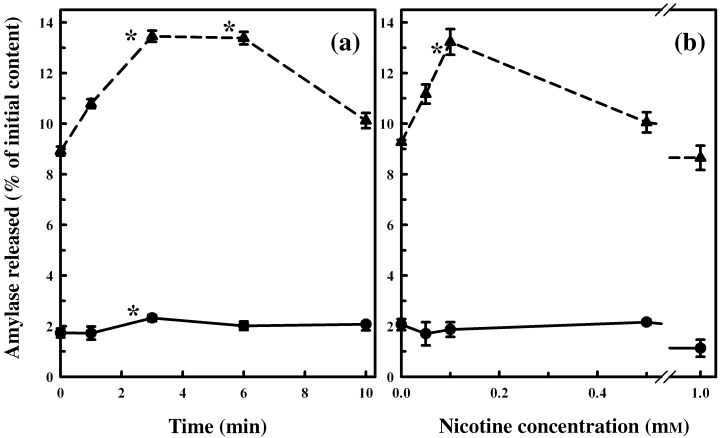
Effect of nicotine treatment on primary cell function. Primary cells were washed, incubated at 37 °C with 100 µm nicotine for 0–10 min (panel a), or with various concentrations of nicotine (panel b), and then were incubated with CCK‐8 (10−9 M, stimulated, triangles with dashed line) or without CCK‐8 (un‐stimulated, circles with solid line) for 30 min. Amylase released into the medium was measured with Procion‐yellow starch as substrate. Data are presented as percentage initial content and represented as the mean ± SEM of four separate experiments. *P < 0.05.
Incubating primary cells with increasing doses of nicotine (50 µm to 1 mm) for 6 min increased amylase secretion, reaching a maximum at 100 µm of nicotine with or without CCK (Fig. 6b), after which the secretion was reduced with the addition of higher doses of nicotine. Thus, we found that the same nicotine dose that produced maximal ERK1/2 also induced the maximal secretory response, as measured by amylase secretion.
Activation of ERK1/2 by nicotine is independent of amylase secretion in primary cells
As mentioned previously, our results indicated that nicotine at a dose of 100 µm induced the activation of ERK1/2‐stimulated and CCK‐stimulated amylase secretion at a maximal rate. To identify whether these two processes were inter‐related, we measured nicotine‐induced amylase secretion in primary cells after incubation with the UO126, an inhibitor of the ERK1/2 pathway. As shown in Fig. 7a, pre‐treatment of cells with 10 µm of UO126 for 30 min had no effect on amylase secretion in the absence or presence of CCK (values for control in the presence of CCK, 9.06 ± 0.33% initial content; with 100 µm nicotine alone in the presence of CCK, 11.50 ± 0.37% initial content; and with UO126 and 100 µm nicotine in the presence of CCK, 11.83 ± 0.30% initial content, respectively). Hence, this study demonstrated that ERK1/2 activation by nicotine is independent of stimulus‐secretion coupling mediating amylase secretion, even though both the MAPK activation and secretory phenomenon were increased in response to the same dose of nicotine.
Figure 7.
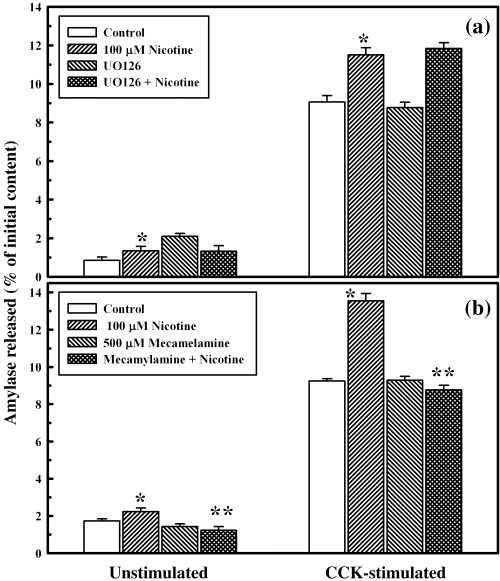
Effect of ERK1/2 inhibitor and mecamylamine on primary cell function, with or without nicotine. Panel a: primary cells (4–6 × 106) were washed and incubated at 37 °C with or without 10 µm UO126 for 30 min, followed by an additional incubation with nicotine for 6 min. The cells were washed and then incubated at 37 °C with or without CCK‐8 (10−9 M) for 30 min. Amylase released into the incubation medium was measured with Procion‐yellow starch as substrate. The data are presented as percentage initial content and represent the mean ± SEM of four experiments. *P < 0.05 between control and nicotine‐added samples. Panel b: primary cells (4–6 × 106) were washed and incubated at 37 °C without or with 500 µm mecamylamine for 30 min, followed by additional 6 min incubation with nicotine. The rest of the procedure is similar to panel a. The data are presented as percentage initial content and represent the mean ± SEM of four experiments. *P < 0.05 between control and nicotine‐added samples; **P < 0.05 between nicotine‐added and mecamylamine + nicotine‐added samples.
Amylase secretion in primary cells by nicotine is receptor‐mediated
To evaluate whether the nicotine‐mediated increase in amylase secretion was via a receptor‐mediated phenomenon, we incubated primary cells with mecamylamine, prior to treatment with nicotine. As shown in Fig. 7b, when cells were incubated with 100 µm of nicotine following incubation with 500 µm of mecamylamine, amylase secretion was significantly lower than for those cells incubated with nicotine alone, both in the presence or absence of CCK (Fig. 7b). These results indicate that the nicotine‐mediated amylase secretion by primary cells was receptor‐mediated.
DISCUSSION
The studies described previously are the first to demonstrate that isolated normal pancreatic primary cells in culture undergo proliferation over a 24‐h period when they are exposed to 100 µm nicotine. Cell proliferation after nicotine treatment has been confirmed by two independent methods as well by cell cycle studies. This response is reversed in the presence of ERK1/2 inhibitor, UO126, suggesting that the proliferation induced in primary cells by nicotine treatment is MAPK‐dependent. The mechanism by which nicotine induced this increase in proliferation of primary cells is mediated via the activation of ERK1/2 signalling pathways, as demonstrated by Western blot analysis using its inhibitor. MAPKs are known to exert several complex functions, such as regulation of cellular growth, proliferation and differentiation (Davis 1993; Marshall 1995; Widman et al. 1999). Activation of these signals and application of its inhibitor in the presence of nicotine do not affect the enhanced enzyme secretion by these cells in response to CCK. Thus, at dose levels studied here, nicotine preserved the physiological response of isolated primary cells as it induced cell proliferation by activating growth signal parameters. Proliferation of primary cells after nicotine treatment thus appears to be independent of stimulus‐secretion coupling response of amylase secretion. At the dose used, nicotine‐induced proliferation of primary cells is MAPK‐pathway specific, particularly by ERK1/2 activation, as a specific inhibitor of the MEK‐ERK pathway, suppressed the proliferation.
The selection of the nicotine dose in the current study was based on published literature in both in‐vivo and other cell culture studies (Majumdar et al. 1986; Chowdhury et al. 1990; Lau et al. 1990; Nakayama et al. 2001; Wang et al. 2004). Dose levels of nicotine used were below peak plasma nicotine concentration in cigarette smokers, which ranges from 10 to 15 mm within 20 min of cigarette smoking (Russel & Fayerabend 1978). In addition, indicated by studies from other laboratories, nicotine doses ranging from 0.75 mm to 25 mm, have been used in isolated rat pancreatic acini (Majumdar et al. 1985). Earlier investigations have shown that nicotine treatment can increase cell numbers in certain cancer cell lines (Cattaneo et al. 1993; Quick et al. 1994; Schuller 1994), suggesting that nicotine exposure can lead to the disruption of the dynamic balance between cell death and cell proliferation, which is required for normal functioning of cells. Also it is well known that pancreatic growth can be affected by gastrointestinal hormones, growth factors and neuropeptides (Charland et al. 2001).
Our study shows that nicotine exposure enhanced cell proliferation of primary cells via a mechanism of early activation of ERK1/2 signalling pathways. However, the sustained increase of cell numbers at 24 h and beyond with nicotine exposure indicates that nicotine might also directly induce the release of autocrine growth factors, as reported in small cell lung cancer proliferation studies (Schuller 1994). Our investigations have indicated that as ERK1/2 is activated by nicotine, the other components of MAPK, JNK1/2 and p38 are not significantly affected. Nakayama et al. (2001) have shown that nicotine treatment at a dose of 100 µm for 5 min induces ERK activation in PC12‐h cells. These data are consistent with our observation in isolated pancreatic acinar cells. It has been further shown that chronic exposure to nicotine via cigarette smoking results in a sustained activation of MAPK (ERK1/2) and in over‐expression of bcl‐2 protein in human lung cancer cells (Heusch & Maneckjee 1998) without affecting the activities of JNK1/2 and p38 MAP kinase. Our results with primary pancreatic acinar cells are consistent with the observation of these cell systems, except that the effect we observed was transient and occurred at a nicotine dose of 100 µm, while in lung cancer cells, these effects occur at a nicotine concentration of 1 µm or less (Heusch & Maneckjee 1998).
In other studies, it has been shown that p42 MAPK is fully activated at 5 min by CCK in freshly isolated pancreatic acini, and JNK is activated maximally at 30 min (and remains significantly elevated at 60 min) (Duan & Williams 1994; Dabrowski et al. 1996) by the same secretagogue. In our study, maximal activation of ERK1/2 by nicotine treatment occurred as early as 3 min after and followed a similar time course to that with CCK treatment, whereas JNK activation was slightly higher than baseline value and occurred between 10 and 30 min (data not shown). On the other hand, p38 MAPK did not show activation with any dose of nicotine in primary cells at any time. These data support the observation that was found in studies of lung cancer cells (Schuller 1994). The results of our studies also demonstrate some parallels in the time‐dependent activation of ERK by CCK and nicotine, although these two secretagogues are completely different from one another in terms of their biological actions on the pancreas (Williams et al. 1978; Yule & Williams 1994; Doi et al. 1995). Furthermore, our results showing activation of ERK1/2, preceding activation of JNK1/2 with nicotine exposure, appear identical to that reported in freshly isolated pancreatic acinar cells in response to CCK (Duan & Williams 1994).
In addition to mitogenesis, we have also focused on the involvement of nicotine in the basal and CCK‐stimulated secretory response in primary cells. CCK can activate MAPK in pancreatic acinar cells in a concentration and time‐dependent manner by a mechanism involving protein kinase C and tyrosine kinase activity (Duan & Williams 1994), via activation of MEK, ras (Duan et al. 1995) and p125 FAK (Kiehne et al. 2002). The dose of CCK that maximally stimulates enzyme secretion in freshly isolated normal pancreatic acini is 10 nm. Although Kiehne et al. (2002) have demonstrated that CCK induces maximal secretion and p42 ERK2 activation in AR42J cells at a dose of 10 nm, other regulatory peptides, such as bombesin, cause only p42 ERK2 activation but not significant enzyme secretion, suggesting that kinase activation and receptor‐mediated signal transduction pathways leading to enzyme secretion might involve separate mechanisms.
In our current study, nicotine treatment maximally increased basal and CCK‐stimulated enzyme secretions at 6 min at the same dose, whereas kinase activation was maximal at 3 min and declined steadily thereafter. Although ERK1/2 activation by nicotine is completely inhibited with specific kinase inhibitor, this inhibitor has no influence on the basal and stimulated secretory responses induced by nicotine. Together, these results suggest that nicotine‐induced proliferation of primary cells is correlated with ERK1/2 activation, which intimates that the ERK1/2 pathway is responsible for the stimulation of primary cell proliferation, as observed in AR42J cells (Bose et al. 2005). Inhibition of ERK1/2 activation by UO126 attenuates nicotine‐induced proliferative response of primary cells, and is further supported by the data. We have also found that this particular inhibitor has no significant effect on the proliferation of control cells in the absence of nicotine (see Table 1).
Stimulation of enzyme secretions by secretagogues in pancreatic acini has been shown to involve several second‐messenger pathways that are rapidly activated by G‐protein‐coupled receptors (Piper et al. 1997; Wang 1997; Wess 1997) and changes in intracellular calcium concentration (Williams 2001). Earlier, we have shown that nicotine at doses of 1–30 mm concentration, either administered in vivo or added in vitro to isolated acini, inhibits CCK‐stimulated enzyme secretion (Chowdhury et al. 1989). The mechanism of this nicotine effect is unclear, although a cell‐mediated intracellular Ca2+ response appears to be involved (Tang et al. 1997; Chowdhury et al. 2002). Additional studies with nicotine doses of lower than 1 mm have been conducted in this cellular system. However, a lower nicotine dose of 100 µm, as used in our current study, has induced a maximal secretory response in primary cells in 3 min, which persisted for 6 min before decreasing. The effects of nicotine on basal level and stimulated secretory response are abolished by mecamylamine, a nicotinic receptor antagonist, suggesting that the secretory response of nicotine is G‐protein‐coupled and receptor‐mediated. ERK1/2 activation by nicotine treatment under similar conditions and in the presence of the nicotine receptor antagonist is unaffected, implying that the kinase and secretory responses induced by nicotine are completely independent of each other and, perhaps, involve a separate mechanism.
It has been reported that in rat sublingual mucous acini, nicotine first triggers the release of acetylcholine from pre‐synaptic nerve terminals, which then activates muscarinic receptors (Zhang & Melvin 1994). In freshly isolated pancreatic acini, we have observed that w‐conotoxin, a potent Q‐type calcium channel blocker, inhibited nicotine‐induced pancreatic secretion completely (Chowdhury P., Udupa K. B., unpublished data), suggesting that the regulation of pancreatic secretion is physiologically regulated by a calcium‐mediated process. The current study has examined the specificity of the nicotinic receptor in primary cells and has clearly demonstrated its effect on downstream events, regulating exocrine secretion.
The importance of linkage of nicotine exposure with lung cancer development has been reported (Jin et al. 2004) but its effects on pancreatic cancer development have remained controversial. Furthermore, studies reporting metabolic, pathological and functional changes of pancreas induced by nicotine (Chowdhury et al. 1990) have been shown to be identical to those reported in patients dying with chronic pancreatitis (Chowdhury & Rayford 2000; Maissonneuve et al. 2005). The current study therefore provides important data showing the ability of nicotine to act like a growth factor for primary pancreatic acinar cells and thus may contribute to the development of pancreatic cancer.
In conclusion, our data demonstrate that at the doses employed, nicotine activates ERK1/2 in isolated primary pancreatic acinar cells. This is associated with cell proliferation, which has been confirmed by three independent assay systems. Activation of kinase signalling does not appear to be correlated with the stimulation of receptor‐mediated secretory responses, suggesting that the mechanisms of these two processes are completely independent.
ACKNOWLEDGEMENTS
The authors thank Mrs Hailing Zhang for her excellent technical skills in performing the work.
Grant Support: Supported, in part by funds from the Central Arkansas Veterans Healthcare System.
REFERENCES
- Bose C, Zhang H, Udupa KB, Chowdhury P (2005) Activation of p‐ERK1/2 by nicotine in pancreatic tumor cell line AR42J: effects on proliferation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G926–G934. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Cattaneo MG, Codignola A, Vicentini LM, Clementi F, Sher E (1993) Nicotine stimulates a serotonergic autocrine loop in human small lung cell carcinoma. Cancer Res. 53, 5566–5568. [PubMed] [Google Scholar]
- Charland S, Boucher M‐J, Houde M, Rivard N (2001) Somatostatin inhibits Akt phosphorylation and cell cycle entry, but not p42/p44 mitogen‐activated protein (MAP) kinase activation in normal and tumoral pancreatic acinar cells. Endocrinology 142, 121–128. [DOI] [PubMed] [Google Scholar]
- Chowdhury P, Rayford PL (2000) Smoking and pancreatic disorders. Eur. J. Gastroenterol. Hepatol. 12, 869–877. [DOI] [PubMed] [Google Scholar]
- Chowdhury P, Hosotani R, Rayford PL (1989) Inhibition of CCK or carbachol‐stimulated amylase release by nicotine. Life Sci. 45, 2163–2168. [DOI] [PubMed] [Google Scholar]
- Chowdhury P, Hosotani R, Chang LW, Rayford PL (1990) Metabolic and pathologic effects of nicotine on the gastrointestinal tract and pancreas of rats. Pancreas 5, 222–229. [DOI] [PubMed] [Google Scholar]
- Chowdhury P, Doi R, Tangoku A, Rayford PL (1995) Structural and functional changes of rat pancreas exposed to nicotine. Int. J. Pancreatol. 18, 257–264. [DOI] [PubMed] [Google Scholar]
- Chowdhury P, MacLeod S, Udupa KB, Rayford PL (2002) Pathophysiological effects of nicotine on the pancreas: an update. Exp. Biol. Med. 227, 445–454. [DOI] [PubMed] [Google Scholar]
- Christophe J (1994) Pancreatic tumoral cell line AR42J: an amphicrine model. Am. J. Physiol. Gastrointest. Liver Physiol. 266, G963–G971. [DOI] [PubMed] [Google Scholar]
- Dabrowski A, Grady T, Logsdon CD, Williams JA (1996) Jun kinases are rapidly activated by cholecystokinin in rat pancreas both in vitro and in vivo . J. Biol. Chem. 271, 5686–5690. [DOI] [PubMed] [Google Scholar]
- Davis R (1993) The mitogen‐activated protein kinase signal transduction pathway. J. Biol. Chem. 268, 14553–14556. [PubMed] [Google Scholar]
- Doi R, Chowdhury P, Nishikawa M, Takaori K, Inoue K, Imamura M, Rayford PL (1995) Carbachol and cholecystokinin enhance accumulation of nicotine in rat acinar cells. Pancreas 10, 154–160. [DOI] [PubMed] [Google Scholar]
- Duan RD, Williams JA (1994) Cholecystokinin rapidly activated mitogen‐activated protein kinase in rat pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 267, G401–G408. [DOI] [PubMed] [Google Scholar]
- Duan RD, Zheng CF, Guan KL, Williams JA (1995) Activation of MAP kinase kinase (MEK) and Ras by cholecystokinin in rat pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 268, G1060–G1065. [DOI] [PubMed] [Google Scholar]
- Harper SJ, Lograsso P (2001) Signaling for survival and death in neurons. The role of stress‐activated kinase, JNK, and p38. Cell Signal. 13, 299–310. [DOI] [PubMed] [Google Scholar]
- Heusch WL, Maneckjee R (1998) Signaling pathways involved in nicotine regulation of apoptosis of human lung cancer cells. Carcinogenesis 19, 551–556. [DOI] [PubMed] [Google Scholar]
- Hosotani R, Chowdhury P, McKay D, Rayford PL (1988) Effect of L‐364718, a new CCK antagonist, on amylase secretion in isolated pancreatic acini. Pancreas 3, 95–98. [DOI] [PubMed] [Google Scholar]
- Hosotani R, Chowdhury P, McKay D, Rayford PL (1989) Mechanism of action of nicotine on amylase release by isolated pancreatic acini. Pharmacol. Biochem. Behav. 3, 663–666. [DOI] [PubMed] [Google Scholar]
- Itano N, Atsumi F, Sawai T, Yamada Y, Miyashi O, Senga T, Hamaguchi M, Kimata K (2002) Abnormal accumulation of hyaluronan matrix diminishes contact inhibition of cell growth and promotes cell migration. Proc. Natl. Acad. Sci. USA 99, 3609–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Gao F, Flagg T, Deng X (2004) Nicotine induces multi‐site phosphorylation of BAD in association with suppression of apoptosis. J. Biol. Chem. 279, 23837–23844. [DOI] [PubMed] [Google Scholar]
- Jung DH (1980) Preparation and application of Procion yellow starch for amylase assay. Clin. Chim. Acta 100, 7–11. [DOI] [PubMed] [Google Scholar]
- Kiehne K, Herzig KH, Folsch UR (2002) Differential activation of p42 ERK and p125 FAK by cholecystokinin and bombesin in the secretion and proliferation of pancreatic amphicrine cell line AR42J. Pancreatology 2, 46–53. [DOI] [PubMed] [Google Scholar]
- Lau PP, Dubick MA, Yu GS, Morrill PR, Geokas MC (1990) Dynamic changes of pancreatic structure and function in rats treated chronically with nicotine. Toxicol. Appl. Pharmacol. 3, 457–465. [DOI] [PubMed] [Google Scholar]
- Longnecker DS, Lilja HS, French J, Kuhlmann E, Noll W (1979) Transplantation of azaserine‐induced carcinomas of pancreas in rats. Cancer Lett. 7, 197–202. [DOI] [PubMed] [Google Scholar]
- Maissonneuve P, Lowenfels AB, Mullhaupt B, Cavallini G, Lankisch PG, Anderson JR, Dimagno EP, Andren‐Sandberg A, Domellof L, Frilloni L, Ammann RW (2005) Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 54, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar APN, Davis GA, Dubick MA, Geokas MC (1985) Nicotine stimulation of protein secretion from isolated rat pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 24, G158–G163. [DOI] [PubMed] [Google Scholar]
- Majumdar APN, Vesenka GW, Dubick MA, Demorrow JM, Geokas MC (1986) Morphological and biochemical changes of the pancreas in rat treated with acetaldehyde. Am. J. Physiol. Gastrointest. Liver Physiol. 250, G598–G606. [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal regulated kinase activation. Cell 180, 179–185. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H (2001) Nicotine‐induced phosphorylation of extracellular signal‐regulated protein kinase and CREB in PC12h cells. J. Neurochem. 9, 489–498. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T (2002) Nicotine‐induced phosphorylation of Akt through epidermal growth factor receptor and Src in PC12h cells. J. Neurochem. 83, 1372–1379. [DOI] [PubMed] [Google Scholar]
- Piper A, Stryjek‐Kaminska D, Klengel R, Zeuzem S (1997) CCK, carbachol and bombesin activate distinct PLC‐isoenzymes via Gq/11 in rat pancreatic acinar membranes. Am. J. Physiol. Gastrointest. Liver Physiol. 272, G135–G140. [DOI] [PubMed] [Google Scholar]
- Quick M, Chan J, Patrick J (1994) Bungarotoxin blocks the nicotine receptor mediated increase in cell number in a neuroendocrine cell line. Brain Res. 655, 161–167. [DOI] [PubMed] [Google Scholar]
- Russel MAH, Fayerabend C (1978) Cigarette smoking. A dependence on high nicotine boli. Drug Metab. Rev. 8, 29–57. [DOI] [PubMed] [Google Scholar]
- Schuller HM (1994) Carbon dioxide potentiates the mitogenic effects of nicotine and its carcinogenic derivative NNK, in normal and neoplastic neuroendocrine lung cells via stimulation of autocrine and protein kinase C‐dependent mitogenic pathways. Neurotoxicology 15, 877–886. [PubMed] [Google Scholar]
- Sharma G, He J, Bazam HE (2003) P38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing. J. Biol. Chem. 278, 21989–21997. [DOI] [PubMed] [Google Scholar]
- Shin YV, Wu WK, Ye YN, So WH, Koo MW, Liu ES, Luo JC, Cho CH (2004) Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal‐regulated kinase and cyclooxygenase‐2. Carcinogenesis 25, 2487–2495. [DOI] [PubMed] [Google Scholar]
- Tang K, Wu H, Mahata SK, Mahata M, Gill BM, Parmer RJ, O’Connor DT (1997) Stimulus coupling to transcription versus secretion in pheochromocytoma cells. Convergent and divergent signal transduction pathways and the crucial roles for route of cytosolic calcium entry and protein kinase C. J. Clin. Invest. 100, 1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Wu H, Mahata SK, O’Connor DT (1998) A crucial role for the mitogen‐activated protein kinase pathway in nicotinic cholinergic signaling to secretory protein transcription in pheochromocytoma cells. Mol. Pharmacol. 54, 59–69. [DOI] [PubMed] [Google Scholar]
- Wang HL (1997) Basic amino acids at the C‐terminus of the third intracellular loop are required for the activation of phospholipase C by cholecystokinin‐B receptors. J. Neurochem. 68, 1728–1735. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen YB, Zhu XN, Chen RZ (2001) Activation of p42/44 mitogen‐activated protein kinase pathway in long‐term potentiation induced by nicotine in hippocampal CA1 region in rats. Acta Pharmacol. 22, 685–690. [PubMed] [Google Scholar]
- Wang XX, Zhu JH, Chen JZ, Shang YP (2004) Effect of nicotine on the number and activity of circulating endothelial progenitor cells. J. Clin. Pharmacol. 44, 881–889. [DOI] [PubMed] [Google Scholar]
- Wess J (1997) G‐protein coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G‐protein recognition. FASEB J. 11, 346–354. [PubMed] [Google Scholar]
- Widman C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen‐activated protein kinase: conservation of a three‐kinase module from yeast to human. Physiol. Rev. 79, 143–180. [DOI] [PubMed] [Google Scholar]
- Williams JA (1995) Signal transduction and intracellular signaling in pancreatic acinar cells. Curr. Opin. Gastroenterol. 11, 397–401. [DOI] [PubMed] [Google Scholar]
- Williams J (2001) Intracellular signaling mechanisms activated by cholecystokinin‐regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu. Rev. Physiol. 63, 77–97. [DOI] [PubMed] [Google Scholar]
- Williams JA, Korc M, Dormer RL (1978) Action of secretagogues on a new preparation of functionally intact isolated pancreatic acini. Am. J. Physiol. 235, E517–E524. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK‐p38 MAP kinase on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yule DI, Williams JA (1994) Stimulus‐secretion coupling in the pancreatic acinus In: Johnson LR, eds. Physiology of the Gastrointestinal Tract, p. 1447–1472. New York: Raven. [Google Scholar]
- Zhang GH, Melvin JE (1994) Nicotine increases [Ca2+]i in rat sublingual mucous acini by stimulating neurotransmitter release from presynaptic terminals. Proc. Soc. Exp. Biol. Med. 204, 292–301. [DOI] [PubMed] [Google Scholar]