

Abstract
The aim of this investigation was to determine whether tumour necrosis factor‐alpha (TNF‐α) has any effect on endothelial progenitor cells (EPCs). Total mononuclear cells were isolated from peripheral blood by Ficoll density gradient centrifugation, and then the cells were plated on fibronectin‐coated culture dishes. After 7 days culture, attached cells were stimulated with tumour necrosis factor‐α (final concentrations: 0, 10, 20, 50 and 100 mg/l) for 0, 6, 12, 24 and 48 h. EPCs were characterized as adherent cells double positive for DiLDL‐uptake and lectin binding, by direct fluorescence staining. EPC proliferation and migration were assayed using the MTT assay and modified Boyden chamber assay, respectively. EPC adhesion assay was performed by re‐plating those cells on fibronectin‐coated dishes, and adherent cells were counted. Tube formation activity was assayed using a tube formation kit. Levels of apoptosis were revealed using an annexin V apoptosis detection kit. Vascular endothelial growth factor Receptor‐1 (VEGF‐R1) and stromal derived factor‐1 (SDF‐1) mRNA, assessed by real‐time RT‐PCR inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) were assayed by western blot analysis. Incubation of EPCs with tumour necrosis factor‐α reduced EPC proliferation, migration, adhesion, tube formation capacity, iNOS and eNOS in concentration‐ and time‐dependent manners. Tumour necrosis factor‐α reduced proliferation, migration, adhesion and tube formation capacity of EPCs. TNF‐α increased EPC apoptosis level, reduced VEGF‐R1 and SDF‐1 mRNA expression; tumour necrosis factor‐α also reduced iNOS and eNOS in the EPCs.
Introduction
Tumour necrosis factor‐alpha (TNF‐α) is a multifunctional cytokine, produced predominantly by activated macrophages and T cells, that plays a key role in several pathophysiological situations involving inflammation and sepsis. In acute coronary syndrome (ACS), TNF‐α is an important proinflammatory cytokine (1). Vascular endothelial progenitor cells (EPCs) are precursors of endothelial cells. Increasing evidence suggests that circulating foetal progenitor cells contribute to postnatal neovascularization. These cells are home to sites of ischaemia, adopt an endothelial phenotype and contribute to new blood vessel formation. Bone‐marrow‐derived haematopoietic progenitor cells can give rise to EPCs and contribute to endothelial recovery and new capillary formation after ischaemia.
At the molecular level, some factors, such as statins and vascular endothelial growth factor (VEGF), can improve the proliferation of EPCs by activating the phosphatidyl‐inositol‐3‐kinase (PI3K)‐Akt‐endothelial nitric oxide synthase (eNOS), suggesting that the PI3K‐Akt‐eNOS signalling pathway may be the important signalling transduction pathway in EPCs (2). Nitric oxide (NO) synthesized from l‐arginine by inducible nitric oxide synthase (iNOS) is a very important signal pathway messenger in human endothelial cells (3). Therefore, we investigated the effects of TNF‐α on activity of EPCs from peripheral blood. We investigated whether TNF‐α can induce EPCs apoptosis. We also investigated the influence of TNF‐alpha on EPCs vascular endothelial growth factor Receptor‐1 (VEGF‐R1) and stromal derived factor‐1 (SDF‐1) mRNA assessed by real‐time RT‐PCR. We detected iNOS and eNOS by western blotting and discuss the role of iNOS and eNOS in these effects.
Materials and methods
Isolation and culture of EPCs
Human EPCs were obtained from six healthy adults and cultured according to previously described techniques (4, 5, 6, 7).Written informed consent was obtained from all people involved in the study. Briefly, total mononuclear cells (MNC) were isolated from blood of the study subjects, by Ficoll density gradient centrifugation. Cells were plated on culture dishes coated with human fibronectin (Chemicon, Pittsburgh, PA, USA) and were maintained in Medium 199 (Sigma, St. Louis, MO, USA) supplemented with 20% foetal calf serum, penicillin (100 U/ml) and streptomycin (100 μg/ml). After 4 days culture, non‐adherent cells were removed by washing in phosphate‐buffered saline (PBS); new medium was added and cultures were maintained until day 7. Attached cells were stimulated with TNF‐α (Sigma; to achieve final concentrations of 10, 20, 50 and 100 mg/l) for 0, 6, 12, 24 and 48 h.
Cell staining
Fluorescence detection of EPCs was performed on attached MNCs after 7 days culture. Direct fluorescence staining was used to detect dual binding of fluorescein isothiocyanate (FITC)‐labelled Ulex europaeus agglutinin (UEA‐1; Sigma) and 1,1‐dioctadecyl‐3,3,3,3‐tetramethylindocarbocyanine (DiI)‐labelled acetylated low‐density lipoprotein (acLDL; Molecular Probe Data Base, Genoa, Italy). Cells were first incubated in acLDL at 37 °C and later fixed in 2% paraformaldehyde for 10 min. After being washed, cells were treated with UEA‐1 (10 μg/ml) for 1 h. Samples were then viewed using an inverted fluorescence microscope (Leica Microsystems AG, Wetzlar, Germany) and a laser scanning confocal microscope (LSCM; Leica). Cells that were doubly fluorescencent were identified as differentiating EPCs (4, 5, 7, 8, 9). Two or three independent investigators evaluated numbers of EPCs per well, by counting 15 randomly selected high‐power fields (×200) using the inverted fluorescence microscope.
Cell migration assay
EPC migration was evaluated using a modified Boyden chamber assay (Jiangsu Qilin Medical Equipment Factory, Shuzhou, China). In brief, isolated EPCs were detached using 0.25% trypsin, harvested by centrifugation, re‐suspended in 500 μl M199 and counted. Then 2 × 104 cells were placed in the upper portion of a modified Boyden chamber. M199 and human recombinant VEGF (50 ng/ml) were introduced into the lower compartment of the chamber. After 24 h incubation at 37 °C, the lower side of the filter was washed in PBS and cells were fixed in 2% paraformaldehyde. For quantification, cells were stained with Giemsa solution. Cells migrating into the lower chamber were counted manually in three random microscope fields (×200) (4, 5).
Cell adhesion assay
EPCs were washed in PBS and gently detached using 0.25% trypsin. After centrifugation and re‐suspension in M199 with 5% foetal bovine serum, identical cell numbers were replated on to fibronectin‐coated culture dishes and incubated for 30 min at 37 °C. Adherent cells were counted blind by independent investigators (4, 5, 10).
EPC proliferation assay
Effects of TNF‐α on EPC proliferation were determined by 3‐(4,5‐dimethyl‐2 thiazoyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide (MTT) assay. After being cultured for 7 days, EPCs were digested using 0.25% trypsin, and then were cultured in serum‐free medium in 96‐well plates (200 μl per well). Cells were supplemented with 10 μl MTT (5 g/l) and incubated for a further span of 6 h. Then supernatant was discarded by aspiration and EPC preparations were shaken with 200 μl Me2SO for 10 min, before optical density (OD) was measured at 490 nm (4, 5).
Tube formation assay
Tube formation assay was performed using a tube formation assay kit (Chemicon) and protocol was followed according to manufacturer’s instructions. Briefly, ECMatrix solution was thawed on ice overnight, then mixed with 10× ECMatrix diluent and placed in a 96‐well tissue culture plate at 37 °C for 1 h, to allow matrix solution to solidify. EPCs were harvested as described earlier and re‐plated (10 000 cells per well) on top of the solidified matrix solution. Cells were incubated at 37 °C for 24 h. Tube formation was inspected using an inverted light microscope at 200× magnification. Formation was defined as development of structures with length at least four times the width (4, 5, 11, 12). Five independent fields were assessed for each well, and average numbers of tubes per 200× field were determined.
Apoptosis assay
Quantitative determination of cells undergoing apoptosis was carried out using an annexin V apoptosis detection kit (Alexis Biochemicals, Farmingdale, NY, USA), according to the manufacturer’s instructions. Percentage of apoptotic cells was measured from EPCs cultured with and without TNF‐α (10, 20, 50 and 100 mg/l) for 4 days. Cells were stained with annexin V FITC and propidium iodide (PI), and subjected to flow cytometric analysis. To exclude necrotic cells, only annexin positive cells were counted. Data are provided as means (SEM) as percentage of annexin V+/PI− cells (representative of apoptotic cells) (13).
EPC VEGF‐R1 and SDF‐1 mRNA assessed by quantitative real‐time RT‐PCR
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) following a standard protocol. Contaminating DNA was eliminated by two sequential steps of DNase (Invitrogen) treatment. mRNA was reverse‐transcribed, and cDNA underwent 40 rounds of amplification (ABI PRISM 7000; Applied Biosystems, Foster City, CA, USA) with the following reaction conditions: 40 cycles of two‐step polymerase chain reaction (95 °C for 15 s and 60 °C for 60 s) after initial denaturation (50 °C for 2 min and 95 °C for 10 min) with 1 mu; l DNA solution in 2× SYBR green PCR master was mixed in reaction buffer. Each sample was tested in triplicate. Authenticity and size of PCR products were confirmed by melting curve analysis (PerkinElmer Inc, San Jose, CA, USA). mRNA levels were normalized using GAPDH as housekeeping gene, and compared to control groups. VEGFR1 primers used for amplification were: Forward, GAACAGAGCAAACAGGAGGC; Reverse, AGGCCAGGAGTGAGAAGTCA, The SDF‐1 primers were: Forward, CACTTTCACTCTCGGTCCAC; Reverse, CTGAAGGGCACAGTTTGGAG (14).
Western blot analysis for iNOS and eNOS
Cell monolayers were washed three times in PBS and lysed with RIPA buffer containing phenylmethylsulphonyl fluoride and aprotinin as protease inhibitors. Lysates (30 μg total protein) were denatured and subjected to 7.5% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and proteins were then transferred to nitrocellulose membranes by electroblotting. Membranes were soaked in blocking solution containing PBS with 5% non‐fat dried milk and 0.05% Tween 20 for 1 h at room temperature. They were then incubated with iNOS or eNOS monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h and then with peroxidase‐conjugated secondary antibodies for 2 h at room temperature. Bands corresponding to iNOS or eNOS were detected using chemiluminescence reagent (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Statistical analysis
Data are expressed as mean ± SD and one‐way ANOVA was used to analyse differences in variables. Differences were considered significant if P‐values were <0.05. All statistical analyses were performed with spss 11.5 (SPSS, Chicago, IL, USA).
Results
Characteristics of human EPCs
Total MNCs isolated and cultured for 7 days were spindle‐shaped, with endothelial cell‐like morphology. Cells were characterized as adherent double‐positive for DiLDL‐uptake and lectin binding by laser scanning confocal microscopy (Fig. 1). We and other investigators have previously demonstrated that EPCs isolated in this fashion also exhibit many other endothelial characteristics, including expression of CD31, von Willebrand factor and vascular endothelial growth factor receptor 2 (4, 5, 7).
Figure 1.
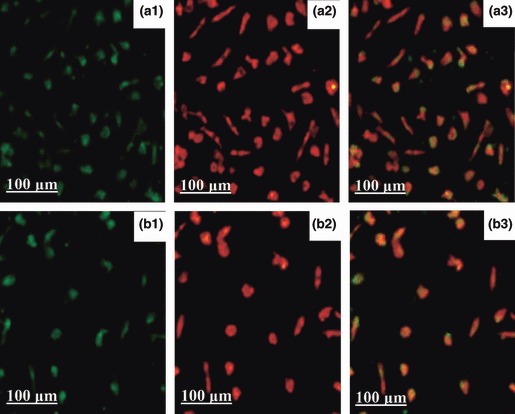
Characteristics of human EPCs.
Mononuclear cells were cultured for 7 days, and adherent cells lectin binding (green, exciting wave‐length 477 nm) and DiLDL uptake (red, exciting wave‐length 543 nm) were assessed under by laser scanning confocal microscopy. Double positive cells appearing yellow in the overlay were identified as differentiating EPCs. Group TNF‐α 100 mg/l (B1, B2 and B3) significantly reduced numbers of EPCs compared to controls (A1, A2 and A3).
Effects of TNF‐α on proliferative capacity of isolated EPCs
We incubated isolated human MNC with different concentrations TNF‐α and results shows that TNF‐α reduced EPCs’ proliferative capacity in concentration‐ and time‐dependent manners (Table 1 and Fig. 2).
Table 1.
Effects of tumour necrosis factor‐α (TNF‐α) on endothelial progenitor cell proliferation
| TNF‐α (mg/l) | A490 nm | ||||
|---|---|---|---|---|---|
| 0 h | 6 h | 12 h | 24 h | 48 h | |
| Control | 0.521 ± 0.049 | 0.514 ± 0.052 | 0.518 ± 0.057 | 0.519 ± 0.062 | 0.523 ± 0.058 |
| 10 | 0.519 ± 0.055 | 0.499 ± 0.046 | 0.468 ± 0.049 | 0.448 ± 0.047*# | 0.422 ± 0.045**## |
| 20 | 0.518 ± 0.059 | 0.458 ± 0.045 | 0.398 ± 0.038**## | 0.338 ± 0.031**## | 0.297 ± 0.031**## |
| 50 | 0.522 ± 0.063 | 0.411 ± 0.047**## | 0.360 ± 0.040**## | 0.259 ± 0.034**## | 0.212 ± 0.026**## |
| 100 | 0.524 ± 0.056 | 0.395 ± 0.051**## | 0.313 ± 0.033**## | 0.202 ± 0.032**## | 0.178 ± 0.022**## |
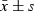 , n = 6. *P < 0.05, **P < 0.01, compared with control group; #
P < 0.05, ##
P < 0.01, compared with 0 h group.
, n = 6. *P < 0.05, **P < 0.01, compared with control group; #
P < 0.05, ##
P < 0.01, compared with 0 h group.
Figure 2.
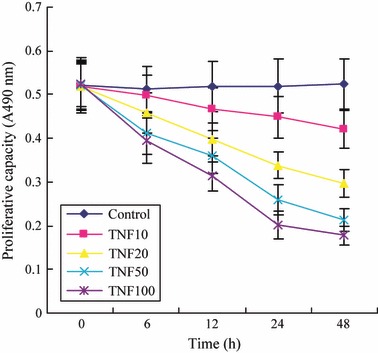
Effects of tumour necrosis factor‐α (TNF‐α) on EPC proliferation.
Effects of TNF‐α on migratory capacity of isolated EPCs
We incubated isolated human MNC with different concentrations TNF‐α and results show that TNF‐α reduced EPC migratory capacity in concentration‐ and time‐dependent manners (Table 2 and Fig. 3).
Table 2.
Effects of TNF‐α on endothelial progenitor cell migration
| TNF‐α (mg/l) | Migratory cells (number) | ||||
|---|---|---|---|---|---|
| 0 h | 6 h | 12 h | 24 h | 48 h | |
| Control | 12.9 ± 3.0 | 13.4 ± 3.2 | 14.1 ± 2.9 | 13.3 ± 2.6 | 13.6 ± 2.7 |
| 10 | 13.1 ± 2.7 | 13.1 ± 2.6 | 11.8 ± 2.4 | 9.9 ± 2.3* | 9.4 ± 2.3*# |
| 20 | 12.9 ± 2.5 | 11.9 ± 3.0 | 10.1 ± 2.8* | 8.8 ± 2.5**# | 8.1 ± 2.3**## |
| 50 | 13.2 ± 2.9 | 10.1 ± 3.1* | 8.0 ± 2.3**## | 7.5 ± 2.4**## | 5.9 ± 2.6**## |
| 100 | 12.8 ± 2.8 | 9.7 ± 3.1* | 7.5 ± 3.5**## | 5.8 ± 2.5**## | 4.6 ± 2.3**## |
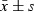 , n = 6. *P < 0.05, **P < 0.01, compared to control group; #
P < 0.05, ##
P < 0.01, compared to 0 h group.
, n = 6. *P < 0.05, **P < 0.01, compared to control group; #
P < 0.05, ##
P < 0.01, compared to 0 h group.
Figure 3.
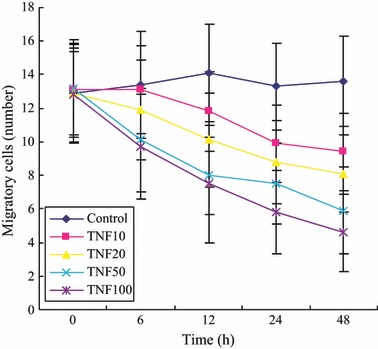
Effects of TNF‐α on EPC migration.
We incubated isolated human MNC with different concentrations of TNF‐α and results show that TNF‐α reduced EPC adhesive capacity in concentration‐ and time‐dependent manners (Table 3 and Fig. 4).
Table 3.
Effects of TNF‐α on endothelial progenitor cell adhesion
| TNF‐α (mg/l) | Adherent cells (number) | ||||
|---|---|---|---|---|---|
| 0 h | 6 h | 12 h | 24 h | 48 h | |
| Control | 32.3 ± 4.9 | 31.5 ± 5.1 | 31.9 ± 5.8 | 30.9 ± 6.2 | 31.7 ± 5.0 |
| 10 | 31.4 ± 5.6 | 30.2 ± 4.8 | 28.5 ± 4.7 | 27.5 ± 5.1 | 25.7 ± 3.4# |
| 20 | 32.5 ± 6.3 | 27.8 ± 5.7 | 26.1 ± 4.3* | 21.9 ± 4.3*## | 19.7 ± 2.9**## |
| 50 | 31.7 ± 5.9 | 23.4 ± 4.9*# | 20.8 ± 4.6**## | 15.4 ± 4.9**## | 11.5 ± 3.1**## |
| 100 | 32.1 ± 5.1 | 20.7 ± 5.4**## | 17.6 ± 4.9**## | 11.4 ± 5.2**## | 8.1 ± 3.7**## |
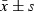 , n = 6. *P < 0.05, **P < 0.01, compared to controls; #
P < 0.05, ##
P < 0.01, compared to 0 h group.
, n = 6. *P < 0.05, **P < 0.01, compared to controls; #
P < 0.05, ##
P < 0.01, compared to 0 h group.
Figure 4.
Effects of TNF‐α on EPC adhesion.
EPC tube formation capacity of control group and TNF‐α 10, 20, 50 and 100 mg/l was 23.3 ± 3.5, 18.3 ± 3.1**, 12.5 ± 3.1**, 8.8 ± 2.7**, 6.5 ± 2.6**, respectively (n = 6, compared to control, **P < 0.01). Incubation of EPC with TNF‐α reduced tube formation capacity of EPCs in concentration‐dependent manner (Fig. 5).
Figure 5.
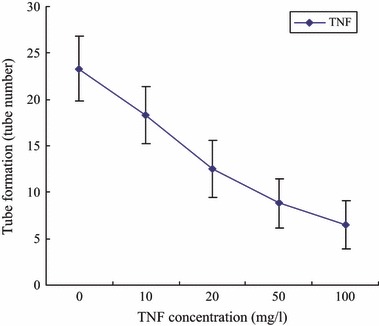
Effects of TNF‐α on EPC tube formation capacity.
Role of TNF‐α‐induced EPC apoptosis
To evaluate potential influence of TNF‐α on apoptosis we analysed binding of annexin V in EPCs from healthy volunteers, cultured for 4 days with and without TNF‐α. Significantly level of apoptosis was found after incubation of EPC with TNF‐α at 50 and 100 mg/l compared to cells cultured in TNF‐α‐free environment (control: 15.8 ± 2.1; 10 mg/l: 16.2 ± 2.7; 20 mg/l: 17.5 ± 3.2; 50 mg/l: 22.5 ± 4.2**; 100 mg/l: 26.5 ± 4.8** as a percentage of total cells, **P < 0.01) (Fig. 6).
Figure 6.
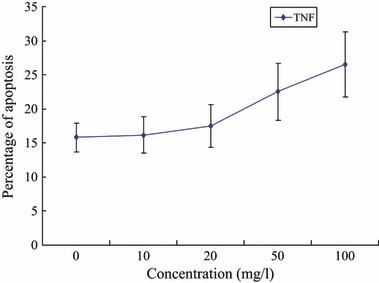
Effects of TNF‐α on EPC apoptosis.
EPC VEGF‐R1 and SDF‐1 mRNA assessed by real‐time RT‐PCR
We found that after 4 days culture, transcription levels of VEGF‐R1 and SDF‐1 were significantly reduced in a concentration‐dependent manner (control: 1.0 ± 0.01; 10 mg/l: 0.8 ± 0.02**; 20 mg/l: 0.7 ± 0.01**; 50 mg/l: 0.5 ± 0.01**; 100 mg/l: 0.3 ± 0.01** fold over control, **P < 0.01) and (control: 1.0 ± 0.02; 10 mg/l: 0.9 ± 0.03; 20 mg/l: 0.7 ± 0.02**; 50 mg/l: 0.6 ± 0.01**; 100 mg/l: 0.5 ± 0.01** fold over control, **P < 0.01), respectively (Fig. 7).
Figure 7.
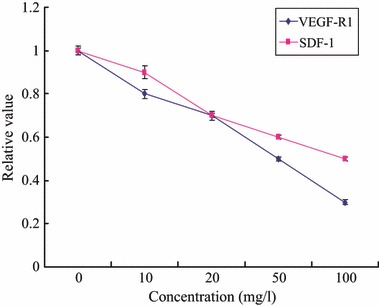
Effects of TNF‐α on EPCs’ apoptosis EPCs VEGF‐R1 and SDF‐1. Line 1: control, Line 2: TNF‐α 10 mg/l, Line 3: TNF‐α 20 mg/l, Line 4: TNF‐α 50 mg/l, Line 5: TNF‐α 100 mg/l.
Effects of TNF‐α on EPC iNOS and eNOS
Western blot analysis showed that iNOS relative light density values of controls and TNF‐α 10, 20, 50 and 100 mg/l were 1.00 ± 0.08, 0.81 ± 0.07, 0.57 ± 0.05, 0.35 ± 0.05 and 0.17 ± 0.03, respectively; eNOS relative light density values were 1.00 ± 0.09, 0.86 ± 0.08, 0.51 ± 0.06, 0.18 ± 0.04 and 0.06 ± 0.03, respectively; expression of iNOS and eNOS were significantly reduced by TNF‐α in a concentration‐dependent manner (Fig. 8).
Figure 8.
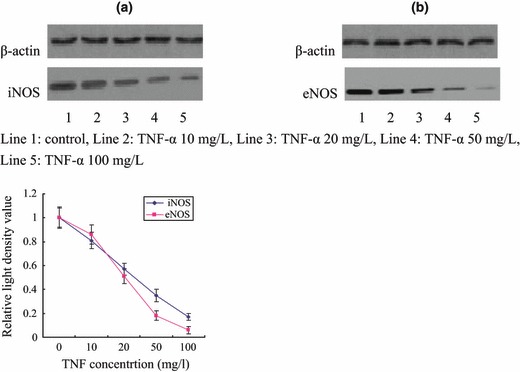
Effects on iNOS (a) and eNOS (b) of EPCs by TNF‐α.
Discussion
TNF‐α is an important proinflammatory cytokine (1). Zhang et al. (15) have shown that myocardial ischaemia/reperfusion (I/R) initiates expression of TNF‐α, which induces activation of xanthine oxidase and production of O2 −, leading to coronary endothelial dysfunction. TNF‐α receptor p75 is required in ischaemia‐induced neovascularization (16). Recent studies have shown that signalling through VEGFR‐1 positively modulates endothelial cell migration and vascular branching. It has been shown that under pathological conditions, activation of VEGFR‐1 by placental growth factor, a VEGFR‐1‐specific ligand, contributes to angiogenesis [Debasree (17)]. Stromal cell‐derived factor‐1 (SDF1) and its receptor CXC chemokine receptor 4 (CXCR4) play a critical role in progenitor cell homing, mobilization and differentiation (18).
Anderson et al. have shown that TNF‐alpha reduced eNOS gene expression through post‐transcriptional regulation of mRNA stability, suggesting a potential mechanism to account for the endothelial dysfunction (19). High dose of simvastatin reduces TNF‐alpha‐induced apoptosis of EPCs, but fails to prevent apoptosis induced by IL‐1βin vitro (13).
There is strong evidence that EPCs play a significant role in neovascularization and re‐endothelialization, particularly during ischaemic conditions. More recently, two groups have documented (in animals and in human subjects) that EPCs contribute up to 25% of endothelial cells in newly formed vessels (20, 21). Thus, increasing numbers of circulating EPCs by transplantation of haematopoietic stem cells, or by injection of in vitro differentiated EPCs, has been shown to improve neovascularization of ischaemic hindlimbs (22), accelerate blood flow in diabetic mice (23) and improve cardiac function (24). Moreover, Vasa et al. (8, 9) have recently reported that patients with coronary heart disease (CHD) have reduced levels and functional impairment of EPCs, which correlated with risk factors for CHD. Thus, stimulation of mobilization and/or differentiation of EPCs may provide a useful novel therapeutic strategy to improve postnatal neovascularization and re‐endothelialization in patients with CHD. Circulating EPCs are constantly exposed to inflammatory factors, such as cytokines and oxidized lipoproteins. Atherosclerosis, in general, is indicative of low‐grade inflammatory processes, and atherosclerotic plaques (that are prone to rupture and result in acute coronary syndromes) reveal an intense inflammatory state (25). TNF‐α and endothelial cells modulate Notch signalling in the bone marrow microenvironment during inflammation (26) and circulating EPCs are not affected by acute systemic inflammation (27). Treatment with TNF‐alpha or bacterial lipopolysaccharide attenuates endocardial endothelial cell‐mediated stimulation of cardiac fibroblasts (28).
The present study shows that TNF‐α reduced proliferative, migratory, adhesive and tube formation capacity of EPCs in time‐ and concentration‐dependent manners. Our in vitro measurements of apoptotic EPCs cultured in TNF‐α‐containing medium demonstrated significantly increased levels of apoptosis compared to in control medium, and this study shows that TNF‐α reduced VEGF‐R1 and SDF‐1 mRNA expression. Thus, we believe that TNF‐α reduces EPC differentiation into endothelial cells and angiogenetic capacity. We are of the opinion that this is an important mechanism of endothelium dysfunction in acute coronary syndromes. We also found that TNF‐α reduced iNOS and eNOS of EPCs in a concentration‐dependent manner. NO can enhance EPC proliferation and function through activating the PI3K/Akt signalling pathway (8, 9). Walter et al. (29) have shown that eNOS mRNA stability and eNOS activity is a common signal transduction pathway in endothelium and EPCs. Therefore, we deduce that decreasing iNOS and eNOS is one of the mechanisms of the inhibitory effect of TNF‐α on EPCs.
References
- 1. Wilson SH, Best PJ, Edwards WD, Holmes DR Jr, Carlson PJ, Celermajer DS et al. (2002) Nuclear factor‐kappaB immunoreactivity is present in human coronary plaque and enhanced in patients with unstable angina pectoris. Atherosclerosis 160, 147–153. [DOI] [PubMed] [Google Scholar]
- 2. Urbich C, Dimmeler S (2005) Risk factors for coronary artery disease, circulating endothelial progenitor cells, and the role of HMG‐CoA reductase inhibitors. Kidney Int. 67, 1672–1676. [DOI] [PubMed] [Google Scholar]
- 3. Guo X, Chen LW, Liu WL, Guo ZG (2000) High glucose inhibits expression of inducible and constitutive nitric oxide synthase in bovine aortic endothelial cells. Acta Pharmacol. Sin. 21, 325–328. [PubMed] [Google Scholar]
- 4. Chen TG, Chen JZ, Xie XD (2006a) Effects of aspirin on number, activity and inducible nitric synthase of endothelial progenitor cells from peripheral blood. Acta Pharmacol. Sin. 27, 430–436. [DOI] [PubMed] [Google Scholar]
- 5. Chen TG, Chen JZ, Wang XX (2006b) Effects of rapamycin on number, activity and eNOS of endothelial progenitor cells from peripheral blood. Cell Prolif. 39, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA et al. (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 348, 593–600. [DOI] [PubMed] [Google Scholar]
- 7. Kalka C, Masuda H, Takahashi T, Kalka‐Moll WM, Silver M, Kearney M et al. (2000) Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. USA 97, 3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM et al. (2001a) Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 103, 2885–2890. [DOI] [PubMed] [Google Scholar]
- 9. Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H et al. (2001b) Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Cir. Res. 89, E1–E7. [DOI] [PubMed] [Google Scholar]
- 10. Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T et al. (2002) Statin therapy accelerate reendothelialization: a novel effect involving mobilization and incorporation of bone marrow‐derived endothelial progenitor cells. Circulation 105, 3017–3024. [DOI] [PubMed] [Google Scholar]
- 11. Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM (2002) Origin of endothelial progenitors in human postnatal bone marrow. J. Clin. Invest. 109, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR et al. (2002) Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 106, 2781–2786. [DOI] [PubMed] [Google Scholar]
- 13. Henrich D, Seebach C, Wilhelm K, Marzi I (2007) High dosage of simvastatin reduces TNF‐alpha‐induced apoptosis of endothelial progenitor cells but fails to prevent apoptosis induced by IL‐1beta in vitro. J. Surg. Res. 142, 13–19. [DOI] [PubMed] [Google Scholar]
- 14. Xu J, Liu X, Chen J, Zacharek A, Cui X, Savant‐Bhonsale S et al. (2010) Cell‐cell interaction promotes rat marrow stromal cell differentiation into endothelial cell via activation of TACE/TNF‐alpha signaling. Cell Transplant. 19, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang C, Xu X, Potter BJ, Wang W, Kuo L, Michael L et al. (2006) TNF‐alpha contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 26, 475–480. [DOI] [PubMed] [Google Scholar]
- 16. Goukassian DA, Qin G, Dolan C, Murayama T, Silver M, Curry C et al. (2007) Tumor necrosis factor‐alpha receptor p75 is required in ischemia‐induced neovascularization. Circulation 115, 752–762. [DOI] [PubMed] [Google Scholar]
- 17. Dutta D, Ray S, Vivian JL, Paul S (2008) Activation of the VEGFR1 chromatin domain an angiogenic signal‐ETS1/HIF‐2α regulatory axis. J. Biol. Chem. 283, 25404–25413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao Q, Ye S, Oberhollenzer F, Mayr A, Jahangiri M, Willeit J et al. (2008) SDF1 gene variation is associated with circulating SDF1alpha level and endothelial progenitor cell number: the Bruneck Study. PLoS One 3, e4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson HD, Rahmutula D, Gardner DG (2004) Tumor necrosis factor‐alpha inhibits endothelial nitric‐oxide synthase gene promoter activity in bovine aortic endothelial cells. J. Biol. Chem. 279, 963–969. [DOI] [PubMed] [Google Scholar]
- 20. Murayama T, Tepper OM, Silver M, Ma H, Losordo DW, Isner JM et al. (2002) Determination of bone marrow‐derived endothelial progenitor cell significance in angiogenic growth factor‐induced neovascularization. Exp. Hematol. 30, 967–972. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki T, Nishida M, Futami S, Fukino K, Amaki T, Aizawa K et al. (2003) Neoendothelialization after peripheral blood stem cell transplantation in humans. A case report of a Tokaimura nuclear accident victim. Cardiovasc. Res. 58, 487–492. [DOI] [PubMed] [Google Scholar]
- 22. Murohara T, Ikeda H, Duan J, Shintani S, Sasaki K, Eguchi H et al. (2000) Transplanted cord blood‐derived endothelial precursor cells augment postnatal neovascularization. J. Clin. Invest. 105, 1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA (2000) Blood‐derived angioblasts accelerate blood‐flow restoration in diabetic mice. J. Clin. Invest. 106, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J et al. (2001) Neovascularization of ischemic myocardium by human bone‐marrowderived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 7, 430–436. [DOI] [PubMed] [Google Scholar]
- 25. Mehta JL, Li DY (1999) Inflammation in ischemic heart disease: response to tissue injury or a pathogenetic villain. Cardiovasc. Res. 43, 291–299. [DOI] [PubMed] [Google Scholar]
- 26. Fernandez L, Rodriguez S, Huang H, Chora A, Fernandes J, Mumaw C et al. (2008) Tumor necrosis factor‐alpha and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Exp. Hematol. 36, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Padfield GJ, Tura O, Haeck ML, Short A, Freyer E, Barclay GR et al. (2010) Circulating endothelial progenitor cells are not affected by acute systemic inflammation. Am. J. Physiol. Heart Circ. Physiol. 298, H2054–H2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuruvilla L, Kartha CC (2009) Treatment with TNF‐alpha or bacterial lipopolysaccharide attenuates endocardial endothelial cell‐mediated stimulation of cardiac fibroblasts. J. Biomed. Sci. 16, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walter DH, Dimmeler S, Zeiher AM (2004) Effects of statins on endothelium and endothelial progenitor cell recruitment. Semin. Vasc. Med. 4, 385–393. [DOI] [PubMed] [Google Scholar]