

Abstract
Abstract. Objectives: CCCTC binding factor (CTCF) is a nuclear protein containing an 11‐zinc‐finger DNA‐binding domain. CTCF plays important roles in the regulation of epigenetics and gene transcription. As a multifunctional protein, CTCF is also involved in the regulation of cell proliferation and of apoptosis. However, mechanisms underlining the regulatory function of CTCF in mediating growth factor‐ and cytokine‐stimulated cell fate are largely unknown. Materials and Methods: The effect of CTCF on insulin‐induced ML‐1 cell proliferation was investigated by studying insulin‐stimulated extracellular signal‐regulated kinase (Erk) and Akt signalling pathways, and the alterations of CTCF activity in these cells. Results: The present study demonstrates that insulin‐induced human haematopoietic myeloblastic ML‐1 cell proliferation requires increased CTCF expression. Inhibition of Erk and Akt pathways with specific blockers or by dominantly negative expression of Erk and Akt mutants markedly suppressed expression of CTCF and resulted in retardation of cell proliferation. Furthermore, insulin‐induced ML‐1 cell proliferation was significantly enhanced by overexpression of cDNA encoding full‐length CTCF. In contrast, ML‐1 cell proliferation was inhibited by knocking down CTCF mRNA using specific small interference RNA. Conclusions: Our results indicate that CTCF is indeed a protein with multifunctional activity that plays a significant role in modulating signalling pathways to mediate insulin‐induced ML‐1 cell proliferation.
INTRODUCTION
Growth factors in serum, such as epidermal growth factor (EGF), insulin and insulin‐like growth factor are known to stimulate proliferation, migration and survival in various cell types (Dragovich et al. 1998; Zhang & Liu 2002). Growth factors bind to specific cognate receptors to activate mitogenic signals with reversible protein phosphorylation; the signals are then transferred and amplified leading to multiple biological effects. Insulin is one of these important cell growth‐related hormones that play major roles in stimulating intracellular signal transduction and cross‐talk through activating the Raf‐MEK‐Erk and phophatidylinsitol 3‐kinase (PI3K)‐Akt pathways. Further downstream events in insulin‐induced signalling cascades include the activation of tyrosine kinase‐signal transducers in the Janus‐family and transcription for the JAK‐STAT pathway (Strobl et al. 1995; Shelton et al. 2004; Steelman et al. 2004). Human haematopoietic myeloblastic ML‐1 cell proliferation/differentiation is dependent on stimulation of growth factors and cytokines (Xu et al. 1996b; Wang et al. 1997, 1999); for example, EGF activates the Erk signalling pathway to regulate cell cycle progression and control their proliferation (Lu et al. 1993; Xu et al. 1996a; Xu et al. 1999), while insulin stimulates their proliferation through activation of the Erk and Akt signalling pathways (Guo et al. 2005). Interestingly, it appears that the Akt pathway, but not the Erk pathway, plays a major role in regulating insulin‐induced ML‐1 cell proliferation (Guo et al. 2005).
CCCTC binding factor (CTCF) is a transcription factor and regulatory protein that interacts with DNA through a DNA‐binding domain composed of 11 zinc fingers (Dunn & Davie 2003). It was first identified as a ubiquitous and highly conserved factor that binds to the promoter of the c‐Myc gene in chickens (Lobanenkov et al. 1990). CTCF mediates transcriptional activation of amyloid β‐protein precursor (Vostrov & Quitschke 1997). It also functions as a repressor, controlling expression of c‐Myc, and as a lysyme silencer, in both humans and chickens (Lobanenkov et al. 1990; Kohne et al. 1993; Filippova et al. 1996). It has been shown that CTCF acts as an insulator protein in the imprinting control region between two imprinted genes, H19 and Igf2 (Bell & Felsenfeld 2000; Hark et al. 2000; Holmgren et al. 2001). The gene encoding CTCF has been localized in human chromosome segment 16q22 (Filippova et al. 1998), and as a phosphoprotein, CTCF can be phosphorylated by protein kinase CK2 (Klenova et al. 2001; Yu et al. 2004; Klenova & Ohlsson 2005). A recent study has demonstrated that there are several CTCF mutations found in breast cancer, prostate cancer and Wilm's tumour cells (Filippova et al. 2002), suggesting that CTCF may be a good candidate for tumour suppressor gene products. In previous studies, we have found that CTCF regulates eye development in mice by binding to repressor elements in the Pax6 gene, which in turn blocks the effect of the ectoderm enhancer on Pax6 P0 promoter activity, resulting in inhibition of Pax6 transcription (Li et al. 2004; Wu et al. 2006). Furthermore, the mitogenic effect of EGF requires increased CTCF and suppressed Pax6 activity in corneal epithelial cells. EGF increases CTCF expression by activating the extracellular signal‐regulated kinase (Erk)‐signalling pathway, resulting in down‐regulation of Pax6 (Li & Lu 2005). These studies further support the notion that CTCF is a multifunctional protein involved with cytokine‐ and stress‐induced cellular responses. Recent reports indicate that CTCF plays a significant role in cell population growth in various types of tissue (Qi et al. 2003; Torrano et al. 2005). However, it is still unclear whether CTCF plays a functional role in insulin‐stimulated cell proliferation. In the present study, we demonstrate for the first time that CTCF involves insulin‐stimulated mitogenic responses in serum starvation‐synchronized human haematopoietic myeloblastic ML‐1 cells. Insulin‐stimulated increase in CTCF expression is one of the important downstream elements resulting from insulin‐induced activation of both the Erk and Akt signalling pathways. These findings provide a new insight into understanding the multifunctional roles of CTCF in regulating cell fate.
MATERIALS AND METHODS
Cell culture and treatment
ML‐1 cells were cultured in Roswell Park Memorial Institute 1640 supplemented with 7.5% heat‐inactivated foetal bovine serum (FBS; InvitrogenTM Life Technologies, Grand Island, NY, USA) in a humidified incubator constantly filled with 5% CO2 at 37 °C. The cells were passed at a seeding density of 3 × 105, and were synchronized by serum starvation in a medium containing 0.3% FBS for 36 h. With different concentrations of insulin, they were induced for 6 h or 24 h. They were then harvested for determination of proliferation parameters by measuring proliferating cell nuclear antigen (PCNA) 6 h after insulin treatment, and by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) analysis 24 h after insulin treatment. PD98059 (20 µm) and wortmannin (1 µm) were added to the culture medium 30 min prior to insulin treatments to inhibit Erk and Akt, respectively.
Cell proliferation index measured by the MTT assay
Cell proliferation was detected by measuring the reduction of a tetrazolium component (MTT) using an established protocol (Li & Lu 2005). Briefly, serum‐starved ML‐1 cells were seeded in six‐well plates at a density of 3 × 105 mL. After treating them with insulin at different concentrations for 24 h, the MTT solution was added to the wells to reach a final concentration of 0.5 mg/mL. Cells were incubated for 2 h in a humidified incubator with 5% CO2 at 37 °C. For the MTT test, ML‐1 cells were harvested and washed once with ice‐cold PBS. Pellets were re‐suspended in 500 µL of 0.04 N HCl in isopropanol in which dark blue crystals of MTT were dissolved. Absorbance of each sample was read at 570 nm wave‐length using a microplate reader (Fisher Scientific, Pittsburgh, PA, USA). Cell proliferation was also evaluated by measuring PCNA expression levels by Western blot analysis. PCNA is a cyclin protein with a molecular weight of 36 kDa, which is strongly associated with DNA synthesis.
Western blot analysis
The cells were treated with the various agents and were harvested by centrifugation at 2000 g for 5 min. Cell pellets were re‐suspended in lysis buffer containing: (in mm) 20 Tris‐HCl, pH 7.5, 137 NaCl, 1.5 MgCl2, 2 ethylenediaminetetraacetic acid, 10 sodium pyrophosphate, 25 β‐glycerophosphate, 10% glycerol, 1% Triton X‐100, 1 sodium orthovanadate, 1 phenylmethylsulfonyl fluoride. After incubation on ice for 30 min, cell lysates were collected by centrifugation at 13 000 g for 15 min and were denatured by adding an equal volume of 2× Laemmli buffer. Cell lysates were boiled for 5 min and then were fractionated using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Proteins were electro‐transferred on to a polyvinylidene fluoride membrane. After blocking with 5% non‐fat milk in Tris‐buffered saline containing 0.1% Tween‐20 (TBST), membranes were incubated with primary antibodies in TBST containing 5% non‐fat milk, overnight at 4 °C. Rabbit polyclonal antibodies against total and phosphorylated (activated) Erk1/2 and Akt were diluted to 1 : 1000 (Cell Signalling Technology, Beverly, MA, USA). Mouse monoclonal antibodies to β‐actin (Sigma, St. Louis, MO, USA) and PCNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were diluted to 1 : 10 000 and 1 : 5000, respectively. Rabbit polyclonal antibody to CTCF (Upstate Cell Signalling Solutions, Lake Placid, NY, USA) was diluted to 1 : 10 000. After washing three times with TBST, membranes were then incubated with horseradish peroxidase (HRP)‐conjugated goat antimouse immunoglobulin G (Santa Cruz Biotechnology) diluted to 1 : 2000 in TBST containing 5% non‐fat milk, or HRP‐conjugated swine antirabbit immunoglobulin G (Dako, Carpinteria, CA, USA) diluted to 1 : 1000 at room temperature for 1 h. Specific signals were developed with Western blotting luminol reagent (Santa Cruz Biotechnology) and were captured on Kodak X‐ray films.
Gene transfection
Expression vector pcDNA3.1 inserted with cDNA encoding full‐length CTCF and dominantly negative constructs containing mutants of Erk and Akt were used in transfection experiments (Han et al. 2002; Li et al. 2004; Lu et al. 2006) ML‐1 cells were transfected with plasmids using a LipofectamineTM kit following manufacture's instructions (InvitrogenTM Life Technologies). Samples containing 5 µg DNA and 10 µL of PlusTM reagent diluted in 90 µL of serum‐free growth medium were incubated with cells at room temperature for 15 min. Cells were continuously cultured in the humidified incubator with 5% CO2 at 37 °C for 6 h, and then they were cultured for different periods (12–30 h). Transfected cells were synchronized by serum‐starvation for 36 h before performing experiments.
RNA interference
Specific primers, complementary to human CTCF mRNA (21 nucleotides plus other eight nucleotides complementary to T7 promoter), were designed in our laboratory (Li & Lu 2005). Designed primers for small interference RNA (siRNA) experiments contained a sense strand sequence ‘AAGGAAUGUCUUCUUUACACC’, and an antisense strand sequence ‘AAGGUGUAAAGAAGACAUUCC’. In addition, a control double strand of siRNA (sense ‘AACAUUCGGUAGAUUCCUCGC’ and antisense ‘AAGCGAGGAAUCUACCGAAUG’) was also synthesized. Sequence homologies of siRNA primers were examined by using an NIH Blast program. SiRNA was synthesized by in vitro transcription using a Silencer® siRNA construction kit (Ambion Inc., Austin, TX, USA). Double‐stranded RNA was synthesized by in vitro transcription using T7 RNA polymerase followed by RNase digestions to obtain siRNA. SiRNAs were transfected into the ML‐1 cells using a LipofectamineTM kit. Non‐specific iRNAs were also synthesized and transfected into ML‐1 cells as controls.
Statistical analysis
For Western blot analysis, signal densities in films were determined by densitometry with Image Calculator software. For MTT assays, relative optical density values were calculated and normalized with control values. Data were shown as mean ± standard errors (SE). Significant differences between the control group and treated groups were determined by one‐way anova and Student's t‐test at P < 0.05.
RESULTS
Insulin‐induced CTCF expression and cell proliferation
ML‐1 cells were had been synchronized in the G1 phase of the cell cycle, by serum starvation for 36 h. Cell population growth rate was revealed in proliferation indices determined by MTT assay. Various dosages of insulin had been applied to stimulate cell proliferation and there were dose‐dependent increases in the cell population growth rate (Fig. 1a). Insulin‐stimulated cell proliferation significantly increased following increased insulin concentrations and reached 2.5 fold compared to control cells at an insulin concentration of 20 µg/mL (n = 6; P < 0.05). This cell proliferation was further verified by examining altered levels of PCNA expression, a marker protein for cells proliferating in early S phase of the cell cycle (Bravo & Macdonald‐Bravo 1987). Western blot analysis revealed that with increased doses of insulin (0, 1, 5, 10 and 20 µg/mL), expression level of PCNA was markedly elevated (Fig. 1b). Next, to determine whether CTCF played a role in proliferation in response to factor stimulation, expression of CTCF was detected using Western blots. As shown in Fig. 1c, the expression level of CTCF sharply increased in response to application of 5 µg/mL insulin. This result indicates that the expression level of CTCF is regulated during insulin‐stimulated ML‐1 cell proliferation, and that it is necessary to further investigate whether CTCF is indeed involved in mediating insulin‐stimulated cell proliferation.
Figure 1.
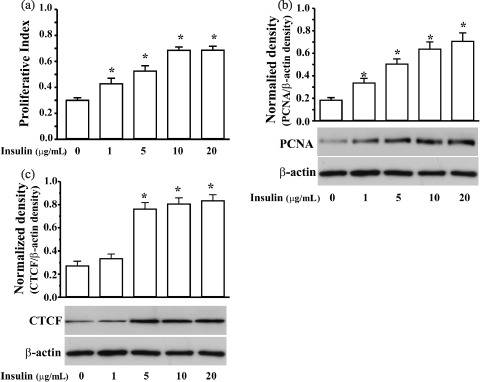
Insulin‐induced proliferation and increased CTCF expression in ML‐1 cells. (a) Dose‐dependent stimulation of proliferation by insulin determined by MTT assay. (b) Dose‐dependent stimulation of proliferation by insulin determined by measuring expression of PCNA. (c) Dose–response increases in expression of CTCF induced by insulin. The cells were synchronized by serum‐starvation for 36 h, and then were stimulated by insulin at indicated dosages for 6 h prior to measurements of CTCF and PCNA, and 24 h prior to MTT assay. Data are presented as mean ± SE and asterisks (*) represent the significant difference compared to controls, analysed by Student's t‐test (n = 4 or 6, P < 0.05).
Insulin‐induced signalling pathways related to CTCF expression
It has been previously shown that insulin stimulates cell proliferation via activation of PI3K‐Akt and Raf‐Ras‐MEK‐Erk pathways in various tissues and cell types (Carel et al. 1996; Liu et al. 2000; Guo et al. 2005). In ML‐1 cells, application of 10 µg/mL of insulin induced fast yet transient phosphorylation of both Erk and Akt (Fig. 2a). Although activation of both Erk and Akt reached peak levels 15 min after insulin stimulation, phosphorylation of Erk fell rapidly to the original level, while phosphorylation of Akt decreased rather slowly and lasted for 2 h. To determine whether CTCF mediates insulin‐stimulated cell proliferation, insulin‐induced activation of Erk and Akt signalling pathways in the cells were suppressed by using PD98059, a blocker of methyl ethyl ketone (MEK), and wortmannin, a blocker for PI3K (Fig. 2b) (Dudley et al. 1995; Duckworth & Cantley 1997). It was noted that there were stronger immunoreactions in insulin‐induced phospho‐Erk and phospho‐Akt compared to those of endogenous Erk and Akt, suggesting that the antibodies specific to phospho‐Erk and phospho‐Akt had higher affinities for the phosphorylated proteins. PD98059 effectively blocked insulin‐induced phosphorylation of Erk, but did not affect activation of Akt. In contrast, wortmannin effectively blocked phosphorylation of both Erk and Akt. Finally, a combination of both blockers was applied to the cells to ensure that insulin‐induced effects on either the Erk or Akt signalling pathway were suppressed. Correspondingly, insulin‐induced increased expression of CTCF was reduced slightly in the PD98059 treated cells, while expression of CTCF was significantly diminished by treating cells with wortmannin or by the combination treatment of PD98059 and wortmannin (Fig. 2c). In addition, both blockers were able to successfully inhibit insulin‐stimulated cell proliferation, measured by both MTT assay and PCNA expression (Fig. 2d,e). There was a weaker effect of PD98059 on suppression of insulin‐induced proliferation compared to either application of wortmannin or application of PD98059 plus wortmannin. To further verify the effects of insulin‐induced activation of Erk and Akt signalling pathways on CTCF expression and cell proliferation, the cells were transfected with dominant negative (DN) constructs of mutated Erk cDNA (DN‐Erk) and/or Akt cDNA (DN‐Akt). Total protein levels of Erk or Akt in the transfected cells, with respective DN‐Erk and/or DN‐Akt, were much higher 72 h after transfection than in control cells transfected with vectors only (Fig. 3a). However, insulin‐induced CTCF expression was significantly lower cells transfected with DN‐Akt and DN‐Akt plus DN‐Erk mutants (Fig. 3b). In addition, insulin‐stimulated proliferation was significantly suppressed in cells transfected with DN‐Erk and/or DN‐Akt (Fig. 3c,d). Comparing the effects of inhibiting Erk and Akt signalling pathways using specific blockers to effect overexpression DN‐Erk and DN‐Akt on insulin‐induced CTCF expression and cell proliferation, the results were fairly consistent. They indicate that increased cellular CTCF activity in response to insulin stimulation is parallel to insulin‐stimulated cell proliferation, further suggesting that CTCF may be a downstream component of the insulin‐induced Akt signalling pathway.
Figure 2.
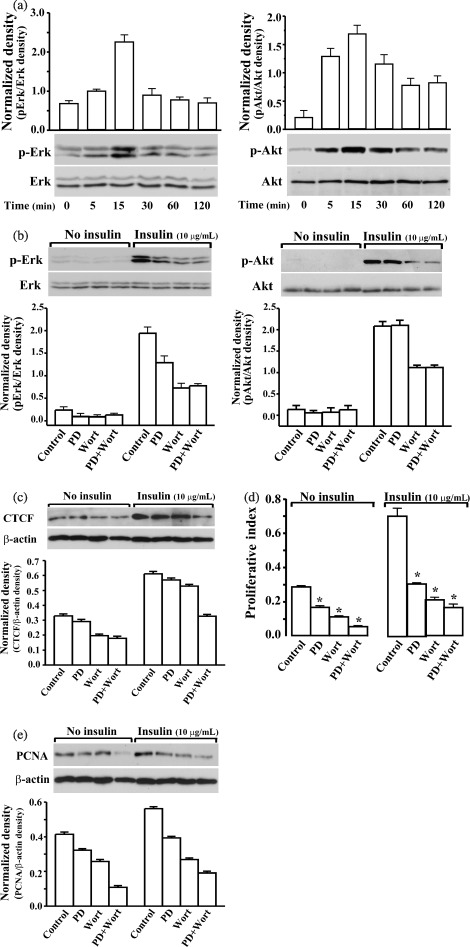
Effects of suppressing insulin‐induced activation of Erk and Akt on CTCT expression, and cell proliferation. (a) Time‐dependent activation of Erk and Akt stimulated by insulin (10 µg/mL). (b) Effects of PD98059 (PD) and wortmannin (Wort) on inhibition of insulin‐induced activation of Erk and Akt, respectively. (c) Effects of PD98059 (PD) and wortmannin (Wort) on insulin‐stimulated CTCF expression determined by Western blot analysis. (d) Effects of PD98059 (PD) and wortmannin (Wort) on insulin‐induced proliferation determined by MTT assay. (e) Effects of PD98059 (PD) and wortmannin (Wort) on insulin‐induced proliferation determined by measuring PCNA expression. Cells were harvested at indicated time points after insulin treatments. PD98059 (20 µm) and wormannin (1 µm) were added to serum starvation‐synchronized cells, individually or together, 30 min prior to insulin treatment (10 µg/mL). Phosphorylated Erk and Akt levels were determined by Western blot analysis. Total amounts of Erk and Akt were measured as loading controls. Cells were harvested at 15 min, 6 h or 24 h after insulin treatment for measurements of Erk and Akt, CTCF and PCNA, or MTT assays, respectively. Asterisks (*) represent significant difference analysed by Student's t‐test (n = 4 or 6, P < 0.05).
Figure 3.

Effects of dominantly negative (DN) expression of Erk and/or Akt mutants on insulin‐induced Erk and Akt activities, CTCT expression and cell proliferation. (a) Effect of DN expression of Erk and/or Akt mutants on Erk and Akt activity. (b) Effects of DN expressions of Erk and/or Akt mutants on CTCF expression. (c) Effects of DN expressions of Erk and/or Akt mutants on insulin‐induced proliferation determined by MTT assay. (d) Effects of DN expression of Erk and/or Akt mutants on insulin‐induced proliferation determined by measuring PCNA expression. DN Erk and/or Akt mutants were transfected or cotransfected into the cells for 72 h before all measurements. The same expression vector minus carrying genes was transfected into cells to provide the controls. Transfected cells were serum‐starved for 36 h and were treated with insulin (10 µg/mL) 6 h prior to Western blot analysis. For MTT assays, transfected cells were harvested 24 h after insulin treatment. Asterisks (*) represent the significant difference analysed by one‐way anova and Student's t‐test (n = 4 or 6, P < 0.05).
Effect of insulin‐induced CTCF expression on cell proliferation
To study the role of CTCF in insulin‐stimulated ML‐1 cell proliferation, the cells were transfected with a pcDNA4‐CTCF construct containing full‐length cDNA encoding CTCF or with a pcDNA4‐To‐A vector for control experiments. The CTCF level in ML‐1 cells transfected with pcDNA4‐CTCF was markedly increased in 72 h (Fig. 4a). The effect of overexpression of CTCF on the insulin‐stimulated cell proliferation was evaluated by MTT assays and by measuring PCNA expression (Fig. 4b,c). Overexpression of CTCF resulted in significant increases in the proliferation index and PCNA expression in insulin‐induced cells (n = 6; P < 0.05). Overexpression of CTCF in transfected cells also markedly promoted proliferation by more than 1.5 times in the absence of insulin stimulation compared to vector‐transfected control cells. The results of promoting insulin‐stimulated proliferation by overexpression of CTCF in the cells indicate that CTCF indeed plays a functional role in the regulation of insulin‐induced signalling pathways.
Figure 4.
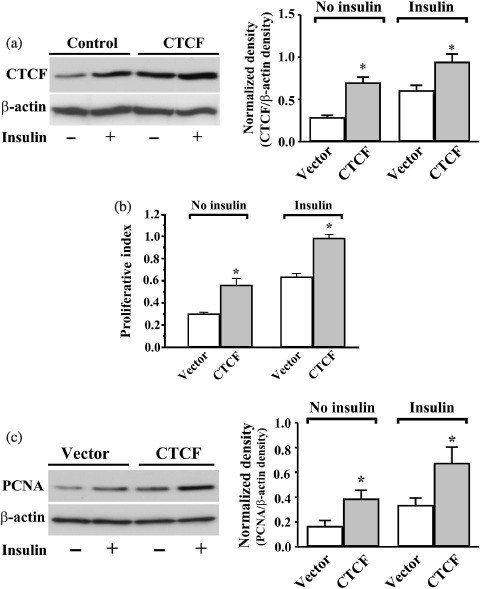
Effects of overexpressing CTCF on insulin‐induced cell proliferation. (a) Effect of insulin on CTCF expression in CTCF transfected cells. Cells were transfected with cDNA encoding full‐length CTCF by using Lipofectamine kit; the expression vector alone was transfected for control experiments. (b) Effect of overexpressing CTCF on insulin‐induced proliferation detected by MTT assay. (c) Effect of overexpressing CTCF on insulin‐induced proliferation determined by measuring PCNA expression. Cells were transfected with CTCF for 30 h and were synchronized by serum starvation for an additional 36 h; they were induced by insulin (10 µg/mL) 6 h prior to Western blot analysis. For MTT assays, cells were stimulated with insulin (10 µg/mL) 24 h prior to measurements. β‐Actin levels were measured as loading controls. Asterisks (*) represent significant difference by using Student's t‐test (n = 4 or 6, P < 0.05).
Effect of suppressing CTCF on insulin‐induced cell proliferation
To further verify the effect of CTCF on insulin‐stimulated proliferation, CTCF mRNA was knocked down by siRNA transfection using specifically designed primers for CTCF; it was effectively suppressed by knocking down CTCF after siRNA transfection. Interestingly, knockdown of CTCF mRNA expression significantly diminished in insulin‐induced cells (Fig. 5a; n = 6; P < 0.01). In addition, application of insulin to cells in which CTCF mRNA was knocked down elicited significant retardation of proliferation measured by both MTT assay and PCNA expression (Fig. 5b,c; n = 6; P < 0.01). The results obtained from CTCF mRNA knockdown experiments are consistent with results shown in CTCF overexpression experiments, indicating that CTCF plays a role, at least partially, in mediating cell proliferation in insulin‐induced signalling pathways.
Figure 5.

Effects of knocking down CTCF mRNA on insulin‐induced cell proliferation. (a) Diminished CTCF expression by knockdown of CTCF determined by Western blot analysis. (b) Effect of knocking down CTCF on insulin‐induced cell proliferation detected by MTT assay. (c) Effect of knocking down CTCF on insulin‐induced proliferation determined by measuring PCNA expression. CTCF expression was suppressed by knocking down CTCF mRNA using the CTCF‐specific siRNA. Cells were harvested 72 h after siRNA transfection and non‐specific siRNA was transfected for control cells. Asterisks (*) and (**) indicate statistically significant differences at confidence interval levels of P < 0.05 and P < 0.01, respectively.
DISCUSSION
Insulin is a hormone found normally in mammalian serum and is well known for its crucial role in glucose metabolism (Buchanan 1976). It is also an important mitogenic stimulator promoting cell proliferation, differentiation and survival in various cell types including ML‐1 cells (Lawlor & Alessi 2001; Guo et al. 2005). Insulin transmits mitogenic signals by activating cognate receptors and triggering reversible phosphorylation. Further downstream, insulin‐induced signals are transduced and amplified to regulate the cell cycle and appropriate gene expression, leading to cell survival, growth, differentiation and/or metabolic changes. The Erk signalling pathway is one of the major signalling pathways responsible for transduction of insulin‐elicited mitogenic signals (Sebolt‐Leopold 2004). These results are consistent in the present study showing that insulin‐induced ML‐1 cell proliferation occurs through activation of the Erk and Akt signalling pathways. New findings in our study indicate that CTCF is a mediator in insulin‐induced mitogenic pathways, downstream of Erk and Akt. Our results suggesting that CTCF plays a role in insulin‐induced mitogenic responses are supported by several studies reporting that CTCF participates in cytokine‐ and stress‐induced cellular responses downstream of various signalling pathways (Klenova et al. 2001; Rasko et al. 2001; Qi et al. 2003). To further reveal the precise role of CTCF in growth factor‐stimulated cell proliferation, we studied regulation of CTCF expression in insulin‐stimulated cell proliferation. Insulin‐induced increases in the expression of CTCF are parallel to insulin‐induced proliferation in the cells. The stimulatory effect of insulin on CTCF expression is fairly consistent with the effect of EGF‐induced increases in CTCF mRNA expression in both ML‐1 cell and in rabbit corneal epithelial cells found in our previous studies (Guo et al. 2005; Li & Lu 2005). It is reasonable to predict that insulin‐induced increase in CTCF expression in ML‐1 cells was the result of increased CTCF mRNA; this supports the notion that CTCF is a mediator for regulating insulin‐induced cell proliferation here.
Involvement of CTCF in insulin‐mediated ML‐1 cell proliferation was studied by detecting the relationship between insulin‐induced activation of the Erk and Akt pathways, and CTCF expression. Our results reveal that application of PD98059 to inhibit MEK activation and wortmannin to inhibit the PI3K pathway was able to suppress insulin‐induced CTCF expression effectively. Furthermore, application of MEK and PI3K inhibitors also suppressed insulin‐induced proliferation. These results provide supportive evidence for the functional role of CTCF in mediating insulin‐stimulated proliferation. Interestingly, blockade of insulin‐induced activation of Erk and Akt pathways yielded different effects of these inhibitors in CTCF expression and cell proliferation; this is possibly because PD98059 effectively blocked phosphorylation of Erk without a cross effect on insulin‐induced Akt activation. However, application of wortmannin to inhibit PI3K blocked insulin‐induced activation of both Akt and Erk in these cells, as shown in Fig. 4. This may be explained by results of recent studies showing that there is a diverging point in insulin‐induced activation of the Ras‐Raf‐MEK‐Erk1/2, and the Akt pathways at the PI3K level, instead of at the receptor level, in malignant tumour cell lines, including those of BEL‐7402, SMMC‐7721 cell, MCF‐7 and LNCaP cells (Liu et al. 2006). In more specific studies, we used dominantly negative expression of Erk and Akt to provide important and reliable information that further supports that CTCF influence is located downstream of insulin‐induced Erk and Akt cascades and affects regulation of proliferation in ML‐1 cells (Fig. 3). Both approaches inhibit insulin‐induced signalling pathways, by using MEK and PI3K inhibitors and by using overexpression of dominantly negative Erk and Akt mutants to suppress insulin‐induced CTCF expression and proliferation. This reveals that CTCF functions as a downstream component of the Erk and Akt cascades mediating insulin‐stimulated proliferation of the cells. A further piece of evidence that supports the role CTCF has in regulating proliferation is that overexpression of CTCF significantly promoted ML‐1 cell proliferation in both the absence and the presence of insulin stimulation (Fig. 4), and that knockdown of endogenous CTCF mRNA using CTCF‐specific siRNA, remarkably diminished the mitogenic effect of insulin (Fig. 5). These results indicate that CTCF indeed plays a regulatory role in insulin‐stimulated ML‐1 cell proliferation.
It has been suggested that CTCF controls many genes that regulate cell proliferation, including p16INK4α, PIM‐1, PLK, p53, p27, E‐cadherin, E2F1, TERT and IGF2 (Klenova et al. 2001). In corneal epithelial cells, CTCF activity is required for suppression of Pax6 transcription during EGF‐induced proliferation because Pax6 regulates differentiation in these cells (Li & Lu 2005). In WEHI‐231 B cells, expression of CTCF is increased during induction of apoptosis by B‐cell receptor ligation, inhibits the PI3K pathway and the treatment of TGF‐β (Qi et al. 2003). In K562 cells, stable constitutive expression of CTCF leads to delay in cell cycle initiation without triggering apoptosis (Torrano et al. 2005). Although they are all haematopoietic cells, ML‐1 cells are earlier stage progenitor cells in the myeloid linage; however, unlike ML‐1 cells, WEHI‐231 B and K562 cells are more differentiated cell subtypes. Our findings in the present study provide new evidence for the multiple functional roles of CTCF in controlling cell population growth in a cell type‐dependent manner. A further explanation for the diverse role of CTCF involving cell growth control in different cell types is that it may be due to the varied involvement of molecular mechanisms in these cells. CTCF is usually studied at the transcriptional regulation level as it is a transcription factor. It has been suggested that in WEHI‐231 B cells, CTCF induces growth arrest and apoptosis by enhancing expression of ARF, p53, p21 and p27, and by inhibiting c‐Myc expression (Qi et al. 2003). CTCF involvement in K562 cell population growth is due to an alternative mechanism as K562 cells are p53‐, ARF‐ and p16INK4A‐negative cells (Torrano et al. 2005). However, it has been shown in previous studies, by changing its expression, that the effect of c‐Myc on regulation of ML‐1 cell proliferation and differentiation is not at the transcription level. Instead, the effect of c‐Myc is determined by re‐distribution of c‐Myc protein in subcellular compartments (Craig et al. 1993). A recent study consistent with our results reveals that CTCF is found during mitosis in the centrosome and midbodies, indicating that CTCF regulates cell proliferation by interacting with specific proteins (Zhang et al. 2004). Our results show for the first time that CTCF regulates insulin‐induced proliferation in ML‐1 cells, and provide the detailed mechanism of CTCF involving insulin‐induced mitogenic signalling pathways. We believe that the results presented in here provide important clues for further exploration of the mechanism that explains the effect of CTCF on regulation of growth factor‐induced cell proliferation.
ACKNOWLEDGEMENTS
This study was supported by National Institutes of Health grants EY12953 and EY15282 to Luo Lu.
REFERENCES
- Bell AC, Felsenfeld G (2000) Methylation of a CTCF‐dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. [DOI] [PubMed] [Google Scholar]
- Bravo R, Macdonald‐Bravo H (1987) Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: association with DNA replication sites. J. Cell Biol. 105, 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan KD (1976) The role of enteric hormones in glucose homeostasis. Horm. Metab. Res. Suppl. 6, 80–84. [PubMed] [Google Scholar]
- Carel K, Kummer JL, Schubert C, Leitner W, Heidenreich KA, Draznin B (1996) Insulin stimulates mitogen‐activated protein kinase by a Ras‐independent pathway in 3T3‐L1 adipocytes. J. Biol. Chem. 271, 30625–30630. [DOI] [PubMed] [Google Scholar]
- Craig RW, Buchan HL, Civin CI, Kastan MB (1993) Altered cytoplasmic/nuclear distribution of the c‐Myc protein in differentiating ML‐1 human myeloid leukemia cells. Cell Growth Differ. 4, 349–357. [PubMed] [Google Scholar]
- Dragovich T, Rudin CM, Thompson CB (1998) Signal transduction pathways that regulate cell survival and cell death. Oncogene 17, 3207–3213. [DOI] [PubMed] [Google Scholar]
- Duckworth BC, Cantley LC (1997) Conditional inhibition of the mitogen‐activated protein kinase cascade by wortmannin. Dependence on signal strength. J. Biol. Chem. 272, 27665–27670. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR (1995) A synthetic inhibitor of the mitogen‐activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 92, 7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KL, Davie JR (2003) The many roles of the transcriptional regulator CTCF. Biochem. Cell Biol. 81, 161–167. [DOI] [PubMed] [Google Scholar]
- Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV (1996) An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c‐myc oncogenes. Mol. Cell. Biol. 16, 2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova GN, Lindblom A, Meincke LJ, Klenova EM, Neiman PE, Collins SJ, Doggett NA, Lobanenkov VV (1998) A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes Chromosomes Cancer 22, 26–36. [PubMed] [Google Scholar]
- Filippova GN, Qi CF, Ulmer JE, Moore JM, Ward MD, Hu YJ, Loukinov DI, Pugacheva EM, Klenova EM, Grundy PE, Feinberg AP, Cleton‐Jansen AM, Moerland EW, Cornelisse CJ, Suzuki H, Komiya A, Lindblom A, Dorion‐Bonnet F, Neiman PE, Morse HC 3rd, Collins SJ, Lobanenkov VV (2002) Tumor‐associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA‐binding specificity. Cancer Res. 62, 48–52. [PubMed] [Google Scholar]
- Guo TB, Lu J, Li T, Lu Z, Xu G, Xu M, Lu L, Dai W (2005) Insulin‐activated, K+‐channel‐sensitive Akt pathway is primary mediator of ML‐1 cell proliferation. Am. J. Physiol. Cell Physiol. 289, C257–C263. [DOI] [PubMed] [Google Scholar]
- Han R, Tsui S, Smith TJ (2002) Up‐regulation of prostaglandin E2 synthesis by interleukin‐1beta in human orbital fibroblasts involves coordinate induction of prostaglandin‐endoperoxide H synthase‐2 and glutathione‐dependent prostaglandin E2 synthase expression. J. Biol. Chem. 277, 16355–16364. [DOI] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM (2000) CTCF mediates methylation‐sensitive enhancer‐blocking activity at the H19/Igf2 locus. Nature 405, 486–489. [DOI] [PubMed] [Google Scholar]
- Holmgren C, Kanduri C, Dell G, Ward A, Mukhopadhya R, Kanduri M, Lobanenkov V, Ohlsson R (2001) CpG methylation regulates the Igf2/H19 insulator. Curr. Biol. 11, 1128–1130. [DOI] [PubMed] [Google Scholar]
- Klenova EM, Chernukhin IV, El‐Kady A, Lee RE, Pugacheva EM, Loukinov DI, Goodwin GH, Delgado D, Filippova GN, Leon J, Morse HC 3rd, Neiman PE, Lobanenkov VV (2001) Functional phosphorylation sites in the C‐terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol Cell. Biol. 21, 2221–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova E, Ohlsson R (2005) Poly (ADP‐ribosyl) ation and epigenetics. Is CTCF PARt of the plot? Cell Cycle 4, 96–101. [DOI] [PubMed] [Google Scholar]
- Kohne AC, Baniahmad A, Renkawitz R (1993) NeP1. A ubiquitous transcription factor synergizes with v‐ERBA in transcriptional silencing. J. Mol. Biol. 232, 747–755. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 114, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Li T, Lu L (2005) Epidermal growth factor‐induced proliferation requires down‐regulation of Pax6 in corneal epithelial cells. J. Biol. Chem. 280, 12988–12995. [DOI] [PubMed] [Google Scholar]
- Li T, Lu Z, Lu L (2004) Regulation of eye development by transcription control of CCCTC binding factor (CTCF). J. Biol. Chem. 279, 27575–27583. [DOI] [PubMed] [Google Scholar]
- Liu H, Kublaoui B, Pilch PF, Lee J (2000) Insulin activation of mitogen‐activated protein (MAP) kinase and Akt is phosphatidylinositol 3‐kinase‐dependent in rat adipocytes. Biochem. Biophys. Res. Commun. 274, 845–851. [DOI] [PubMed] [Google Scholar]
- Liu L, Xie Y, Lou L (2006) PI3K is required for insulin‐stimulated but not EGF‐stimulated ERK1/2 activation. Eur J. Cell Biol. 85, 367–374. [DOI] [PubMed] [Google Scholar]
- Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH (1990) A novel sequence‐specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC‐motif in the 5′‐flanking sequence of the chicken c‐myc gene. Oncogene 5, 1743–1753. [PubMed] [Google Scholar]
- Lu J, Lu Z, Reinach PS, Zhang J, Dai W, Lu L, Xu M (2006) TGF‐beta2 inhibits AKT activation and FGF‐2‐induced corneal endothelial cell proliferation. Exp. Cell Res. 312, 3631–3640. [DOI] [PubMed] [Google Scholar]
- Lu L, Yang T, Markakis D, Guggino WB, Craig RW (1993) Alterations in a voltage‐gated K+ current during the differentiation of ML‐1 human myeloblastic leukemia cells. J. Membr. Biol. 132, 267–274. [DOI] [PubMed] [Google Scholar]
- Qi CF, Martensson A, Mattioli M, Dalla‐Favera R, Lobanenkov VV, Morse HC, 3rd (2003) CTCF functions as a critical regulator of cell‐cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc. Natl. Acad. Sci. USA 100, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko JE, Klenova EM, Leon J, Filippova GN, Loukinov DI, Vatolin S, Robinson AF, Hu YJ, Ulmer J, Ward MD, Pugacheva EM, Neiman PE, Morse HC 3rd, Collins SJ, Lobanenkov VV (2001) Cell growth inhibition by the multifunctional multivalent zinc‐finger factor CTCF. Cancer Res. 61, 6002–6007. [PubMed] [Google Scholar]
- Sebolt‐Leopold JS (2004) MEK inhibitors: a therapeutic approach to targeting the Ras‐MAP kinase pathway in tumors. Curr. Pharm. Des. 10, 1907–1914. [DOI] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, White ER, McCubrey JA (2004) Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF‐1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle 3, 372–379. [PubMed] [Google Scholar]
- Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA (2004) JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR‐ABL in cell cycle progression and leukemogenesis. Leukemia 18, 189–218. [DOI] [PubMed] [Google Scholar]
- Strobl JS, Wonderlin WF, Flynn DC (1995) Mitogenic signal transduction in human breast cancer cells. Gen. Pharmacol. 26, 1643–1649. [DOI] [PubMed] [Google Scholar]
- Torrano V, Chernukhin I, Docquier F, D’Arcy V, Leon J, Klenova E, Delgado MD (2005) CTCF regulates growth and erythroid differentiation of human myeloid leukemia cells. J. Biol. Chem. 280, 28152–28161. [DOI] [PubMed] [Google Scholar]
- Vostrov AA, Quitschke WW (1997) The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta‐protein precursor promoter. Evidence for a role in transcriptional activation. J. Biol. Chem. 272, 33353–33359. [DOI] [PubMed] [Google Scholar]
- Wang L, Xu B, White ER, Lu L (1997) Growth factor‐mediated K+ channel activity associated with human myeloblastic ML‐1 cell proliferation. Am. J. Physiol. 273, C1657–C1665. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhou P, Craig RW, Lu L (1999) Protection from cell death by mcl‐1 is mediated by membrane hyperpolarization induced by K (+) channel activation. J. Membr. Biol. 172, 113–120. [DOI] [PubMed] [Google Scholar]
- Wu D, Li T, Lu Z, Dai W, Xu M, Lu L (2006) Effect of CTCF‐binding motif on regulation of PAX6 transcription. Invest Ophthalmol. Vis. Sci. 47, 2422–2429. [DOI] [PubMed] [Google Scholar]
- Xu D, Wang L, Dai W, Lu L (1999) A requirement for K+‐channel activity in growth factor‐mediated extracellular signal‐regulated kinase activation in human myeloblastic leukemia ML‐1 cells. Blood 94, 139–145. [PubMed] [Google Scholar]
- Xu B, Wilson BA, Lu L (1996a) Induction of human myeloblastic ML‐1 cell G1 arrest by suppression of K+ channel activity. Am. J. Physiol. 271, C2037–C2044. [DOI] [PubMed] [Google Scholar]
- Xu B, Wilson BA, Lu L (1996b) Induction of human myeloblastic ML‐1 cell G1 arrest by suppression of K+ channel activity. Am. J. Physiol. 271, C2037–C2044. [DOI] [PubMed] [Google Scholar]
- Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, Farrar D, Tavoosidana G, Mukhopadhyay R, Kanduri C, Oshimura M, Feinberg AP, Lobanenkov V, Klenova E, Ohlsson R (2004) Poly(ADP‐ribosyl)ation regulates CTCF‐dependent chromatin insulation. Nat. Genet. 36, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Zhang R, Burke LJ, Rasko JE, Lobanenkov V, Renkawitz R (2004) Dynamic association of the mammalian insulator protein CTCF with centrosomes and the midbody. Exp. Cell Res. 294, 86–93. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18. [DOI] [PubMed] [Google Scholar]