

Abstract
Objectives: Erythroid differentiation is a dynamic process in which a pluripotent stem cell undergoes a series of developmental changes that commit it to a specific lineage. These alterations involve changes in gene expression profiles. In this study, gene expression profiles during differentiation of human erythroid cells of a normal blood donor were evaluated using SAGE.
Materials and methods: Global gene expression was evaluated in cells collected immediately before addition of erythropoietin (0 h) and 192 and 336 h after addition of this hormone. Real‐time PCR was used to evaluate activation of differentially expressed genes.
Results: The data indicate that global aspects of the transcriptome were similar during differentiation of the majority of the genes and that a relatively small set of genes is probably involved in modification of erythroid cells during differentiation. We have identified 93 differentially expressed genes during erythroid development, and expression of some of these was confirmed by qPCR. Various genes including EYA3, ERH, HES6, TIMELESS and TRIB3 were found to be homologous to those of Drosophila melanogaster and here are described for the first time during erythroid development. An important and unique carboxypeptidase inhibitor described in mammalians, LXN, was also identified.
Conclusions: The results of this study amplify previously published data and may contribute to comprehension of erythroid differentiation and identification of new target genes involved in some erythroid concerning diseases.
Introduction
Haematopoiesis is maintained by pluripotent, long‐term repopulating stem cells that generate progenitors capable of differentiating into all three haematopoietic lineages. Erythroid cell maturation, known as erythropoiesis, is mediated by a combination of regulatory proteins acting in concert. These direct development of progenitor cells into mature erythrocytes, which are one of the most highly specialized cell types in the human body (1, 2). This process can be reproduced in an in vitro study using a two‐phase liquid culture. Using this technique, stem cells differentiate into erythroid cells by addition of the hormone erythropoietin (EPO) in the culture (3).
Extensive studies have led to a considerable understanding of the cellular and molecular control of haemoglobin production during red blood cell differentiation (4, 5, 6, 7); however, identification of the genes expressed as part of the erythroid differentiation programme remains an important goal because of the insights that these data will bring to erythrocyte biology and disease (8). One of the first studies evaluating gene expression in human erythroid cells was carried out by Gubin et al. (9). These authors made a subtractive library before and after addition of erythropoietin in a two‐phase liquid culture, and obtained a transcriptional profile of genes arising only in response to EPO. Following this study, several related ones on similar themes evaluated global gene expression in haematopoietic stem cells (10, 11, 12, 13) and in reticulocytes (14, 15).
Using a microarray strategy, Komor et al. (16) evaluated gene expression during differentiation of erythroid cells, megakaryocytes and platelets. This work identified several genes that were differentially expressed during differentiation of cells. However, during microarray analysis, knowledge of presence and sequence of genes to be analysed is required and, thus, only genes spotted on slides are studied, making it difficult to find new genes not evaluated in the analysis (17).
Although several studies have been performed on haematopoietic cells, global gene expression during erythroid differentiation has been poorly evaluated. As such, identification of all genes expressed as part of the erythroid differentiation programme remains an important goal (8).
Here, we report global gene expression during differentiation of human erythroid cells from a normal blood donor, in a two‐phase liquid culture, using Serial Analysis of Gene Expression (SAGE) (18). Global gene expression was evaluated in cells collected immediately before addition of erythropoietin (0 h) and 192 and 336 h after addition of this hormone. We identified 93 differentially expressed genes and development and expression of some of these genes was confirmed by qPCR.
Our data amplify previously published research and will contribute to understanding the pattern of gene expression during erythroid differentiation. In addition, these results contribute to the comprehension of erythroid differentiation and identification of new target genes involved in haematopoietic diseases.
Materials and methods
Erythroid cell cultures
Blood from normal volunteers was cultured using a two‐phase liquid culture procedure, as described previously (3). Briefly, mononuclear cells were isolated from peripheral blood samples by centrifugation over a Ficoll‐Hypaque gradient and cultured for 7 days (phase I) in IMDM medium (Invitrogen, Rockville, MS, USA) supplemented with 20% foetal calf serum (Invitrogen), 1 μg/ml cyclosporin A (Sandoz, Holzkirchen, Germany) and 10% conditioned medium, collected from culture of the human bladder carcinoma 5637 cell line. Cells were incubated at 37 °C in an atmosphere of 5% CO2 and 92% extra humidity. After 7 days, non‐adherent cells were harvested and re‐cultured in phase II medium, IMDM supplemented with 30% foetal calf serum (Invitrogen), 1% deionized bovine serum albumin (BSA; Sigma, St Louis, MO, USA), 10−5 m 2‐mercaptoethanol (Sigma), 1.5 nmol/l glutamine (Invitrogen), 300 μg iron‐saturated transferrin (Sigma), 10−6 m dexamethasone, 5 ng/ml human stem cell factor (SCF; Calbiochem, Darmstadt, Germany), 1 U/ml human recombinant erythropoietin (Cilag, Beerse, Belgium), 2.5 μg/ml funzigone (Invitrogen), 50 μg/ml streptomycin (Invitrogen) and 25 μg/ml glutamicin (Invitrogen). Cell samples were collected from phase II cultures at 0, 192 and 336 h after erythropoietin addition. Cell numbers and viability were determined by trypan blue exclusion. Samples of 5 × 106 cells were pelleted and resuspended in Trizol (Invitrogen) and stored at −80 °C for total RNA extraction and cDNA synthesis. For morphological analyses of cell differentiation stages, cytospin slides were prepared and stained with Leishman’s stain before examination using an Eclipse E‐600 microscope (Nikon, Tokyo, Japan) with Image Pro‐Express 4.0 software (Media Cybernetic, Bethesda, MD, USA).
RNA extraction
Total RNA was extracted with TRIzol reagent (Invitrogen, Rockville, MS, USA), according to the manufacturer’s protocol. Samples were quantified using a NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies Inc, Wilmington, DE, USA).
SAGE libraries and data analysis
Libraries were constructed using the I‐SAGE kit (Invitrogen) with Nla III enzyme, as described by the manufacturer. To produce libraries, 10 μg of total RNA was prepared. Sequencing was carried out in a Dynamic ET Terminator cycle sequencer (GE Healthcare, Uppsala, Sweden) and MEGA‐BACE automated DNA sequencer (Amersham Pharmacia, Bucks, UK). Vector sequences were trimmed with Phred/Phrap software. Automatic tag detection and differential gene expression analyses were performed using eSAGE software v1.2 (19). Only tags presenting P < 0.01 and fold ≥10 between comparisons were considered to be differently expressed. Data bank ‘Best Gene for a tag’, from SAGEGenie, CGAP (http://cgap.nci.nih.gov/SAGE), downloaded on April 2007, was used for tag‐to‐gene mapping. According to their identification, tags were further classified as ‘no match’ (no correspondence found in the data bank), ‘known genes’ or ‘putative genes/proteins’, including ESTs (expressed sequence tags), ORFs (open reading frames), cDNA clones and hypothetical proteins. Functional classification of transcripts was performed according to Gene Ontology Consortium criteria (http://www.geneontology.org). Hierarchical clustering analysis by Spearman’s confidence correlation was used to identify gene clusters. The separation ratio was set at 0.5.
Quantitative real time polymerase chain reaction
RNA samples were subjected to DNAse I treatment (Invitrogen) and reverse transcription using SuperScript III (Invitrogen). Primers were designed using PrimerExpressTM programme (Applied Biosystems, Foster City, CA, USA) (Table S1). Ideal concentration for use was determined for each pair of primers and amplification efficiency was calculated according to the equation E (−1/slope), to confirm accuracy and reproducibility of the reactions (Table S1). Amplification specificity was verified by running a dissociation protocol. Quantitative real time polymerase chain reaction (qRT‐PCRs) were performed in duplicate, using 12.5 μl SYBR Green Master Mix (Applied Biosystems), 25 ng cDNA and ideal quantities of each primer, in a final volume of 25 μl. Samples were run in MicroAmp Optical 96‐well plates (Applied Biosystems) in a 5700 Sequence Detection System (Applied Biosystems). To validate SAGE profiles, GAPDH was used as a reference gene. Gene expressions in SAGE samples are presented as mean ± SEM.
Results
We performed a large‐scale gene expression study of erythroid differentiation using SAGE. Samples of cultures were collected at 0 (SAGE‐0H), 192 (SAGE‐192H) and 336 (SAGE‐336H) hours after erythropoietin addition and typical morphology was detectable during cell differentiation (Fig. S1). Cells were collected at these points and their RNA was prepared for SAGE library construction.
After sequencing and tag extraction, 30 512 tags for SAGE‐0H, 30 117 tags for SAGE‐192H and 30 189 for SAGE‐336H profiles were generated, representing 12 026, 11 709 and 11 337 unique tags respectively. Identification of tags in the libraries demonstrated that 28%, 26.2% and 26.7% respectively, had no correspondence in the data bank (no matches) and could represent novel genes. A complete list of tags is available for download at http://www.lge.ibi.unicamp.br/~anderf.
To investigate reliability of the profiles designed by SAGE, we arbitrarily selected 18 genes to be studied by qRT‐PCR in the same samples used to generate the libraries. Both techniques were consistent in identifying expression of 17 of 18 genes studied (HBA, HBB, HBG, RNAseI, TIMP1, TIMP2, LYZ, B2M, MMP9, NFE2, AHSP, S100A8, S100A9, BCR, GATA1, PFN1 and CEBPB). Only expression of the STAT5A gene demonstrated discordant results between the techniques (Fig. 1).
Figure 1.
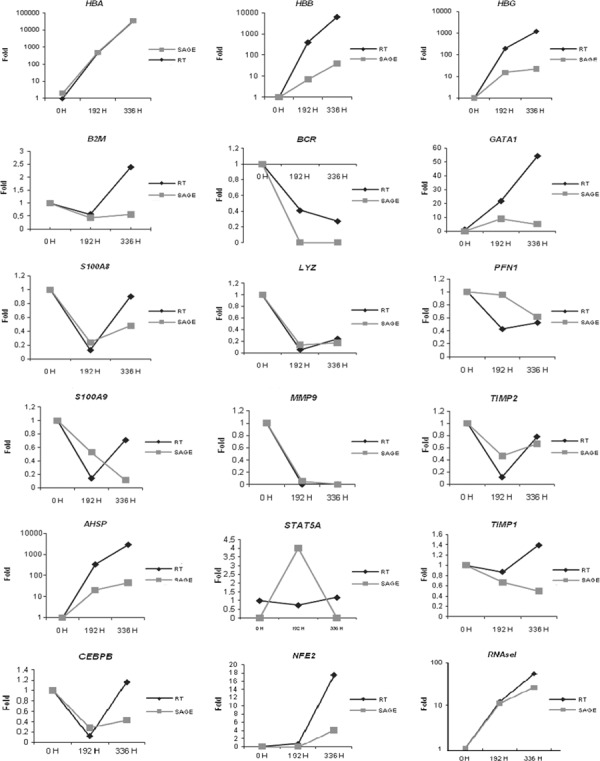
Validation of SAGE technique – eighteen genes arbitrarily selected for study by qRT‐PCR in the same samples used to generate libraries. Results showed a 95% concordance (17 of 18).
For subsequent analysis, only tags present at least five times in one of the libraries were considered (20, 21, 22). Using these data, expression profiles of libraries were compared using the Gene Ontology Consortium Database. Most abundant genes expressed at the beginning of differentiation were found to be related to various pathways including immune response, lysozyme activity, iron homeostasis, cell proliferation and apoptosis. At 192 h after erythropoietin addition, the most abundant genes were related to ribosomal activity, reflecting intense and dynamic protein production in this intermediate phase. At the end of differentiation we observed high expression of genes involved in haemoglobin synthesis, such as HBA, HBB and HBG, and these represent the most expressed proteins in reticulocytes and in red cells. Summaries of the most expressed genes in each library are described in Table 1.
Table 1.
The most expressed genes (more than 100 copies) in 0H, 192 and 336H library respectively
| Tag | 0H | 192H | 336H | Hs | Symbol | Description | Ontology |
|---|---|---|---|---|---|---|---|
| GGGCATCTCT | 203 | 32 | 39 | Hs.520048 | HLA‐DRA | Major histocompatibility complex, class II, DR alpha | Immune response |
| ATCAAGAATC | 238 | 26 | 27 | Hs.14623 | IFI30 | Interferon, gamma‐inducible protein 30 | Lysozyme activity |
| ATGTAAAAAA | 253 | 37 | 45 | Hs.524579 | LYZ | Lysozyme (renal amyloidosis) | Lysozyme activity |
| GTTGTGGTTA | 304 | 131 | 166 | Hs.534255 | B2M | Beta‐2‐microglobulin | Immune response |
| CCCTGGGTTC | 361 | 115 | 156 | Hs.433670 | FTL | Ferritin, light polypeptide | Cellular iron ion homeostasis |
| TTGGGGTTTC | 386 | 274 | 458 | Hs.524910 | FTH1 | Ferritin, heavy polypeptide 1 | Cellular iron ion homeostasis |
| GTTCACATTA | 420 | 99 | 116 | Hs.436568 | CD74 | CD74 molecule, major histocompatibility complex, class II invariant chain | Cell proliferation/negative regulation of apoptosis/signal transduction |
| GAAATACAGT | 648 | 121 | 148 | Hs.67201 | NT5C | 5′, 3′‐nucleotidase, cytosolic | 5′‐nucleotidase activity |
| GGATTTGGCC | 174 | 152 | 101 | Hs.437594 | TSPAN4 | Tetraspanin 4 | Membrane fraction |
| CACAAACGGT | 140 | 66 | 71 | Hs.504517 | TSPAN9 | Tetraspanin 9 | Membrane fraction |
| TTGGTGAAGG | 169 | 18 | 45 | Hs.522584 | TMSB4X | Thymosin, beta 4, X‐linked | Cytoskeleton organization and biogenesis |
| CTGACCTGTG | 128 | 26 | 50 | Hs.77961 | HLA‐B | Major histocompatibility complex, class I, B | Immune response |
| CCACTGCACT | 139 | 22 | 46 | Hs.107003 | CCNB1IP1 | Cyclin B1 interacting protein 1 | Apoptosis |
| ACATTCTTTT | 103 | 21 | 41 | Hs.190495 | GPNMB | Glycoprotein (transmembrane) nmb | Negative regulation of cell proliferation |
| AGGGCTTCCA | 105 | 99 | 43 | Hs.534404 | RPL10 | Ribosomal protein L10 | Ribosomal subunit |
| GTGAAACCCC | 107 | 62 | 86 | Hs.590913 | PAFAH2 | Platelet‐activating factor acetylhydrolase 2, 40 kDa | Phospholipid binding |
| AGTTTCTTGT | 108 | 33 | 45 | Hs.647419 | CD68 | CD68 molecule | Transmembrane glycoprotein |
| CCTGTAATCC | 108 | 34 | 50 | Hs.591920 | NT5C2 | 5′‐nucleotidase, cytosolic II | 5′‐nucleotidase activity |
| CCCATCGTCC | 192 | 279 | 102 | Hs.559716 | Transcribed locus, weakly similar to XP_220207.3 similar to serine/arginine repetitive matrix 2 [Rattus norvegicus] | RNA/protein binding | |
| GAGGGAGTTT | 151 | 191 | 98 | Hs.523463 | RPL27A | Ribosomal protein L27a | Ribosomal subunit |
| GAAAAATGGT | 80 | 183 | 80 | Hs.449909 | RPSA | Ribosomal protein SA | Ribosomal subunit |
| GCATAATAGG | 139 | 178 | 89 | Hs.381123 | RPL21 | Ribosomal protein L21 | Ribosomal subunit |
| CTGGGTTAAT | 96 | 172 | 135 | Hs.438429 | RPS19 | Ribosomal protein S19 | Ribosomal subunit |
| ATAATTCTTT | 153 | 174 | 120 | Hs.156367 | RPS29 | Ribosomal protein S29 | Ribosomal subunit |
| GGGCTGGGGT | 85 | 163 | 72 | Hs.425125 | RPL29 | Ribosomal protein L29 | Ribosomal subunit |
| TTGGTCCTCT | 116 | 149 | 105 | Hs.632703 | RPL41 | Ribosomal protein L41 | Ribosomal subunit |
| TTCAATAAAA | 92 | 145 | 71 | Hs.356502 | RPLP1 | Ribosomal protein, large, P1 | Ribosomal subunit |
| CAATAAATGT | 91 | 141 | 71 | Hs.558601 | RPL37 | Ribosomal protein L37 | Ribosomal subunit |
| TGCACGTTTT | 92 | 141 | 70 | Hs.265174 | RPL32 | Ribosomal protein L32 | Ribosomal subunit |
| TGTGTTGAGA | 125 | 135 | 80 | Hs.644639 | EEF1A1 | Eukaryotic translation elongation factor 1 alpha 1 | Translational elongation/GTPase activity |
| TAATAAAGGT | 72 | 130 | 65 | Hs.512675 | RPS8 | Ribosomal protein S8 | Ribosomal subunit |
| TGTACCTGTA | 43 | 111 | 61 | Hs.524390 | TUBA3 | Tubulin, alpha 3 | Microtubule‐based movement/GTPase activity |
| GCAAGAAAGT | 36 | 253 | 1391 | Hs.523443 | HBB | Haemoglobin, beta | Haemoglobin synthesis |
| CTTCTTGCCC | 20 | 147 | 1264 | Hs.449630 | HBA1 | Haemoglobin, alpha 1 | Haemoglobin synthesis |
| CCCAACGCGC | 7 | 26 | 473 | Hs.449630 | Haemoglobin, alpha 1 | Haemoglobin synthesis | |
| TAGGTTGTCT | 198 | 191 | 211 | Hs.374596 | TPT1 | Tumour protein, translationally controlled 1 | Anti‐apoptosis/cellular calcium ion homeostasis |
| ATGCAGAGCT | 4 | 120 | 178 | Hs.295459 | HBG1 | Haemoglobin, gamma A | Haemoglobin synthesis |
| ATTCAGAGCT | 2 | 105 | 154 | Hs.295459 | Haemoglobin, gamma A | Haemoglobin synthesis | |
| TTAACCCCTC | 5 | 56 | 130 | Hs.78224 | RNASE1 | Ribonuclease, RNase A family, 1 (pancreatic) | RNA binding/endonuclease activity |
Differential gene expression between the libraries was further analysed using P < 0.01 criterium, and fold higher than 10 to select tags that presented differential expression with a statistically significant level. Ninety‐three genes were identified and these were hierarchically clustered by Spearman’s confidence correlation, with a separation ratio set at 0.5. We identified 32 up‐regulated genes in the 0H library, 29 in 196H and 32 in 336H (Fig. 2). Tag number found for each gene is displayed in Table 2. Differentially expressed genes were categorized by Molecular Function and Biological Process using the gene ontology consortium. At the beginning of differentiation (OH), processes such as cell adhesion, cell proliferation, cell development and apoptosis regulation were found to be up‐regulated. After 192 h of erythropoietin addition, processes like structural constituents of ribosomes, transcription factor activity and RNA polymerase II activity were up‐regulated. At the end of differentiation, these processes were down‐regulated and cells demonstrated restriction of expression of pathways, like transport, biosynthetic processes, oxygen binding pathways plus ion, tetrapyrrole, nucleotide, protein and cofactors (Fig. S2).
Figure 2.
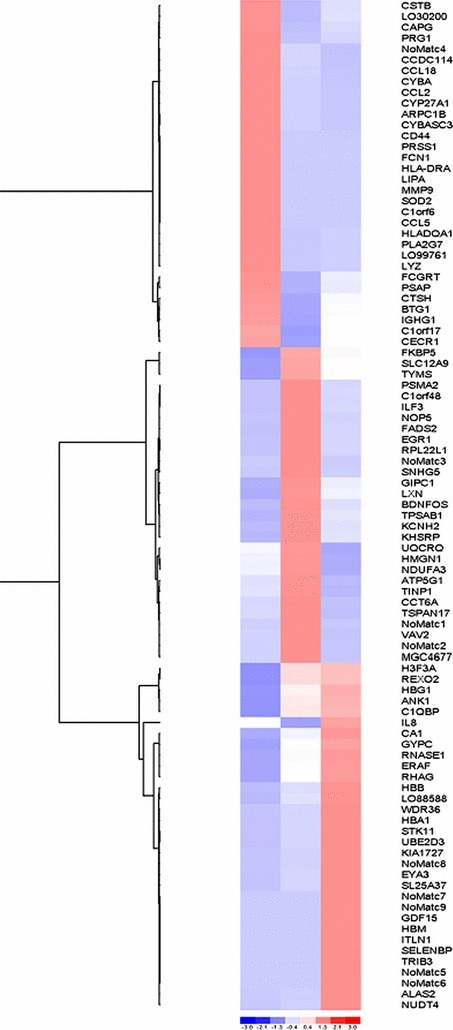
Cluster analysis of differentially expressed genes associated with erythroid differentiation. Three clusters were found according to up‐regulation of each stage of development. Colour code: blue, low expression; red, high expression. Intensity of colour reflects reliability of expression data.
Table 2.
Differentially expressed genes found during erythroid development
| Gene symbol | Hs number | Description | Number of tags | ||
|---|---|---|---|---|---|
| 0H | 192H | 336H | |||
| No Match 1 | 5 | 107 | 0 | ||
| No Match 2 | 1 | 14 | 0 | ||
| No Match 3 | 0 | 10 | 0 | ||
| No Match 4 | Hs.605719 | CDNA clone IMAGE:3927515 | 10 | 1 | 0 |
| No Match 5 | Unclustered ESTs | 0 | 0 | 25 | |
| No Match 6 | Hs.623908 | Transcribed locus, strongly similar to XP_001072910.1 similar to Oligodendrocyte transcription factor 3 (Oligo3) ‐(Oligodendrocyte‐specific bHLH transcription factor 3) (Basic helix‐loop‐helix domain‐containing class B protein 7) [Rattus norvegicus] | 0 | 0 | 15 |
| No Match 7 | 0 | 0 | 13 | ||
| No Match 8 | Unclustered ESTs | 0 | 3 | 36 | |
| No Match 9 | 0 | 0 | 11 | ||
| ALAS2 | Hs.522666 | Aminolevulinate, delta‐, synthase 2 (sideroblastic/hypochromic anaemia) | 0 | 2 | 71 |
| ANK1 | Hs.491558 | Ankyrin 1, erythrocytic | 0 | 18 | 25 |
| ARPC1B | Hs.489284 | Actin‐related protein 2/3 complex, subunit 1B, 41 kDa | 18 | 1 | 0 |
| ATP5G1 | Hs.80986 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit C1 (subunit 9) | 2 | 10 | 0 |
| BDNFOS | Hs.577179 | Brain‐derived neurotrophic factor opposite strand | 2 | 20 | 5 |
| BTG1 | Hs.255935 | B‐cell translocation gene 1, anti‐proliferative | 38 | 5 | 19 |
| C11orf17 | Hs.131180 | Chromosome 11 open reading frame 17 | 10 | 1 | 6 |
| C19orf48 | Hs.256301 | Chromosome 19 open reading frame 48 | 0 | 9 | 1 |
| C19orf6 | Hs.515003 | Chromosome 19 open reading frame 6 | 10 | 0 | 0 |
| C1QBP | Hs.555866 | Complement component 1, q subcomponent binding protein | 3 | 12 | 15 |
| CA1 | Hs.23118 | Carbonic anhydrase I | 0 | 21 | 61 |
| CAPG | Hs.516155 | Capping protein (actin filament), gelsolin‐like | 41 | 3 | 8 |
| CCDC114 | Hs.112645 | Coiled‐coil domain containing 114 | 11 | 1 | 0 |
| CCL18 | Hs.143961 | Chemokine (C‐C motif) ligand 18 (pulmonary and activation‐regulated) | 12 | 1 | 0 |
| CCL2 | Hs.303649 | Chemokine (C‐C motif) ligand 2 | 17 | 1 | 0 |
| CCL5 | Hs.514821 | Chemokine (C‐C motif) ligand 5 | 11 | 0 | 0 |
| CCT6A | Hs.82916 | Chaperonin‐containing TCP1, subunit 6A (zeta 1) | 2 | 10 | 1 |
| CD44 | Hs.502328 | CD44 molecule (Indian blood group) | 10 | 0 | 0 |
| CECR1 | Hs.170310 | Cat eye syndrome chromosome region, candidate 1 | 17 | 1 | 9 |
| CSTB | Hs.695 | Cystatin B (stefin B) | 53 | 5 | 13 |
| CTSH | Hs.148641 | Cathepsin H | 20 | 0 | 8 |
| CYBA | Hs.513803 | Cytochrome b‐245, alpha polypeptide | 14 | 1 | 0 |
| CYBASC3 | Hs.22546 | Cytochrome b, ascorbate dependent 3 | 18 | 1 | 0 |
| CYP27A1 | Hs.516700 | Cytochrome P450, family 27, subfamily A, polypeptide 1 | 19 | 1 | 0 |
| EGR1 | Hs.326035 | Early growth response 1 | 0 | 11 | 1 |
| ERAF | Hs.274309 | Erythroid associated factor | 1 | 20 | 45 |
| EYA3 | Hs.185774 | Eyes absent homologue 3 (Drosophila) | 1 | 7 | 69 |
| FADS2 | Hs.502745 | Fatty acid desaturase 2 | 0 | 10 | 1 |
| FCGRT | Hs.111903 | Fc fragment of IgG, receptor, transporter, alpha | 21 | 4 | 9 |
| FCN1 | Hs.440898 | Ficolin (collagen/fibrinogen domain containing) 1 | 18 | 0 | 0 |
| FKBP5 | Hs.407190 | FK506 binding protein 5 | 1 | 19 | 12 |
| GDF15 | Hs.616962 | Growth differentiation factor 15 | 0 | 0 | 11 |
| GIPC1 | Hs.631639 | GIPC PDZ domain containing family, member 1 | 1 | 11 | 4 |
| GYPC | Hs.59138 | Glycophorin C (Gerbich blood group) | 0 | 10 | 20 |
| H3F3A | Hs.533624 | H3 histone, family 3A | 11 | 23 | 25 |
| HBA1 | Hs.449630 | Haemoglobin, alpha 1 | 0 | 176 | 1780 |
| HBB | Hs.523443 | Haemoglobin, beta | 36 | 270 | 1489 |
| HBG1 | Hs.295459 | Haemoglobin, gamma A | 6 | 225 | 332 |
| HBM | Hs.647389 | Haemoglobin, mu | 0 | 0 | 15 |
| HLA‐DQA1 | Hs.387679 | Major histocompatibility complex, class II, DQ alpha 1 | 39 | 3 | 5 |
| HLA‐DRA | Hs.520048 | Major histocompatibility complex, class II, DR alpha | 10 | 1 | 1 |
| HMGN1 | Hs.356285 | High‐mobility group nucleosome‐binding domain 1 | 5 | 13 | 1 |
| IGHG1 | Hs.510635 | Immunoglobulin heavy constant gamma 1 (G1m marker) | 11 | 2 | 6 |
| IL8 | Hs.443948 | Interleukin 8 | 42 | 11 | 73 |
| ILF3 | Hs.465885 | Interleukin enhancer binding factor 3, 90 kDa | 1 | 10 | 2 |
| ITLN1 | Hs.50813 | Intelectin 1 (galactofuranose binding) | 0 | 0 | 10 |
| KCNH2 | Hs.647099 | Potassium voltage‐gated channel, subfamily H (eag‐related), member 2 | 0 | 20 | 4 |
| KHSRP | Hs.646750 | KH‐type splicing regulatory protein (FUSE binding protein 2) | 1 | 10 | 3 |
| KIAA1727 | Hs.132629 | KIAA1727 protein | 0 | 1 | 13 |
| LIPA | Hs.643030 | Lipase A, lysosomal acid, cholesterol esterase (Wolman disease) | 10 | 1 | 1 |
| LOC388588 | Hs.22047 | Hypothetical gene supported by BC035379; BC042129 | 1 | 13 | 59 |
| LOC399761 | Hs.647203 | Hypothetical protein LOC399761 | 23 | 0 | 1 |
| LOC730200 | Hs.553015 | Hypothetical protein LOC730200 | 17 | 0 | 3 |
| LXN | Hs.478067 | Latexin | 0 | 15 | 5 |
| LYZ | Hs.524579 | Lysozyme (renal amyloidosis) | 280 | 38 | 49 |
| MGC4677 | Hs.446688 | Hypothetical protein MGC4677 | 1 | 15 | 0 |
| MMP9 | Hs.297413 | Matrix metallopeptidase 9 (gelatinase B, 92 kDa gelatinase, 92 kDa type IV collagenase) | 16 | 0 | 0 |
| NDUFA3 | Hs.198269 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3, 9 kDa | 3 | 9 | 0 |
| NOP5/NOP58 | Hs.471104 | Nucleolar protein NOP5/NOP58 | 2 | 15 | 3 |
| NUDT4 | Hs.591008 | Nudix (nucleoside diphosphate‐linked moiety X)‐type motif 4 | 0 | 3 | 57 |
| PLA2G7 | Hs.584823 | Phospholipase A2, group VII (platelet‐activating factor acetylhydrolase, plasma) | 30 | 1 | 2 |
| PRG1 | Hs.1908 | Proteoglycan 1, secretory granule | 12 | 1 | 2 |
| PRSS1 | Hs.622865 | Protease, serine, 1 (trypsin 1) | 23 | 1 | 1 |
| PSAP | Hs.523004 | Prosaposin (variant Gaucher disease and variant metachromatic leukodystrophy) | 111 | 14 | 41 |
| PSMA2 | Hs.333786 | Proteasome (prosome, macropain) subunit, alpha type, 2 | 1 | 10 | 2 |
| REXO2 | Hs.7527 | REX2, RNA exonuclease 2 homologue (S. cerevisiae) | 0 | 12 | 14 |
| RHAG | Hs.120950 | Rh‐associated glycoprotein | 0 | 12 | 28 |
| RNASE1 | Hs.78224 | Ribonuclease, RNase A family, 1 (pancreatic) | 5 | 56 | 130 |
| RPL22L1 | Hs.380933 | Ribosomal protein L22‐like 1 | 0 | 11 | 1 |
| SELENBP1 | Hs.632460 | Selenium‐binding protein 1 | 0 | 0 | 12 |
| SLC12A9 | Hs.521087 | Solute carrier family 12 (potassium/chloride transporters), member 9 | 2 | 24 | 14 |
| SLC25A37 | Hs.122514 | Solute carrier family 25, member 37 | 0 | 2 | 23 |
| SNHG5 | Hs.292457 | Small nucleolar RNA host gene (non‐protein coding) 5 | 3 | 27 | 4 |
| SOD2 | Hs.487046 | Superoxide dismutase 2, mitochondrial | 10 | 1 | 1 |
| STK11 | Hs.515005 | Serine/threonine kinase 11 | 0 | 1 | 10 |
| TINP1 | Hs.482526 | TGF beta‐inducible nuclear protein 1 | 3 | 10 | 1 |
| TPSAB1 | Hs.405479 | Tryptase alpha/beta 1 | 0 | 45 | 11 |
| TRIB3 | Hs.516826 | Tribbles homologue 3 (Drosophila) | 0 | 0 | 11 |
| TSPAN17 | Hs.532129 | Tetraspanin 17 | 2 | 9 | 1 |
| TYMS | Hs.592338 | Thymidylate synthetase | 0 | 13 | 7 |
| UBE2D3 | Hs.518773 | Ubiquitin‐conjugating enzyme E2D 3 (UBC4/5 homologue, yeast) | 0 | 1 | 10 |
| UQCRQ | Hs.146602 | Ubiquinol‐cytochrome c reductase, complex III subunit VII, 9.5 kDa | 4 | 9 | 1 |
| VAV2 | Hs.369921 | Vav 2 oncogene | 1 | 12 | 0 |
| WDR36 | Hs.533237 | WD repeat domain 36 | 0 | 3 | 32 |
Of the differentially expressed genes, we found several with homology to Drosophila melanogaster genes (Fig. 3). These genes have been identified in humans, however, most of them do not have any function yet described. We also found high expression of TIMELESS, HES6, EYA3, ERH and TRIB3 genes during the intermediate phase and at the end of differentiation.
Figure 3.
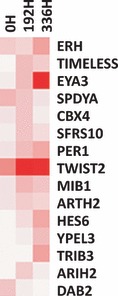
Cluster analysis of 15 differentially expressed genes homologous to D. melanogaster found during erythroid differentiation. These genes have been identified in humans; however, most of them do not have any described function. Genes HES6, EYA3, ERH and TRIB3 were found with high expression at the end of differentiation, while TIMELESS showed high expression in the intermediate phase. With the exception of ERH, expression of these genes were hardly observed at the beginning of differentiation. Intensity of colour reflects reliability of expression data.
To understand whether expressions of these genes are related to erythroid lineage expression, we evaluated them in further two‐phase liquid cultures (Fig. 4a) and in CD34+ culture (Fig. 4b). CD34+ cell culture was used as contamination with other cell types such as lymphocytes and monocytes/macrophages is lower than that seen in two‐phase culture and all cells are committed to the erythroid lineage. Results confirmed SAGE data in both cultures and demonstrated that probably, differences observed are related to erythroid lineage and not to other cell types. We also evaluated expression of LXN gene, the only known carboxypeptidase inhibitor in mammals (23), because its expression was observed only after the intermediate stage of differentiation and was lower at the end of differentiation.
Figure 4.
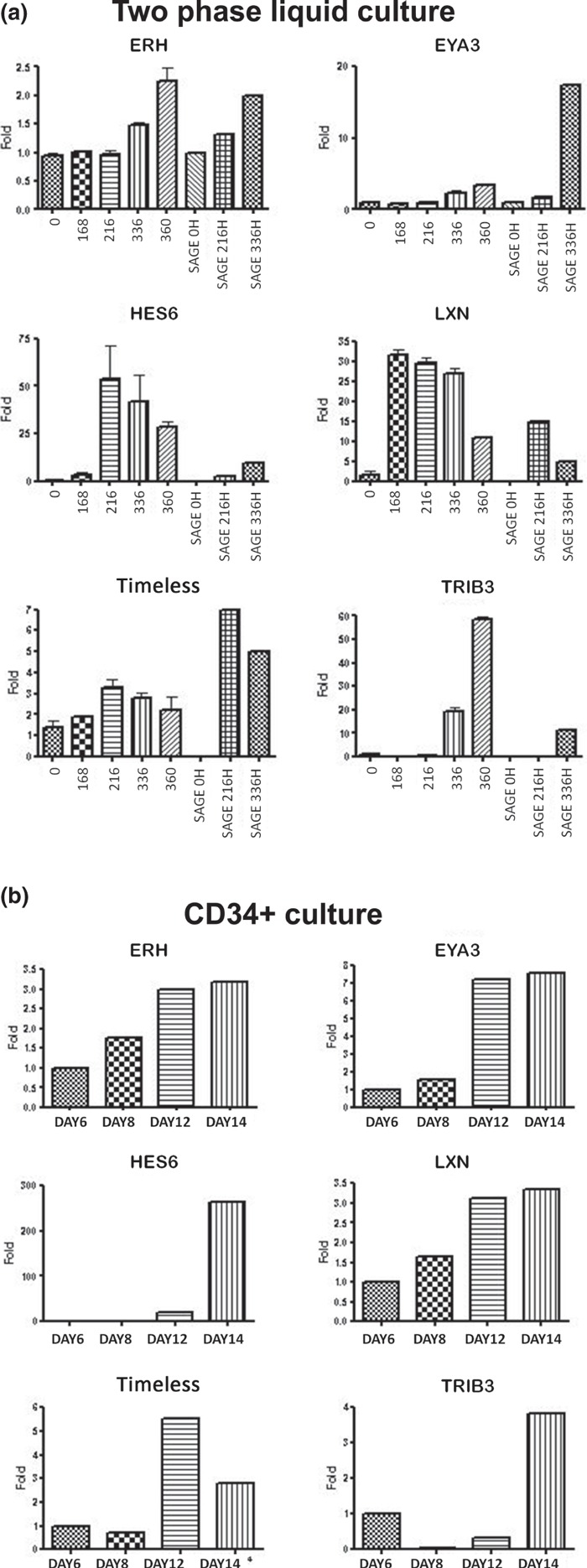
Gene expression of selected genes during erythroid differentiation. Gene expressions of six selected genes were evaluated by qPCR in three different two‐phase liquid cultures (a) and in a CD34+ culture (b). Expressions observed in both cultures are the same as those identified by SAGE analysis. The pattern observed in SAGE libraries is displayed together with two‐phase culture.
To verify whether expressions of these genes were ubiquitous, we also evaluated them in several tissues using a cDNA tissue library (Clontech Laboratories Inc., Mountain View, CA, USA). We observed high expression of EYA3 and LXN in bone marrow, and for genes ERH, TRIB3 and TIMELESS, we observed high expression in other haematopoietic islands such as placenta and liver. Exceptionally, expression of HES6 gene was not observed in these tissues and highest expression was observed in intestine and brain (Fig. 5).
Figure 5.
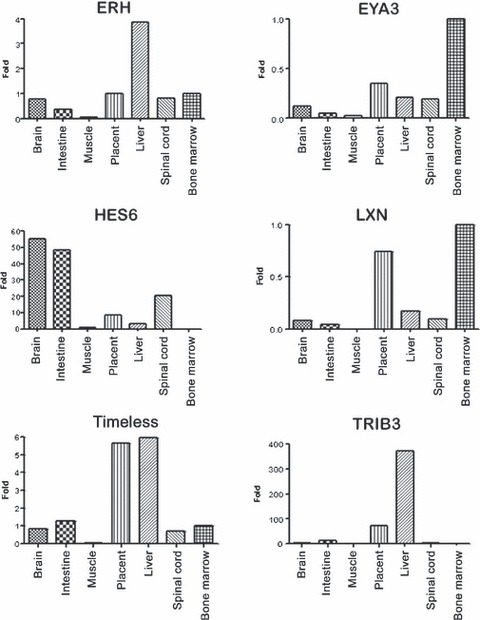
Differential expression of selected genes in several tissues using a cDNA tissue library (Clontech Laboratories Inc).
Discussion
Gene expression during erythroid differentiation is poorly understood. Study of the global pattern of gene expression that accompanies erythroid differentiation could help improve understanding of erythroid‐specific mechanisms that are required for optimal function of erythrocytes and therefore, identify targets for treatment of erythrocyte disorders (8).
To understand this mechanism, global gene expression during erythroid differentiation was evaluated using SAGE. By this strategy, 93 genes were identified that presented differential expression at statistically significant levels. As such, these genes may easily be involved in several important processes that lead to differentiation of haematopoietic stem cells into erythrocytes and may constitute therapeutic targets for haematopoietic diseases.
Several genes found in this study as differentially expressed are well described in the literature; these include ALAS2, ANK1, GDF15, NUDT4 and AHSP (16, 24), and validate the results found in our libraries. In addition, some genes are described for the first time. Among them, an interesting finding was presence of some genes homologous to genes of D. melanogaster and that were highly and differentially expressed during erythroid differentiation here (Fig. 3). Most differentially expressed genes were TIMELESS, TRIB3, EYA3, HES6 and ERH identified in humans, but some of them do not have any described function in people and none has been reported during erythroid differentiation.
Timeless protein is mainly known for its essential role in circadian rhythm in Drosophila; however, a recent study in humans suggests an intimate connection between the circadian cycle and DNA damage checkpoints that is partly mediated by Timeless protein. Timeless protein interacts with Chk1 kinase, which regulates DNA damage‐induced G2/M arrest and is mainly activated by BRCA1 (25, 26). The gene was also identified among a common prognostic signature of 29 genes that are associated with patient survival in breast cancer (27) and as a candidate to predict response to tamoxifen, the most common endocrine agent used to treat women at all stages of breast cancer (28). To date, there are no studies demonstrating the relationship of this gene with erythropoiesis, and our data suggest its participation during erythroid maturation, as increase in its expression was observed from the intermediate stage of differentiation onwards, being more evident in CD34+ cells (Fig. 4).
Tribbles 3 homologue (TRIB3), is a putative protein kinase that, in Drosophila, appears to play a role in regulation of the cell cycle and cell migration. In mammals, TRIB3 was initially cloned as an inducible gene in neuronal PC‐3 cells following NGF withdrawal. The protein is emerging as a negative regulator of various signal transducers and has been implicated in several processes, including apoptosis regulation, cell survival, regulation of adipocyte differentiation and insulin resistance (29, 30, 31, 32), and also acts as an important participant in tumour cell growth (33). Overexpression of this gene at the end of erythroid differentiation (Fig. 4) demonstrates that this process is finely regulated, as the cells are almost fully differentiated and intense proliferation typically observed in previous stages is controlled. Deregulation of expression of this could be implicated in increase in cell proliferation, in turn inducing a tumour development.
EYA3 (Eyes absent 3) is another gene that demonstrated increase in expression at the end of differentiation, suggesting a possible role of this transcription factor in maturation of erythroid cells. Li et al. (34) demonstrated that the Eya family (EYA1, EYA2 and EYA3) has protein phosphatase function, and its enzymatic activity is required for regulating genes that encode growth control and signalling molecules, modulating precursor cell proliferation. Studies with Eya1‐deficient mice show that the gene controls critical early inductive signalling events involved in ear and kidney formation and integrate Eya1 into the genetic regulatory cascade controlling kidney formation upstream of Gdnf, which is required to direct ureteric bud outgrowth via activation of c‐ret Rtk (35). Occasionally, anaemic embryos of these mice are seen, suggesting a haematopoietic defect (12). In a study analysing gene expression of purified haematopoietic stem cells (HSC), the authors identified expression of EYA1 and EYA2 and suggested that they could be involved in HSC self‐renewal (12); however, in our study, expression of EYA3 was not identified. EYA3 is mapped to chromosome 1 and no studies have been carried out on it in humans. Recently, Soker et al. (36) studied pleiotropic effects in Eya3‐knockout mice and showed that homozygous mutants displayed decreased bone mineral content and shorter body length; furthermore, apparently no haematopoietic effects were observed. Our results suggest that this transcription factor could be important at the end of differentiation as its expression was observed to be high at the end of differentiation and high expression was found in bone marrow.
HES6 (Hairy/Enhancer of Split 6) is another Drosophila homologous gene that encodes a member of a subfamily of basic helix‐loop‐helix transcription repressors (37). The protein encoded by this gene functions as a cofactor, interacting with other transcription factors through a tetrapeptide domain in its C‐terminus (38), and may be involved in neurogenesis (39) and cell proliferation in promyelocytic leukaemia (40). However, precise molecular mechanism of Hes6‐mediated control of differentiation remains to be elucidated (40). This transcription factor was found to be highly expressed at the end of differentiation here, but was not observed at the beginning of CD34+ differentiation (Fig. 4) in bone marrow (Fig. 5), showing that its expression is stage‐specific and finely regulated.
ERH (Expression of Enhancer of Rudimentary) gene was found to be continuously regulated during erythropoiesis and its expression increased during differentiation (Fig. 4). The product of this gene is a small, highly conserved, nuclear protein with a unique three‐dimensional structure. Involvement of ERH in fundamental processes such as regulation of pyrimidine metabolism, cell cycle progression, transcription and cell growth control has been suggested (41, 42, 43, 44); however, none of these interactions has been verified experimentally. To date, the mechanism of action of ERH remains unclear, and our result needs to be studied in detail to identify its function in erythroid differentiation.
In addition to these Drosophila homologous genes, LXN (latexin) gene was observed to be continuously expressed from the beginning of differentiation and was highly expressed in bone marrow (4, 5). LXN is the only known carboxypeptidase inhibitor in mammals and despite several structure–function studies of latexin, there is little knowledge of its biological roles in stem cells and ageing. Recent studies have shown that LXN is a negative regulator of stem cell number and acts through at least two mechanisms to modulate stem cell pool size: (i) it decreases HSC cell replication and (ii) it increases HSC apoptosis. Thus, in the haematopoietic system, and perhaps other organs, latexin influences ageing and lifespan through its action on stem cells (23). Continuous expression of the gene, found in this study, showed that its regulation was directly related to differentiation of the cells; during cell proliferation and consequent maturation, expression of the gene increased then began to decline. Further studies on gene expression using inhibition and superexpression of these genes in CD34+ cultures are being carried out and results will provide new insights to the relationship of its the expression to haematopoiesis.
Another important finding in our study was the number of tags that had no correspondence in the data bank and that were denominated ‘no matches’ (27% approximately); these tags could represent novel genes. Several studies observed the same results and have shown that approximately 35% of total SAGE tags are unmapped or unidentified. Several authors have suggested that this could be explained by several reasons: for instance, tags overlapping two exons, tags extended into the polyA tail and tags that differ from the genome sequence due to polymorphism. These tags could also correspond to antisense transcripts or new variants of known transcripts, suggesting that many transcripts are still to be annotated and that the human transcriptome seems to be more complex than shown in current genome annotations (45). Study on non‐identified SAGE tags could help improve the annotation process and identify genes with important functions that could potentially be used as targets for disease therapies.
One of these tags (No Match 1 –Table 2) demonstrated a large increase in expression during the intermediate phase of differentiation and could be very important in metabolic pathways involved in differentiation of erythroid cells. Two other tags (No Match 5 and 8 –Table 2) demonstrated increases at the end of differentiation and could be involved in maturation of haematopoietic cells. Identification of these tags could identify new genes or new isoforms of genes involved in differentiation of erythroid cells.
Results shown in this study amplify previously published data and present new clues concerning gene regulation and dynamic organization of genes in chromosomes of cells contributing to comprehension of erythroid differentiation, and to identification of new target genes involved in some erythroid diseases.
Supporting information
Fig. S1 Morphology of cells during erythroid differentiation using two‐phase liquid culture after erythropoietin addition. Typical morphology was detectable during differentiation (0 h, proerythroblast; 192 h, basophilic erythroblasts and 336 h, orthocromatic erythroblasts).
Fig. S2 Gene ontology categorization. Differentially expressed genes were categorized by Molecular Function and Biological Process using the gene ontology classification.
Table S1 Sequence and ideal concentration for the primers used in qPCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This study was supported by Grants from FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, São Paulo, Brazil, 02/13801‐7). A.F.C. was also supported by FAPESP (05/51222‐7).The authors thank Dr. Nicola Conran, HEMOCENTRO‐UNICAMP, for help with English revision.
References
- 1. Baron MH (1997) Transcriptional control of globin gene switching during vertebrate development. Biochim. Biophys. Acta 1351, 51–72. [DOI] [PubMed] [Google Scholar]
- 2. Shivdasani RA, Orkin SH (1996) The transcriptional control of hematopoiesis. Blood 87, 4025–4039. [PubMed] [Google Scholar]
- 3. Pope SH, Fibach E, Sun J, Chin K, Rodgers GP (2000) Two‐phase liquid culture system models normal human adult erythropoiesis at the molecular level. Eur. J. Haematol. 64, 292–303. [DOI] [PubMed] [Google Scholar]
- 4. Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C et al. (1997) Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 11, 774–785. [DOI] [PubMed] [Google Scholar]
- 5. Jimenez G, Griffiths SD, Ford AM, Greaves MF, Enver T (1992) Activation of the beta‐globin locus control region precedes commitment to the erythroid lineage. Proc. Natl. Acad. Sci. USA 89, 10618–10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ford AM, Bennett CA, Healy LE, Navarro E, Spooncer E, Greaves MF (1992) Immunoglobulin heavy‐chain and CD3 delta‐chain gene enhancers are DNase I‐hypersensitive in hemopoietic progenitor cells. Proc. Natl. Acad. Sci. USA 89, 3424–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heyworth C, Pearson S, May G, Enver T (2002) Transcription factor‐mediated lineage switching reveals plasticity in primary committed progenitor cells. EMBO J. 21, 3770–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ney PA (2006) Gene expression during terminal erythroid differentiation. Curr. Opin. Hematol. 13, 203–208. [DOI] [PubMed] [Google Scholar]
- 9. Gubin AN, Njoroge JM, Bouffard GG, Miller JL (1999) Gene expression in proliferating human erythroid cells. Genomics 59, 168–177. [DOI] [PubMed] [Google Scholar]
- 10. Georgantas RW III, Tanadve V, Malehorn M, Heimfeld S, Chen C, Carr L et al. (2004) Microarray and serial analysis of gene expression analyses identify known and novel transcripts overexpressed in hematopoietic stem cells. Cancer Res. 64, 4434–4441. [DOI] [PubMed] [Google Scholar]
- 11. Zhou G, Chen J, Lee S, Clark T, Rowley JD, Wang SM (2001) The pattern of gene expression in human CD34(+) stem/progenitor cells. Proc. Natl. Acad. Sci. USA 98, 13966–13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Terskikh AV, Miyamoto T, Chang C, Diatchenko L, Weissman IL (2003) Gene expression analysis of purified hematopoietic stem cells and committed progenitors. Blood 102, 94–101. [DOI] [PubMed] [Google Scholar]
- 13. Steidl U, Kronenwett R, Rohr UP, Fenk R, Kliszewski S, Maercker C et al. (2002) Gene expression profiling identifies significant differences between the molecular phenotypes of bone marrow‐derived and circulating human CD34+ hematopoietic stem cells. Blood 99, 2037–2044. [DOI] [PubMed] [Google Scholar]
- 14. Bonafoux B, Commes T (2009) Serial analysis of gene expression adapted for downsized extracts (SAGE/SADE) analysis in reticulocytes. Methods Mol. Biol. 496, 299–311. [DOI] [PubMed] [Google Scholar]
- 15. Bonafoux B, Lejeune M, Piquemal D, Quere R, Baudet A, Assaf L et al. (2004) Analysis of remnant reticulocyte mRNA reveals new genes and antisense transcripts expressed in the human erythroid lineage. Haematologica 89, 1434–1438. [PubMed] [Google Scholar]
- 16. Komor M, Guller S, Baldus CD, De Vos S, Hoelzer D, Ottmann OG et al. (2005) Transcriptional profiling of human hematopoiesis during in vitro lineage‐specific differentiation. Stem Cells 23, 1154–1169. [DOI] [PubMed] [Google Scholar]
- 17. Wang SM (2007) Understanding SAGE data. Trends Genet. 23, 42–50. [DOI] [PubMed] [Google Scholar]
- 18. Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 19. Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res. 7, 986–995. [DOI] [PubMed] [Google Scholar]
- 20. Hashimoto S, Nagai S, Sese J, Suzuki T, Obata A, Sato T et al. (2003) Gene expression profile in human leukocytes. Blood 101, 3509–3513. [DOI] [PubMed] [Google Scholar]
- 21. Chabardes‐Garonne D, Mejean A, Aude JC, Cheval L, Di Stefano A, Gaillard MC et al. (2003) A panoramic view of gene expression in the human kidney. Proc. Natl. Acad. Sci. USA 100, 13710–13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Chaldee M, Gaillard MC, Bizat N, Buhler JM, Manzoni O, Bockaert J et al. (2003) Quantitative assessment of transcriptome differences between brain territories. Genome Res. 13, 1646–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang Y, Van Zant G (2008) Aging stem cells, latexin, and longevity. Exp. Cell Res. 314, 1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pina C, Enver T (2007) Differential contributions of haematopoietic stem cells to foetal and adult haematopoiesis: insights from functional analysis of transcriptional regulators. Oncogene 26, 6750–6765. [DOI] [PubMed] [Google Scholar]
- 25. Yarden RI, Pardo‐Reoyo S, Sgagias M, Cowan KH, Brody LC (2002) BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet. 30, 285–289. [DOI] [PubMed] [Google Scholar]
- 26. Unsal‐Kacmaz K, Mullen TE, Kaufmann WK, Sancar A (2005) Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 25, 3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naderi A, Teschendorff AE, Barbosa‐Morais NL, Pinder SE, Green AR, Powe DG et al. (2007) A gene‐expression signature to predict survival in breast cancer across independent data sets. Oncogene 26, 1507–1516. [DOI] [PubMed] [Google Scholar]
- 28. Tozlu‐Kara S, Roux V, Andrieu C, Vendrell J, Vacher S, Lazar V et al. (2007) Oligonucleotide microarray analysis of estrogen receptor alpha‐positive postmenopausal breast carcinomas: identification of HRPAP20 and TIMELESS as outstanding candidate markers to predict the response to tamoxifen. J. Mol. Endocrinol. 39, 305–318. [DOI] [PubMed] [Google Scholar]
- 29. Prudente S, Scarpelli D, Chandalia M, Zhang YY, Morini E, Del Guerra S et al. (2009) The TRIB3 Q84R polymorphism and risk of early‐onset type 2 diabetes. J. Clin. Endocrinol. Metab. 94, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi Y, Ohoka N, Hayashi H, Sato R (2008) TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J. Lipid Res. 49, 880–892. [DOI] [PubMed] [Google Scholar]
- 31. Zhou Y, Li L, Liu Q, Xing G, Kuai X, Sun J et al. (2008) E3 ubiquitin ligase SIAH1 mediates ubiquitination and degradation of TRB3. Cell. Signal. 20, 942–948. [DOI] [PubMed] [Google Scholar]
- 32. Rzymski T, Paantjens A, Bod J, Harris AL (2008) Multiple pathways are involved in the anoxia response of SKIP3 including HuR‐regulated RNA stability, NF‐kappaB and ATF4. Oncogene 27, 4532–4543. [DOI] [PubMed] [Google Scholar]
- 33. Bowers AJ, Scully S, Boylan JF (2003) SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 22, 2823–2835. [DOI] [PubMed] [Google Scholar]
- 34. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK et al. (2003) Eya protein phosphatase activity regulates Six1‐Dach‐Eya transcriptional effects in mammalian organogenesis. Nature 426, 247–254. [DOI] [PubMed] [Google Scholar]
- 35. Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R (1999) Eya1‐deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat. Genet. 23, 113–117. [DOI] [PubMed] [Google Scholar]
- 36. Soker T, Dalke C, Puk O, Floss T, Becker L, Bolle I et al. (2008) Pleiotropic effects in Eya3 knockout mice. BMC Dev. Biol. 8, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qian D, Radde‐Gallwitz K, Kelly M, Tyrberg B, Kim J, Gao WQ et al. (2006) Basic helix‐loop‐helix gene Hes6 delineates the sensory hair cell lineage in the inner ear. Dev. Dyn. 235, 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bae S, Bessho Y, Hojo M, Kageyama R (2000) The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development 127, 2933–2943. [DOI] [PubMed] [Google Scholar]
- 39. Jacobsen KX, Vanderluit JL, Slack RS, Albert PR (2008) HES1 regulates 5‐HT1A receptor gene transcription at a functional polymorphism: essential role in developmental expression. Mol. Cell. Neurosci. 38, 349–358. [DOI] [PubMed] [Google Scholar]
- 40. Eun B, Lee Y, Hong S, Kim J, Lee HW, Kim K et al. (2008) Hes6 controls cell proliferation via interaction with cAMP‐response element‐binding protein‐binding protein in the promyelocytic leukemia nuclear body. J. Biol. Chem. 283, 5939–5949. [DOI] [PubMed] [Google Scholar]
- 41. Isomura M, Okui K, Fujiwara T, Shin S, Nakamura Y (1996) Cloning and mapping of a novel human cDNA homologous to DROER, the enhancer of the Drosophila melanogaster rudimentary gene. Genomics 32, 125–127. [DOI] [PubMed] [Google Scholar]
- 42. Pogge von Strandmann E, Senkel S, Ryffel GU (2001) ERH (enhancer of rudimentary homologue), a conserved factor identical between frog and human, is a transcriptional repressor. Biol. Chem. 382, 1379–1385. [DOI] [PubMed] [Google Scholar]
- 43. Wojcik E, Murphy AM, Fares H, Dang‐Vu K, Tsubota SI (1994) Enhancer of rudimentaryp1, e(r)p1, a highly conserved enhancer of the rudimentary gene. Genetics 138, 1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gelsthorpe M, Pulumati M, McCallum C, Dang‐Vu K, Tsubota SI (1997) The putative cell cycle gene, enhancer of rudimentary, encodes a highly conserved protein found in plants and animals. Gene 186, 189–195. [DOI] [PubMed] [Google Scholar]
- 45. Keime C, Semon M, Mouchiroud D, Duret L, Gandrillon O (2007) Unexpected observations after mapping LongSAGE tags to the human genome. BMC Bioinformatics 8, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Morphology of cells during erythroid differentiation using two‐phase liquid culture after erythropoietin addition. Typical morphology was detectable during differentiation (0 h, proerythroblast; 192 h, basophilic erythroblasts and 336 h, orthocromatic erythroblasts).
Fig. S2 Gene ontology categorization. Differentially expressed genes were categorized by Molecular Function and Biological Process using the gene ontology classification.
Table S1 Sequence and ideal concentration for the primers used in qPCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item