

Abstract
Objectives
Glioblastoma multiforme (GBM) is the most aggressive brain tumour type in humans. Its poor prognosis is largely attributed to its invasiveness and high rate of recurrence. Recurring GBM is commonly resistant to chemotherapeutic drugs, making it specially difficult to treat. Recent studies have revealed that matricellular glycoprotein SPOCK1 to be upregulated in several cancer types and to be specifically expressed in invasive GBM, but not in other types of non‐invasive brain tumour, which prompted us to study the mechanism of action of SPOCK1 in invasion, recurrence and drug resistance of GBM cells.
Materials and methods
SPOCK1 expression in GBM tissues was evaluated using qPCR, Western blotting and immunohistochemical staining. Cell migration was tested by the wound healing method and cell invasion was assessed using transwell plates with Matrigel coating. Western blotting was performed for E‐cadherin, vimentin, N‐cadherin, p‐Akt and Akt. Cell viability was examined using the MTT assay.
Results
We found that the expression of SPOCK1 was significantly upregulated in recurrent GBM. We also demonstrated that SPOCK1 positively regulated migration, invasion and EMT process of GBM cells. Furthermore, SPOCK1 mediated TMZ resistance in GBM, as knockdown of SPOCK1 expression in TMZ‐resistant GBM cells substantially sensitized these cells to TMZ.
Conclusion
SPOCK1 results were positive and it mediated TMZ resistance in GBM. In addition, SPOCK1 regulated invasion and TMZ resistance in GBM cells via the Akt signalling pathway.
Introduction
Glioblastoma multiform (GBM) is the most aggressive brain tumour type in humans. It involves the malignancy of glial cells and accounts for a large portion of brain tumour cases. Despite recent advancement in surgical treatment and clinical prevention, the prognosis of glioblastoma remains poor 1. The poor prognosis of GBM is largely attributed to their invasiveness and high rate of recurrence 2. Currently, treatment for GBM combines surgery, radiotherapy and chemotherapy 3. Temozolomide (TMZ) is currently the standard of care for the treatment of GBM 4. TMZ is an alkylating agent that promotes cell apoptosis by inducing DNA strand breaks during cell replication 5. Clinical use of this drug has been shown to be effective in delaying tumour progression and leading to prolongation of survival 6. Despite the frontline status of TMZ, many GBM patients exhibit resistance to TMZ treatment when GBM recurs 7. The resistance to TMZ has become a major obstacle to GBM therapy. Understanding the pathogenesis and drug resistance of GBM will facilitate the development of novel strategies for effective treatment of the disease. Unfortunately, although several reports tried to understand TMZ resistance in GBM on molecular and cellular levels, the mechanisms remain elusive.
A recent study comparing gene expression in invasive versus non‐invasive brain tumours has revealed that the expression levels of the gene SPARC/osteonectin, CWCY and Kazal‐like domains proteoglycan 1 (SPOCK1) were significantly upregulated in invasive brain tumours 8. SPOCK1 encodes a matricellular glycoprotein (de‐adhesive extracellular matrix glycoprotein) belonging to a novel Ca2+‐binding proteoglycan family, and has been found to play a critical role in cell cycle control, apoptosis, DNA repair and metastasis 9, 10, 11. Interestingly, another member of the same proteoglycan family, SPARC, has been well studied in a variety of cancer types. Accumulating evidence has confirmed the involvement of SPARC in the regulation of cell adhesion, proliferation, apoptosis as well as cell cycle progression 12. Importantly, SPOCK1 itself has also been demonstrated by a number of studies to play critical roles in the recurrence of prostate cancer 13, the metastasis of gallbladder cancer 14, the invasion of hepatocellular carcinoma 10 and the epithelial–mesenchymal transition (EMT) of lung cancer cells 15. In this context, although the detailed mechanism of action by which SPOCK1 contributes to cancer development and progression is not clear, it is possible that SPOCK1 could also regulate the metastasis, the recurrence and the drug resistance in GBM.
In this study, we demonstrated that both the mRNA levels and the protein levels of SPOCK1 were significantly upregulated in recurrent GBM. We also found that SPOCK1 positively regulated the migration, invasion and EMT process of GBM cells. Furthermore, we reported that SPOCK1 mediated TMZ resistance in GBM, as knockdown of SPOCK1 expression in TMZ‐resistant GBM cells substantially sensitized these cells to TMZ. In addition, we found that the effects of SPOCK1 in regulating the invasion and the TMZ resistance in GBM cells were via the Akt signalling pathway.
Materials and methods
Glioblastoma (GBM) samples
The study protocol was approved by the Medical Ethics and Human Clinical Trial Committee of First Hospital of Jilin University. Diagnosis of four GBM patients was histopathologically confirmed by two pathologists according to WHO carcinoma standards. These four GBM cases underwent primary GBM resection and regularly took TMZ, followed by recurrent GBM resection after 12–15 months.
Cell culture and transfection
The U87 and U251 GBM cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in Dulbecco's modified Eagle's media (DMEM) (Gibco, Grand Island, NY, USA) containing 10% (v/v) foetal bovine serum (FBS, Gibco), 2 mm L‐glutamine (Sigma‐Aldrich, St. Louis, MO, USA), 100 U/ml penicillin and 100 U/ml streptomycin (Gibco). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Full‐length SPOCK1 gene was amplified from U87 cell cDNA library and constructed into pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) at NheI and XbaI sites to generate SPOCK1 overexpression plasmid SPOCK1‐pcDNA3.1. U87 and U252 cells were seeded into six‐well plate and cultured to 70% confluence. Then SPOCK1‐pcDNA3.1 was transfected into cells using X‐tremeGENE HP DNA Transfection Reagent (Roche, Mannheim, Germany) according to the manufacturer's instructions. SPOCK1 siRNA were purchased from GenePharma (Shanghai, China). After cells reached 70% confluency, SPOCK1 siRNA was transfected into cells using X‐tremeGENE siRNA Transfection Reagent (Roche according to the manufacturer's instructions. The cells were subjected to functional assays 2 days after siRNA or plasmid transfection.
Immunohistochemistry staining
Immunohistochemitry studies were performed on 5 μm sections of formalin fixed, paraffin‐embedded tissue. Antigen retrieval was carried out with 0.01 mol/l citrate buffer at pH 6.0 in an 800 W microwave oven for 15 min before immunostaining. The sections were blocked for endogenous protein binding and peroxidase activity with an application of Dual Endogenous Block (Dako, Santa Clara, CA, USA) for 10 min. The sections were then incubated with a monoclonal antibody specific for SPOCK1 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a 1:25 dilution overnight at 4 °C. The next day, sections were incubated with biotinylated secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. Vectstain Elite ABC complex (Vector lab, Burlingame, CA, USA) and AEC+ Substrate Chromogen (Dako) were used for primary antibody detection. Negative control sections were incubated in PBS instead of primary antibody.
Real‐time PCR (RT‐PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription involved the Superscript III First Strand Synthesis kit (Invitrogen). Expression of genes was detected by use of SYBR® Green Real‐Time PCR Master (Invitrogen) and normalization involved the 2‐Ct method relative to β‐actin. The primers used for SOPCK1: sense, 5′‐CAACTGCTTGTTCCCAGAGG‐3′ and antisense, 5′‐GCCAATGACTTCCCTATCCA‐3′; E‐cadherin: sense, 5′‐AATCCAAAGCCTCAGGTCATAAACA‐3′ and antisense, 5′‐TTGGGTCGTTGTACTGAATGGTC‐3′; Vimentin: sense, 5′‐GTTTCCCCTAAACCGCTAGG ‐3′ and antisense, 5′‐AGCGAGAGTGGCAGAGGA‐3′; N‐cadherin: sense, 5′‐TGGATGGGCTGCCTCCAGGTGAC‐3′ antisense, 5′‐ACCAGCCCACCCCTCGAGCCC‐3′; β‐actin: sense, 5′‐GTGGGGCGCCCCAGGCACCA‐3′ and antisense, 5′‐CTCCTTAATGTCACGCACGATTT‐3′.
Western blot
GBM cells and tissues were harvested and lysed with RIPA buffer containing phosphatase and protease inhibitor cocktail (Santa Cruz Biotechnology). Lysates were centrifuged at maximum speed for 10 min at 4 °C. The supernatants were collected and the protein concentrations were determined using Bradford protein assay (Bio‐Rad, Irvine, CA, USA). Proteins were separated by electrophoresis on 10% sodium dodecyl sulphate (SDS)‐polyacrylamide gels, and transferred onto nitrocellulose membranes (Bio‐Rad) for Western blotting. Membranes were blocked (with 3% BSA in TBS) and then incubated overnight with primary antibody at 4 °C. After HRP‐conjugated secondary antibody incubation for 1 h, the bound antibodies were detected using the Supersignal West Pico ECL chemiluminescence kit (Thermo Scientific, Rockford, IL, USA). The antibodies for SPOCK1 (1:1000), E‐cadherin (1:500), Vimentin (1:1000), N‐cadherin (1:1000), p‐AKT (1:400), AKT (1:1000), β‐actin (1:2000) and secondary antibodies were purchased from Santa Cruz Biotechnology.
Wound healing assay
Transfected U87 or U251 cells were seeded into each well of a six‐well plate and incubated overnight to reach near confluence. The cell monolayers were wounded with a 1 ml pipette tip and cultured with DMEM medium supplemented with 10% FBS at 37 °C with 5% CO2 for 48 h. The gaps in cell monolayers were photographed and quantified using Image J software.
Transwell assay
Invasion chambers (Corning Inc., Corning, NY, USA) in a 24‐well plate coated with 50 ml matrigel in DMEM were incubated at 37 °C for 2 h. Transfected U87 or U251 cells (1 × 105) were suspended in 200 μl serum‐free DMEM medium and seeded into the upper layer of chamber. Cell culture medium (700 μl) was added to each lower layer and then the chambers incubated for 24 h at 37 °C with 5% CO2. The chambers were washed with PBS three times, fixed with methanol and finally stained with crystal violet. Percentage of invaded cell number was calculated.
Fluorescent staining
GBM cells were seeded in a chamber slide and incubated for 24 h. The cells were then washed with PBS after removing the medium. Subsequently, cells were washed three times with PBS, fixed in 4% formaldehyde for 20 min and permeabilized in 0.1% Triton X‐100 for 30 min followed by PBS washing three times. Slides were blocked with solution (10% normal goat serum plus 3% Triton X‐100) for 30 min at room temperature, followed by incubation with primary antibodies against SPOCK1 (Santa Cruz Biotechnology) in the blocking solution for 2 h at room temperature. Slides were then washed three times with PBS and incubated with secondary antibody in the blocking solution. Nuclei were stained with the DAPI. Slides were washed three times with PBS and examined with a fluorescence microscope.
Cell proliferation and viability assay
Cell proliferation and viability was analysed by MTT assay (Promega, Madison, WI, USA). Briefly, transfected glioblastoma cells (5 × 103 per well) were seeded into 96‐well plates and incubated overnight. Then, the cells were treated with various concentrations of TMZ (0, 100, 250, 500, 1000 and 2000 μm) and incubated further. After 48 h incubation, MTT solution was added to the treated cell cultures, incubated for 2 h, and then the absorption at 570 nm was read using a microplate reader.
Colony formation assay
GBM cells (500 per well) were seeded on six‐well plate. After incubated overnight, the cells were treated with TMZ (30 μm). Cells were incubated at 37 °C, and the medium was replaced with fresh medium every 3 days. Colonies were fixed and then stained with 1% crystal violet 14 days later and counted.
Apoptosis assay
Transfected U87 or U251 cells were treated with TMZ (250 μm) for 24 h. The cells were then harvested and immediately fixed in 75% ethanol at 4 °C overnight, treated with 50 mg/l RNase A (Sigma, St. Louis, MO, USA) for 1 h at 37 °C, and stained with 50 mg/l PI (Sigma) for 15 min. Samples were analysed for their DNA content by flow cytometry (BD Biosciences, San Jose, CA, USA). The apoptosis analysis was performed by an Annexin V‐FITC kit (Sigma) according to the manufacturer's instructions. The data were analysed with flowjo software (BD Biosciences).
Statistics
Values were expressed as mean ± SD of samples measured in triplicate. Statistical analysis was performed using SPSS V19.0. (SPSS, Inc., Chicago, IL, USA) Quantitative data from two groups were analysed using the Student's t‐test and two‐tailed distribution. Three or more groups were analysed using analysis of variance (ANOVA). A P value <0.05 was considered significant.
Results
SPOCK1 is upregulated in recurrent GBM
In order to investigate the role of SPOCK1 during GBM recurrence, we first examined the mRNA expression of SPOCK1 in paired primary and recurrent GBM tissue samples from four patients. Compared to those in primary tumour tissue samples, the expression levels of SPOCK1 were significantly upregulated in recurrent tumour tissue samples (Fig. 1a). We next performed Western blot analysis to detect SPOCK1 protein in these samples. Consistent with the gene expression data, the protein levels of SPOCK1 were also higher in recurrent samples, compared to primary samples (Fig. 1b). We randomly selected GBM samples from two patients and performed immunohistochemistry staining, and we observed significantly stronger SPOCK1 signals in both recurrent GBM tissue samples, compared to their respective primary tissue counterparts (Fig. 1c). Taken together, these findings indicated that the expression of SPOCK1 was upregulated in recurrent GBM tissues and therefore SPOCK1 might play important roles in the development and recurrence of GBM.
Figure 1.
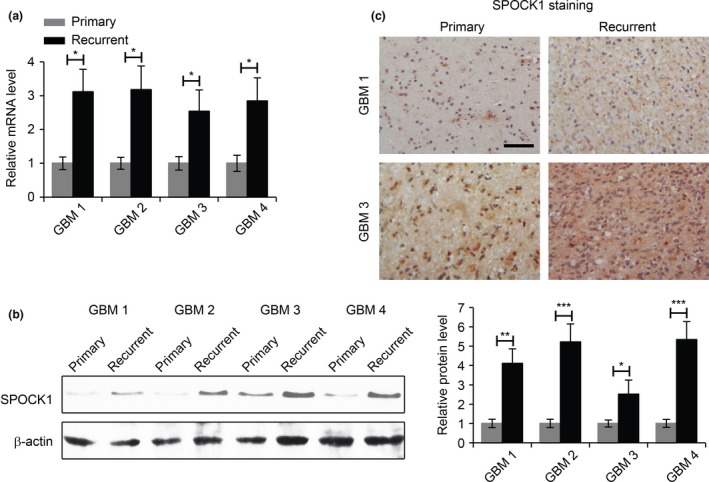
The expression of SPOCK1 is upregulated in recurrent GBM. (a) Relative levels of SPOCK1 mRNA in primary and recurrent GBM samples from four GBM patients. (b) Western blot analysis of the levels of SPOCK1 protein in primary and recurrent GBM samples of four GBM patients. β‐actin was used as a loading control. The expression of SPOCK1 was qualified (right panel). (c) Immunohistochemistry staining of SPOCK1 in primary and recurrent GBM samples 1 and 3. Scale bar: 100 μm. *P < 0.05, **P < 0.01 compared to primary samples.
SPOCK1 regulates GBM cell migration and invasion
To investigate the roles of SPOCK1 in the migration and invasion of GBM cells, we introduced SPOCK1 siRNA into two GBM cell lines, U87 and U251, to reduce the expression of SPOCK1 (Fig. 2a). We found that the reduction of SPOCK1 expression dramatically inhibited cell migration in both cell lines, as revealed by a wound healing assay (Fig. 2b). In addition, SPOCK1 knockdown also led to significantly reduced invasion in both cell lines in a transwell invasion assay (Fig. 2c). Furthermore, we increased the expression of SPOCK1 in U87 and U251 cells by overexpression (Fig. 3a), and performed the migration and the invasion assays. As expected, overexpression of SPOCK1 promoted both the migration (Fig. 3b) and invasion (Fig. 3c) in both cell lines.
Figure 2.
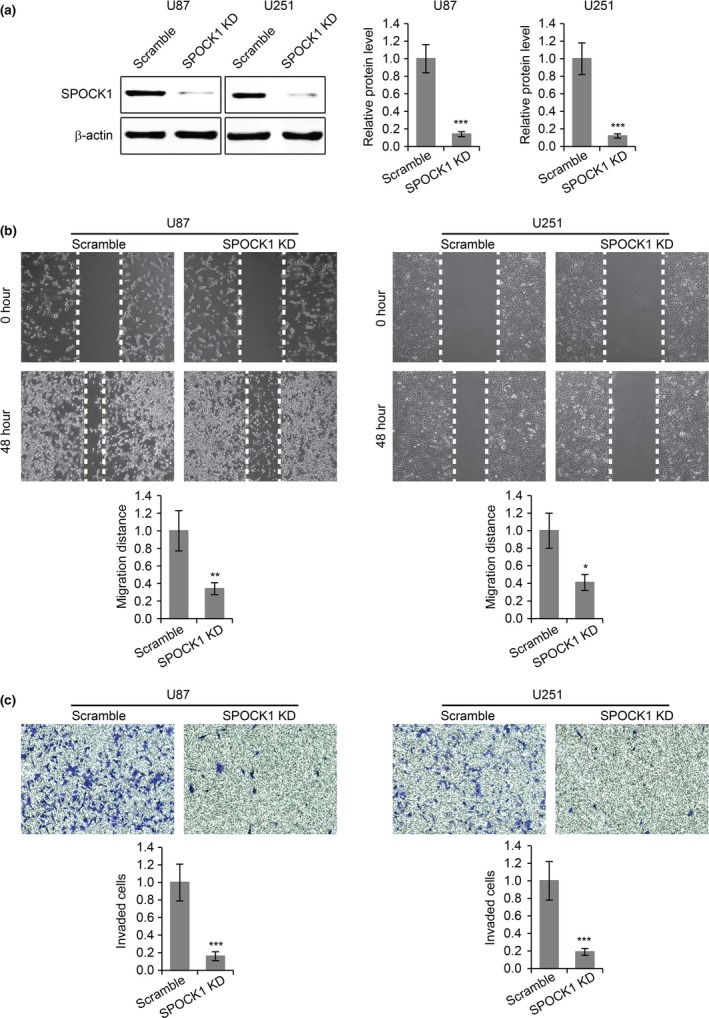
Knockdown of SPOCK1 inhibits GBM cell migration and invasion. (a) Western blot for SPOCK1 in U87 and U251 cells transfected with scramble siRNA or SPOCK1 siRNA (SPOCK1 KD). (b) Wound healing cell migration assay and (c) transwell cell invasion assay in U87 and U251 cells transfected with scramble siRNA or SPOCK1 siRNA (SPOCK1 KD). *P < 0.05, **P < 0.01, ***P < 0.001 compared to scramble control cells.
Figure 3.
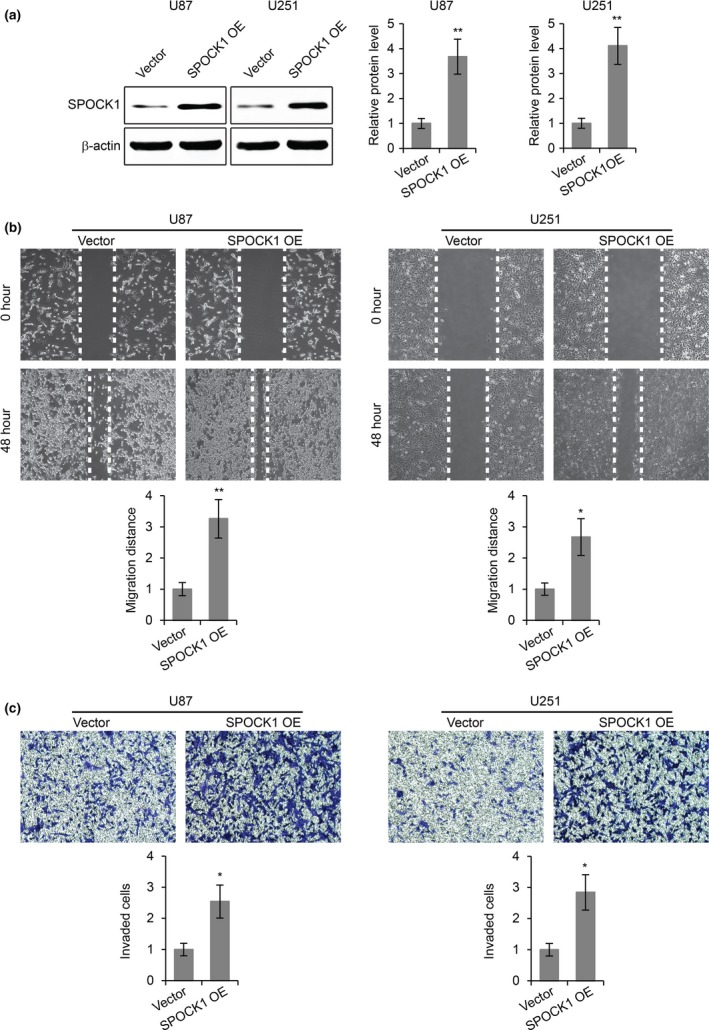
SPOCK1 overexpression promotes GBM cell migration and invasion. (a) Western blot for SPOCK1 in U87 and U251 cells transfected with empty vector or plasmid overexpressing SPOCK1 (SPOCK1 OE). (b) Wound healing cell migration assay and (c) transwell cell invasion assay in U87 and U251 cells transfected with empty vector or plasmid overexpressing SPOCK1 (SPOCK1 OE). *P < 0.05, **P < 0.01 compared to vector control cells.
Epithelial‐to‐mesenchymal transition (EMT) is an important process during GBM migration and invasion 16. Therefore, we sought to determine the effects of SPOCK1 on the EMT process of GBM cells. To this end, we examined the gene expression of three EMT marker proteins (E‐cadherin, Vimentin and N‐cadherin) in GBM cells with SPOCK1 knockdown or overexpression. We found that SPOCK1 knockdown led to increased E‐cadherin expression and reduced Vimentin and N‐cadherin expression (Fig. 4a), while SPOCK1 overexpression resulted in reduced E‐cadherin expression and increased Vimentin and N‐cadherin expression (Fig. 4b). These results were further confirmed by Western blot analysis (Fig. 4c,d). Collectively, these findings indicated that SPOCK1 positively regulated the migration, invasion and EMT process of GBM cells.
Figure 4.
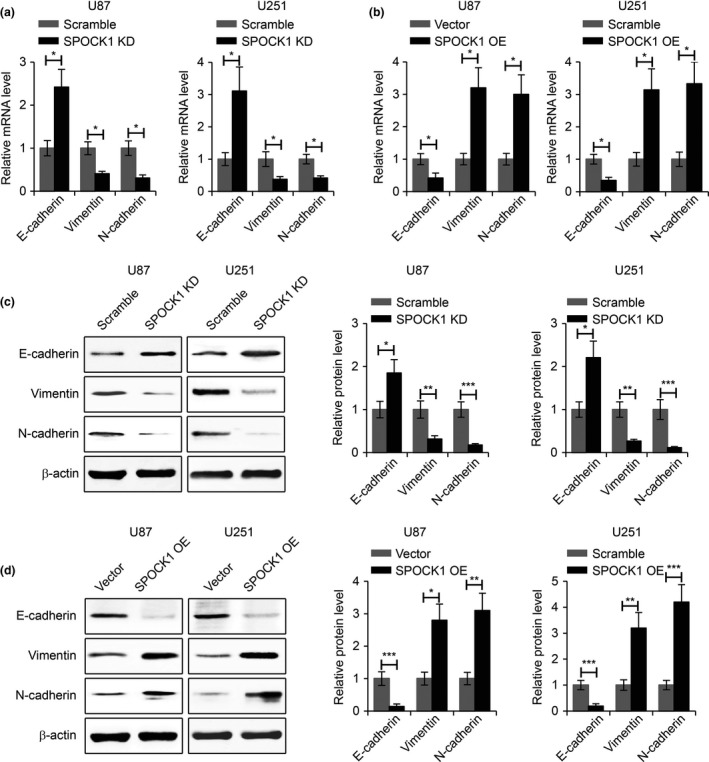
SPOCK1 regulates the EMT process of GBM cells. (a) Relative mRNA levels of E‐cadherin, Vimentin and N‐cadherin in SPOCK1 knockdown (SPOCK1 KD) or (b) SPOCK1 overexpressing (SPOCK1 OE) U87 and U251 cells. (c) Western blot for E‐cadherin, Vimentin and N‐cadherin in SPOCK1 knockdown (SPOCK1 KD) or (d) SPOCK1 overexpressing (SPOCK1 OE) U87 and U251 cells. *P < 0.05, **P < 0.01, ***P < 0.001 compared to control cells.
SPOCK1 mediates TMZ resistance in GBM
The invasive behaviour of GBM is a major factor for its recurrence and its resistance to the chemotherapy drug TMZ 17. In order to investigate the potential roles of SPOCK1 in TMZ sensitivity of GBM cells, we established TMZ‐resistant subclones of U87 and U251 cells by exposing these cells to TMZ for 6 months. We then analysed SPOCK1 expression levels in TMZ‐resistant (RE) versus TMZ‐sensitive (control) GBM cells. We found that both the mRNA (Fig. 5a) and the protein levels (Fig. 5b) of SPOCK1 were significantly increased in TMZ‐resistant U87 and U251 cells. Immunofluorescence staining of SPOCK1 also revealed elevated levels of SPOCK1 in TMZ‐resistant GBM cells (Fig. 5c).
Figure 5.
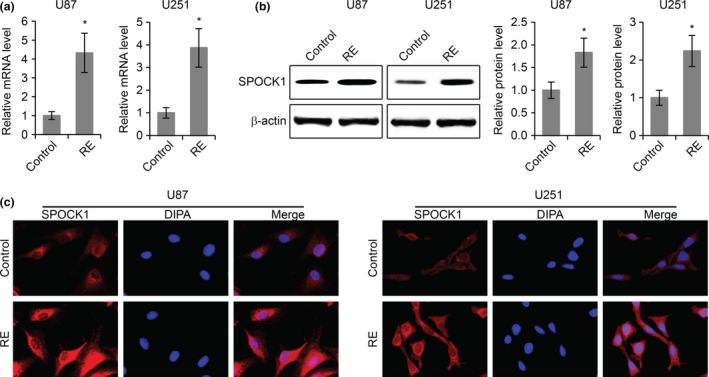
The expression of SPOCK1 is upregulated in TMZ‐resistant GBM cells. (a) Relative levels of SPOCK1 mRNA in TMZ‐resistant (RE) U87 and U251 cells. (b) Western blot for SPOCK1 protein levels in TMZ‐resistant (RE) U87 and U251 cells compared to TMZ‐sensitive control cells. (c) Immunofluorescence staining of SPOCK1 in TMZ‐resistant (RE) and TMZ‐sensitive control U87 and U251 cells. *P < 0.05 compared to TMZ‐sensitive control cells.
To determine whether or not the TMZ resistance of GBM cells was mediated by SPOCK1, we knocked down SPOCK1 in TMZ‐resistant U87 and U251 cells (Fig. 6a) and performed a variety of assays on these cells. MTT cell proliferation assays were first performed. Cell growth was enhanced in RE cell, whereas knockdown of SPOCK1 in RE cells significantly reduced cell proliferation (Fig. 6b). In the chemotoxicity assay, while increased cell viability against TMZ was present in TMZ‐resistant GBM cells (IC50: 616.33 μm in U87‐control cells versus 1291.19 μm in U87‐RE cells; 470.15 μm in U251‐control cells versus 903.99 μm in U251‐RE cells), knockdown of SPOCK1 in these TMZ‐resistant cells significantly increased their sensitivity to TMZ (IC50: 1291.19 μm in U87‐RE cells versus 691.95 μm in U87‐RE cells with SPOCK1 knockdown; 903.99 μm in U251‐RE cells versus 605.71 μm in U251‐RE cells with SPOCK1 knockdown) (Fig. 6c). In addition, we also performed colony formation assay and confirmed that the knockdown of SPOCK1 led to increased TMZ sensitivity in both U87 and U251 TMZ‐resistant cells (Fig. 6d). Furthermore, in the cell apoptosis assay, we found that compared to TMZ‐sensitive cells, there were less apoptotic cells in TMZ‐resistant GBM cell lines, but SPOCK1 knockdown in these TMZ‐resistant cells significantly increased the number of apoptotic cells (Fig. 6e). Taken together, these data indicated that SPOCK1 mediated TMZ resistance in GBM cells, and that loss of SPOCK1 could sensitize GBM cells to TMZ.
Figure 6.
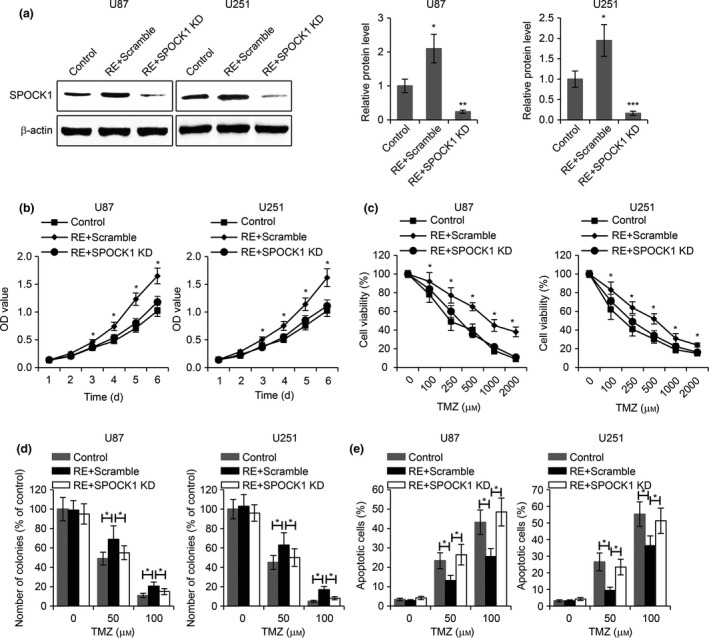
Knockdown of SPOCK1 sensitizes GMB cells to TMZ. (a) Western blot for SPOCK1 in TMZ‐sensitive control cells and TMZ‐resistant U87 and U251 cells transfected with scramble siRNA (RE+scramble) or SPOCK1 siRNA (RE+SPOCK1 KD). (b) MTT cell proliferation, (c) MTT viability assay, (d) colony formation assay, and (e) apoptosis assay in TMZ‐sensitive control cells and TMZ‐resistant U87 and U251 cells transfected with scramble siRNA (RE+scramble) or SPOCK1 siRNA (RE+SPOCK1 KD). *P < 0.05, **P < 0.01 compared to control cells.
SPOCK1 regulates GBM cell metastasis and TMZ resistance via the Akt pathway
The Akt signalling pathway was recently demonstrated to participate in the regulation of SPOCK1‐mediated cancer metastasis 14, and to contribute to TMZ resistance in GBM 18. In order to investigate whether or not SPOCK1 regulated cell invasion and TMZ resistance in GBM via the Akt signalling pathway, we first examined active p‐Akt levels in SPOCK1 knockdown U87 cells, and we found that the p‐Akt levels were significantly decreased in SPOCK1 knockdown cells compared to those in control cells (Fig. 7a). The total Akt expression was not regulated by SPOCK1 downregulation. Importantly, inhibition of Akt signalling pathway activity by MK‐2206 significantly reduced U87 cell migration (Fig. 7b) and invasion (Fig. 7c), and when Akt signalling was inhibited, SPOCK1 knockdown was no longer able to further inhibit cell migration and invasion (Fig. 7b,c). We next examined p‐Akt and Akt levels in SPOCK1 overexpressing cells, and we found that SPOCK1 overexpression induced elevated levels of p‐Akt (Fig. 7d). Although SPOCK1 overexpression led to increased levels of cell migration and invasion, these effects were completely abolished by MK‐2206 treatment (Fig. 7e,f). In addition, we also examined the levels of p‐Akt in TMZ‐resistant GBM cells with or without SPOCK1 knockdown. Consistent with previous findings, the activity of the Akt signalling pathway was inhibited by SPOCK1 knockdown (Fig. 7g). Significantly, inhibition of the Akt signal pathway by MK‐2206 significantly reduced cell viability and colony formation in TMZ‐resistant U87 cells, and SPOCK1 failed to further affect cell viability and colony formation when the Akt signal pathway was inhibited (Fig. 7h,i). Taken together, these results demonstrated that the SPOCK1 regulation of GBM cell invasion and TMZ resistance was via the Akt signalling pathway.
Figure 7.
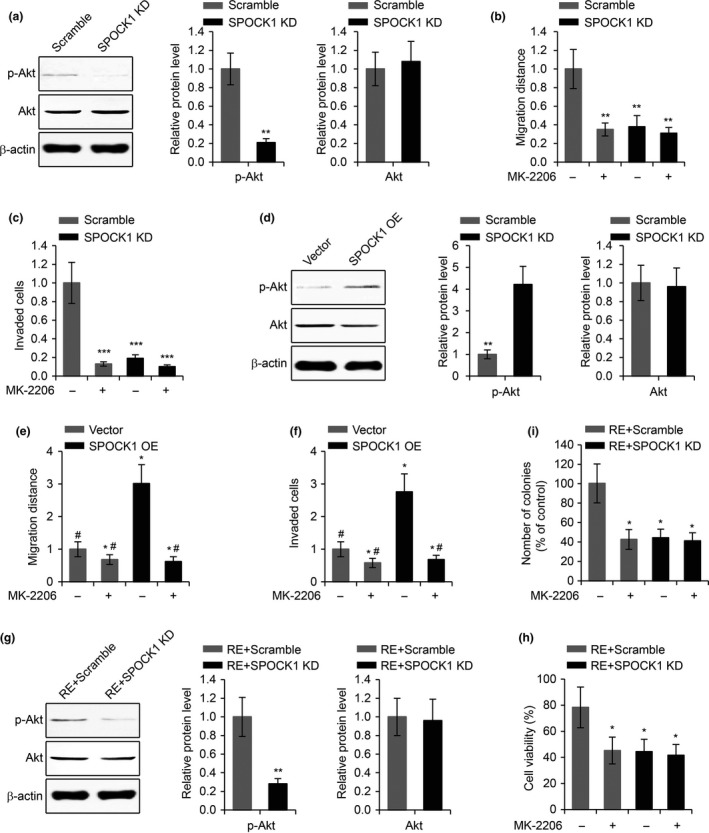
SPOCK1 regulates GBM cell invasion and TMZ resistance via the Akt pathway. (a) Western blot of phosphorylated Akt (p‐Akt) and total Akt levels in U87 cells transfected with scramble siRNA or SPOCK1 siRNA (SPOCK1 KD). p‐Akt and total Akt levels related to β‐actin were quantified in right panel. (b) Wound healing cell migration assay and (c) transwell cell invasion assay in U87 cells transfected with scramble siRNA or SPOCK1 siRNA (SPOCK1 KD) subjected to the treatment of Akt inhibitor MK‐2206. (d) Western blot of phosphorylated Akt (p‐Akt) and total Akt levels in U87 cells transfected with empty vector or plasmid overexpressing SPOCK1 (SPOCK1 OE). (e) Wound healing cell migration assay and (f) transwell cell invasion assay in U87 cells transfected with empty vector or plasmid overexpressing SPOCK1 (SPOCK1 OE) subjected to the treatment of Akt inhibitor MK‐2206. (g) Western blot of phosphorylated Akt (p‐Akt) and total Akt levels in TMZ‐resistant U87 cells transfected with scramble siRNA (RE+scramble) or SPOCK1 siRNA (RE+SPOCK1 KD). (h) MTT cell viability assay and (i) colony formation assay in TMZ‐resistant U87 cells transfected with scramble siRNA (RE+scramble) or SPOCK1 siRNA (RE+SPOCK1 KD) subjected to the treatment of Akt inhibitor MK‐2206. *P < 0.05, **P < 0.01 and ***P < 0.01 compared to scramble or empty vector control cells. #P < 0.05 compared to SPOCK1 OE cells without MK‐2206 treatment.
Discussion
GBM is a highly lethal disease, and most individuals with this disease die because of tumour invasion and recurrence. Recurring GBM is commonly resistant to chemotherapy drugs, making it especially difficult to treat. Recent studies revealed that SPOCK1 was upregulated in several cancer types including lung cancer 15, hepatocellular cancer 10 and gallbladder cancer 14, and was specifically expressed in invasive GBM but not other types of non‐invasive brain tumours 8. It is therefore intriguing to find out the mechanism of action of SPOCK1 in the invasion, recurrence and drug resistance of GBM cells. In this study, we reported that the expression levels of SPOCK1 were significantly upregulated in recurrent GBM. We also demonstrated that SPOCK1 positively regulated the migration, invasion and EMT process of GBM cells. Significantly, we found that SPOCK1 mediated TMZ resistance in GBM, and that the effects of SPOCK1 in regulating the invasion and the TMZ resistance in GBM cells were via the Akt signalling pathway.
As there was no study that focused on the differential expression of SPOCK1 between primary and recurring GBM, we first characterized the mRNA and the protein levels of SPOCK1 in these two types of GBM. Surprisingly, from the same patients, the levels of SPOCK1 were significantly upregulated in the recurring tumour, compared to those in the primary tumour. This observation led us to associate the functions of SPOCK1 to the resistance to chemotherapy drug TMZ, as TMZ resistance was also a unique property of recurring GBM.
In investigating the functional roles of SPOCK1, we first found that SPOCK1 was capable of inducing the migration and the invasion of GBM cells, suggesting that SPOCK1 was likely involved in the metastasis of GBM cells. Metastasis is a complicated molecular and cellular process that contributes to the most common cause of mortality in GBM patients. During metastasis, a series of cellular events happen, including cell migration, degradation of base membrane, cell invasion, cell adhesion and angiogenesis. It is important to note that, although in this study we have demonstrated that SPOCK1 was involved in the regulation of cell migration and cell invasion, it is possible that SPOCK1 also played roles in other cellular processes that facilitated metastasis. Additionally, in this study, we used systematic approach to show a role for SPOCK1 in the resistance of GBM to TMZ. The expression levels of SPOCK1 were significantly different in TMZ‐sensitive versus TMZ‐resistant GBM cells. Knocking down SPOCK1 sensitized TMZ‐resistant cells to this drug. These findings supported a theory that SPOCK1 was essential for the protection of GBM cells against chemotherapy drugs, which would potentially poise SPOCK1 as a novel therapeutic target in the treatment of TMZ‐resistant GBM.
Further investigation in this study revealed that the effects of SPOCK1 in the regulation of cell invasion and TMZ resistance were dependent on the activity of the Akt signalling pathway, as Akt inhibition abolished SPOCK1 effects. Our findings were consistent with previous studies regarding the general functions of the Akt signalling pathway in cancer development. The Akt pathway was reported to play important roles in regulating apoptosis 19. Dysfunction of apoptosis could lead to the expansion of neoplastic cells with deregulated proliferation and accumulation of genetic mutations 20, and is therefore one of the major mechanisms in the onset and the progression of cancer. Indeed, the Akt signalling pathway was demonstrated by a large body of literatures to involve in many aspects of cancer development 21, 22. For example, its activation induced cell growth 23, promotes EMT 24 and regulates MYC‐mediated signalling for apoptosis progression 25. In this context, the inhibition of cell invasion and the sensitization of GBM cells by SPOCK1 knockdown could be explained, at least in part, by the inactivation of the Akt signalling pathway.
It is worth noting that SPOCK1 is a matricellular glycoprotein family member, and that there are other members in this family, including SPARC, TESTICAN‐2 and TESTICAN‐3. The function of these glycoproteins in the regulation of GBM development was not characterized. Given the structural similarity between them, it is interesting in our future study to examine whether or not SPARC, TESTICAN‐2 and TESTICAN‐3 could also contribute in the regulation of TMZ resistance in GBM.
Conflict of interest
All authors have no conflict of interest to declare.
Acknowledgements
This work was supported by Natural Science Foundation of Heilongjiang Province of China (Support No. H2015083).
References
- 1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. [DOI] [PubMed] [Google Scholar]
- 2. Giese A, Bjerkvig R, Berens ME, Westphal M (2003) Cost of migration: invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 21, 1624–1636. [DOI] [PubMed] [Google Scholar]
- 3. Park CK, Lee SH, Kim TM, Choi SH, Park SH, Heo DS et al (2013) The value of temozolomide in combination with radiotherapy during standard treatment for newly diagnosed glioblastoma. J. Neurooncol. 112, 277–283. [DOI] [PubMed] [Google Scholar]
- 4. Dehdashti AR, Hegi ME, Regli L, Pica A, Stupp R (2006) New trends in the medical management of glioblastoma multiforme: the role of temozolomide chemotherapy. Neurosurg. Focus 20, E6. [DOI] [PubMed] [Google Scholar]
- 5. Zhang J, Stevens MF, Bradshaw TD (2012) Temozolomide: mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 5, 102–114. [DOI] [PubMed] [Google Scholar]
- 6. Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P et al (2002) Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J. Clin. Oncol. 20, 1375–1382. [DOI] [PubMed] [Google Scholar]
- 7. Johannessen TC, Bjerkvig R (2012) Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 12, 635–642. [DOI] [PubMed] [Google Scholar]
- 8. Colin C, Baeza N, Bartoli C, Fina F, Eudes N, Nanni I et al (2006) Identification of genes differentially expressed in glioblastoma versus pilocytic astrocytoma using Suppression Subtractive Hybridization. Oncogene 25, 2818–2826. [DOI] [PubMed] [Google Scholar]
- 9. Chen L, Hu L, Chan TH, Tsao GS, Xie D, Huo KK et al (2009) Chromodomain helicase/adenosine triphosphatase DNA binding protein 1‐like (CHD1 l) gene suppresses the nucleus‐to‐mitochondria translocation of nur77 to sustain hepatocellular carcinoma cell survival. Hepatology 50, 122–129. [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Chen L, Chan TH, Liu M, Kong KL, Qiu JL et al (2013) SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice. Gastroenterology 144(179–191), e174. [DOI] [PubMed] [Google Scholar]
- 11. Murphy‐Ullrich JE, Sage EH (2014) Revisiting the matricellular concept. Matrix Biol. 37, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang L, Feng J (2014) SPARC in Tumor Pathophysiology and as a Potential Therapeutic Target. Curr. Pharm. Des. 20, 6182–6190. [DOI] [PubMed] [Google Scholar]
- 13. Yang C, Fischer‐Keso R, Schlechter T, Strobel P, Marx A, Hofmann I (2015) Plakophilin 1‐deficient cells upregulate SPOCK1: implications for prostate cancer progression. Tumour Biol. 36, 9567–9577. [DOI] [PubMed] [Google Scholar]
- 14. Shu YJ, Weng H, Ye YY, Hu YP, Bao RF, Cao Y et al (2015) SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol. Cancer. 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miao L, Wang Y, Xia H, Yao C, Cai H, Song Y (2013) SPOCK1 is a novel transforming growth factor‐beta target gene that regulates lung cancer cell epithelial‐mesenchymal transition. Biochem. Biophys. Res. Commun. 440, 792–797. [DOI] [PubMed] [Google Scholar]
- 16. Kubelt C, Hattermann K, Sebens S, Mehdorn HM, Held‐Feindt J (2015) Epithelial‐to‐mesenchymal transition in paired human primary and recurrent glioblastomas. Int. J. Oncol. 46, 2515–2525. [DOI] [PubMed] [Google Scholar]
- 17. Hatzikirou H, Basanta D, Simon M, Schaller K, Deutsch A (2012) ‘Go or grow’: the key to the emergence of invasion in tumour progression? Math. Med. Biol 29, 49–65. [DOI] [PubMed] [Google Scholar]
- 18. Mu Q, Wang L, Yu F, Gao H, Lei T, Li P et al (2015) Imp2 regulates GBM progression by activating IGF2/PI3K/Akt pathway. Cancer Biol. Ther. 16, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hemmings BA (1997) Akt signaling: linking membrane events to life and death decisions. Science 275, 628–630. [DOI] [PubMed] [Google Scholar]
- 20. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 21. Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3‐kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu X, Zhen Y, Yang H, Wang H, Zhou Y, Wang E et al (2013) Loss of connective tissue growth factor as an unfavorable prognosis factor activates miR‐18b by PI3K/AKT/C‐Jun and C‐Myc and promotes cell growth in nasopharyngeal carcinoma. Cell Death Dis. 4, e634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong L, Schafer G, Bu H, Zhang Y, Klocker H (2012) Lamin A/C protein is overexpressed in tissue‐invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 33, 751–759. [DOI] [PubMed] [Google Scholar]
- 24. Ha GH, Park JS, Breuer EK (2013) TACC3 promotes epithelial‐mesenchymal transition (EMT) through the activation of PI3K/Akt and ERK signaling pathways. Cancer Lett. 332, 63–73. [DOI] [PubMed] [Google Scholar]
- 25. Gill RM, Gabor TV, Couzens AL, Scheid MP (2013) The MYC‐associated protein CDCA7 is phosphorylated by AKT to regulate MYC‐dependent apoptosis and transformation. Mol. Cell. Biol. 33, 498–513. [DOI] [PMC free article] [PubMed] [Google Scholar]