

Abstract
Objectives: Isolation of mouse mesenchymal stem cells (mMSCs), by the approach of plastic adherence, has been difficult due to persistent contamination by haematopoietic cells (HCs); we have observed that this contamination was due to engagement between HCs and mMSCs. The HCs can be lifted together with the mMSCs despite their insensitivity to trypsin digestion. Herein, we provide a single‐step procedure to rapidly segregate mMSCs from HC contaminants using transient lower‐density plastic adherence (tLDA).
Materials and methods: The tLDA was performed by replating bone marrow adherent cells at lower density (1.25 × 104 cells/cm2) than usual, allowing for transient adherence of no more than 3 h, followed by trypsin digestion. tLDA‐isolated cells were evaluated by immunophenotyping, multi‐differentiation potentials, immunosuppressive properties, and therapeutic potential as demonstrated by symptoms of osteoporosis.
Results: The single‐step tLDA method can effectively eliminate the persistent HC contaminants; tLDA‐isolated cells were phenotypically equivalent to those reported as mMSCs. The isolated cells possessed classic tri‐lineage differentiation potential into osteogenic, adipogenic and chondrogenic lineages and had immunosuppressive properties. After intravenous transplantation, they migrated into the allogeneic bone marrow and rescued hosts from osteoporosis symptoms, demonstrating their therapeutic potential.
Conclusions: We have developed a simple and economical method that effectively isolates HC‐free, therapeutically functional mMSCs from bone marrow cell adherent cultures. These cells are suitable for various mechanistic and therapeutic studies in the mouse model.
Introduction
Postnatal mesenchymal stem cells (MSCs) possess self‐renewal capacity and have the potential to differentiate along multiple mesenchymal cell lineages (1, 2, 3). These properties qualify MSCs to be an ideal system to study lineage commitment and differentiation in vitro. Recent data also suggest that MSCs are immunoprivileged, and more importantly, can display immunosuppressive properties (4, 5). MSCs are capable of migrating to some tissues, particularly when such tissues are injured or suffer pathological conditions (2, 3, 6). These properties make MSCs attractive for a number of therapeutic applications (7, 8, 9, 10, 11, 12).
As demonstrated by Friedenstein and colleagues, MSCs are relatively easy to isolate from the bone marrow by their physical propensity of adherence to tissue culture plates and flasks (13, 14). However, in some species such as mice (a valuable animal model for human diseases), MSCs can be difficult to isolate from contaminating haematopoietic cells (HCs) (2, 15). In culture, HCs adhere randomly, with some of them adhering to the plastic surface, while others engage with MSCs (16, 17, 18, 19, 20). In addition, the persistent contamination of HCs in mMSC cultures can be maintained due to expression of growth factors and cytokines from MSCs which support development and proliferation of hematopoietic progenitor/stem cells during extended culture periods (21, 22).
The conventional approach to isolate mMSCs from HCs is by repeatedly passaging the adherent marrow cells with repeated trypsin digestion. Trypsin‐insensitive HCs eventually become depleted from the mMSCs cultures (23, 24, 25). However, this method is not optimal for isolation of mMSCs, as their proliferation and differentiation potentials are lost along the passages (26, 27, 28). Recently, improvements in mMSCs isolation protocols have been made, all of which aim to solve the problem of HC contamination prominently observed in mouse marrow cultures. These improvements include (i) magnetic selection (16, 29, 30); (ii) cell sorting (31); (iii) limiting dilutions (32, 33); (iv) exposure to cytotoxic materials (34, 35); and (v) other unique culture systems (25, 36). However, despite these advances, the methods still have inconvenient effects on proliferation and differentiation properties of isolated mMSCs, thus restricting their widespread utility (37).
To overcome these limitations, we have developed an improved single‐step plastic‐adherent method to isolate mMSCs from heterogeneous bone marrow cultures using the following transient lower‐density plastic adherence (tLDA) strategy: cells are plated at a lower density (1.25 × 104 cells/cm2) than ususal, to allow attachment of all HCs on the plastic surface and avoid them overlapping mMSCs. After a short‐term adherent period, less than 3 h, followed by trypsin digestion, HCs are eliminated because unlike mMSCs, plastic‐adhering HCs cannot be lifted after trypsinization. In this study, we demonstrate that these tLDA‐isolated mMSCs (which we call ‘tLDA‐mMSCs’), have classic tri‐lineage differentiation potential, have immunosuppressive effects, and show homing and therapeutic properties in a model of osteoporosis. The data presented here demonstrate that MSCs from mouse bone marrow can be isolated with a single‐step plastic‐adherent procedure.
Materials and methods
Animals
The C57BL/6 and ICR mice were purchased from the Laboratory Animal Center of National Taiwan University College of Medicine (Taipei, Taiwan). Mice were kept under standard conditions, and all experimental procedures were approved by the Institutional Animal Care and Use Committee.
Isolation and culture of mouse bone marrow adherent cells
Bone marrow cells were obtained from 6‐ to 8‐week‐old C57BL/6 mice as described previously (23, 25) with the following modifications. Initially, we plated cells and four residual bone fragments on to 60‐cm2 tissue culture dishes (TPP, Trasadingen, Switzerland) at a density of 2 × 105 cells/cm2 in MEM alpha (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 20% foetal bovine serum (FBS; Hyclone, Logan, UT, USA), 2 mm l‐glutamine (Invitrogen, Carlsbad, CA, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen); no cytokines were added at any stage. All non‐adherent cells were removed after 72 h. When these primary cultures reached 70% confluence, cells were lifted by incubation with 0.025% trypsin/0.1 mm ethylenediaminetetraacetic acid (trypsin/EDTA; Invitrogen) for 5 min at 37 °C. They were then ready for tLDA purification and immunodepletion as described below. Regular subsequent passages were performed by replating the cells at concentration of 5 × 104 cells/cm2.
Purification of mMSCs by tLDA approach
To isolate mMSCs using the tLDA approach, lifted cells from the primary culture were passed through a 30 μm mesh (Miltenyi Biotec, Auburn, CA, USA) to remove cell clumps containing both mMSCs and HCs. Then filtered single cells were replated on to 60‐cm2 tissue culture dishes at density of 1.25 × 104 cells/cm2. These cells were allowed to adhere for 2–3 h at 37 °C with 95% air and 5% CO2. Then, they were lifted after incubation with trypsin/EDTA for 5 min at 37 °C. The cells that did not separate in 5 min (which will be HCs) were discarded. Instead of flushing the cells repeatedly after trypsin digestion, dishes were only gently shaken to avoid HCs being removed from the plastic surface. Then lifted cells were seeded at 5 × 104 cells/cm2 and were defined as tLDA‐mMSCs.
Immunodepletion
To isolate mMSCs using immunodepletion, primary cultured cells were lifted by incubation with trypsin/EDTA, resuspended in washing buffer composed of phosphate‐buffered saline (PBS) with 0.5% bovine serum albumin (BSA; Sigma) and 2 mm EDTA, and passed through 30 μm mesh (Miltenyi Biotec) to remove cell clumps. Cell suspensions were centrifuged at 400 g for 10 min and supernatant was removed completely; cell pellets were resuspended in 90 μl of washing buffer per 107 cells. They were then incubated at 4 °C for 15 min on magnetic microbeads conjugated with antibodies either against CD11b or CD45 (Miltenyi Biotec), according to the manufacturer’s instructions.
Population doubling time
Mean population doubling time (PDT) was calculated according to the equation: TD = t plog2/(logNt − logNo), where Nt is number of cells harvested, No is number of cells inoculated and t was time in culture (in h). Trypan blue exclusion was used to count cells, with the help of a haemocytometer (38).
Immunofluorescence
For staining of cell surface proteins, cells were washed in PBS, fixed in 4% paraformaldehyde for 10 min, and blocked in 0.1% BSA (Sigma) in PBS with 0.1% Tween 20 (Sigma) for 30 min. Cells were then incubated in phycoerythrin (PE)‐coupled antibodies against mouse CD11b and CD45 (BD Biosciences, San Jose, CA, USA) for 1 h in the dark. For counterstaining, cells were incubated in 0.1 μg/ml 4,6 diamidino‐2‐phenylindole (DAPI; Invitrogen) for 1 min, rinsed in PBS and fluorescence was observed microscopically.
Fluorescence‐activated cell sorting analysis
Cells were detached using trypsin/EDTA and were stained with antibodies against CD11b, CD29, CD31, CD34, CD44, CD45, CD80 (B7‐1), CD86 (B7‐2), CD105, CD117, stem cell antigen (Sca‐1), class I (H‐2Kb) and class II (I‐A) MHC molecules (eBioscience, San Diego, CA, USA) for 30 min in the dark according to product instructions. After fixation, 10 000 events were acquired on a FACSCalibur (Becton Dickinson, San Jose, CA, USA) and analysed using the FlowJo software (Tree Star, Ashland, OR, USA). All experiments included negative controls without antibodies or with isotype controls (eBioscience).
Tri‐lineage differentiation assays
To induce adipogenic differentiation, cells were cultured in MEM alpha (Sigma) supplemented with 10% FBS, 10 μg/ml insulin (Sigma), 1 μm dexamethasone (Sigma), 0.5 mm isobutyl‐methylxanthine (Sigma) and 100 μm indomethacin (Sigma) for 2 weeks, and oil red O (Sigma) was used for lipid droplet staining (39). Osteogenic differentiation was induced by MEM alpha supplemented with 10% FBS, 0.1 μm dexamethasone, 10 mmβ‐glycerolphosphate (Sigma) and 50 μm ascorbic acid (Sigma) for 3 weeks. At day 14, committed osteogenic cells were characterized by alkaline phosphatase (ALPase) assay, and bone matrix mineralization was determined using alizarin red S (ARS; Sigma) staining at day 21 (39). To induce chondrogenic differentiation, a pellet culture system was used (40). Chondrogenic induction medium consisted of MEM alpha supplemented with 1% FBS, 6.25 μg/ml insulin, 50 μm ascorbic acid and 10 ng/ml TGF‐β1 (R&D Systems, Minneapolis, MN, USA). Production of proteoglycan was characterized by toluidine blue (Sigma) staining.
Mitogen proliferation assay and allogeneic mixed lymphocyte reaction
Mononuclear spleen cells from C57BL/6 and BALB/c mice were prepared using Ficoll‐Paque density centrifugation (1.077 g/ml; Amersham Pharmacia Biotech, Piscataway, NJ, USA), followed by two washes in RPMI 1640 (Invitrogen) supplemented with 50 μm 2‐mercaptoethanol (Invitrogen), 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. A graded number of mitomycin C (Sigma)‐treated mMSCs was seeded in triplicate in flat‐bottom 96‐well plates and maintained at 37 °C for 6 h. In mitogen proliferation assays, splenocytes (2 × 105 cells/well) from C57BL/6 mice containing 5 μg/ml concanavalin‐A (ConA; Sigma) were cultured with or without mMSCs. In mixed lymphocyte reaction (MLR), responding splenocytes (2 × 105 cells/well) and an equal number of mitomycin C‐treated allogeneic stimulating splenocytes from BALB/c mice were added into the mMSC culture. Proliferation assays were performed after 4 or 5 days using CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay kit (Promega, Madison, WI, USA), according to the manufacturer’s instructions. Absorbance was measured using a SpectraMax 190 ELISA plate reader (Molecular Devices, Sunnyvale, CA, USA) at 490 nm.
Generation of GFP‐expressing transgenic mice as mMSC donors for cell transplantation studies
To provide traceable mMSCs in the transplantation studies, we generated 11 EGFP‐expressing transgenic mouse lines by pronuclear microinjection of ICR strain zygotes with ScaI‐PstI DNA fragment containing β‐actin promoter‐driven EGFP from pCX‐EGFP plasmid (41, 42, 43). The best transgenic line that homogenously expressed high levels of GFP in mMSCs from bone marrow was selected by tLDA approach as described above.
Therapeutic studies of mMSCs in osteoporotic mice
To generate osteoporotic mice, 6‐week‐old female C57BL/6 mice were ovariectomized (OVX) by dorsal approach as described previously (44). Six months later, GFP‐labelled mMSCs were injected into the mice intravenously (n = 7 mice/group). For intravenous injection, each mouse received 1.5 × 106 GFP‐mMSCs in 200 μl PBS on day 0, 6, 12, 18, 24, and 30. An additional group of OVX mice was injected with PBS for comparison (sham control). Two months later, mice were killed for femoral microcomputerized tomography (μCT) analysis; then bone marrow was flushed from left femurs for GFP signal detection. The right femur bone marrow was fixed in 10% phosphate‐buffered formalin for 24 h and transferred to 70% ethanol for histological evaluation.
Histological evaluation
Femurs were dehydrated in ethanol, embedded in methylmethacrylate and sectioned longitudinally using a Leica/Jung 2255 microtome (Leica, Wetzlar, Germany) at an 8 μm thickness. Sections were processed for haematoxylin–eosin staining. For immunohistological staining of GFP protein, diluted rabbit anti‐GFP polyclonal antibody (Abcam, Cambridge, UK) and goat anti‐rabbit immunoglobulin G conjugated with biotin (Abcam) respectively were used. Sections were then incubated with horseradish peroxidase (HRP)‐conjugated streptavidin (Abcam), 3‐Amino‐9‐ethylcarbazole (AEC; Sigma) was added as substrate, and samples were washed and mounted.
Microcomputerized tomography
Femoral μCT was measured as described using NI Labview and MS Visual C/C++ (45). A designed graphical user interface was used for controlling all aspects of CT system. Quantification method of the 3‐dimensional bone density from femur head of osteoporotic mice was designed specifically for this study (Y.‐T. Fang, M.‐L. Jan, H.‐C. Lu, Y.‐C. Ni, S.‐Y. Peng, W.T.‐K. Cheng and L.‐H. Shen, unpublished data).
Statistical analysis
Data were expressed as mean ± standard deviation. Student’s t‐test was used to assess significance of the differences; P‐values <0.05 were considered statistically significant.
Results
Development of mouse bone marrow adherent cell cultures
Bone marrow cells were harvested from C57BL/6 mice (2.25 ± 0.17 × 107 bone marrow nucleate cells/mouse, n = 6), seeded at a density of 2 × 105 cells/cm2 and non‐adherent cells were removed by changing medium on day 3. Approximately, 2.03 ± 0.37 mMSC colonies (CFU‐F/cm2, n = 12) were developed on day 8. In addition to mMSCs, a large number of contaminating small spherical cells, characterized as lymphohaematopoietic cells, engaged on mMSCs colonies as a second layer (Fig. 1a). Fluorescence‐activated cell sorting (FACS) analysis revealed that more than 30% of cells that arose under CFU‐F growth conditions were HCs as shown by CD11b and CD45 expression (Fig. 1b). After trypsin digestion, some plastic‐adherent HCs could not be lifted by trypsinization (Fig. 1c,d), however, trypsin‐lifted cells still contained many HCs when seeded at density of 5 × 104 cells/cm2 in the next passage. These HCs not only adhered to the plastic surface, but also engaged with plastic‐adherent mMSCs (Fig. 1e,f).
Figure 1.
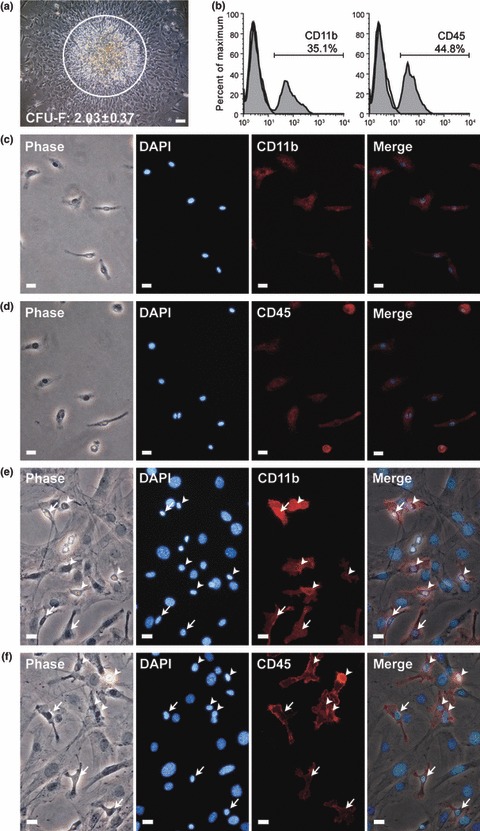
HCs contaminating MSCs from mouse bone marrow adherent cultures. (a) Phase‐contrast image of mMSC colony from primary bone marrow adherent culture. A large number of small spherical cells, characteristic of lymphohaematopoietic cells (circled), engaged upon mMSC colonies as a second layer. (b) Primary marrow adherent culture was analysed for expression of haematopoietic markers, CD11b and CD45, by flow cytometry. (c, d) Immunofluorescence staining (red) showed CD11b (c) and CD45 (d) expression on unlifted marrow adherent cells. (e) Phase‐contrast image of mouse bone marrow plastic‐adherent cells, first passage (seeding density: 5 × 104 cells/cm2). Immunofluorescence staining (red) showed CD11b+ and CD45+ HCs either adhered to plastic (arrow) or engaged on mMSCs (arrowhead). Nuclei counterstained with DAPI (blue). Scale bars represent 100 μm (a) and 50 μm (c–f).
Engaged HCs persistently contaminate mMSC cultures
To clarify whether persistent HC contamination resulted from HCs engaged with mMSCs, microscope time‐lapse recording analysis was carried out. This clearly showed that HCs engaged with mMSCs could be lifted together with mMSCs by trypsin digestion, despite being insensitive to trypsin themselves; on the other hand, HCs adhered adjacent to, but not engaged with mMSCs were not lifted during this process (Fig. 2a–f). Using conventional serial passage methods, mouse bone marrow adherent cultures were persistently contaminated with HCs. Nonetheless, these HCs gradually declined in number with increase in passages, and were barely detected at the 10th passage (Fig. S1a). However, although mMSCs acquired after 10 passages were pure enough to perform experiments that required a HC‐free cell population, proliferative activity of the mMSCs at this stage was significantly lower and had PDT of approximately 2–3 days (Fig. S1b). These cells became flat and polygonal in shape (Fig. S1c) and lacked general multi‐differentiation potential towards adipogenic, osteogenic and chondrogenic pathways (Fig. S1d–f). Therefore, the serial passage method was not suitable for experiments that required HC‐free population of mMSCs.
Figure 2.
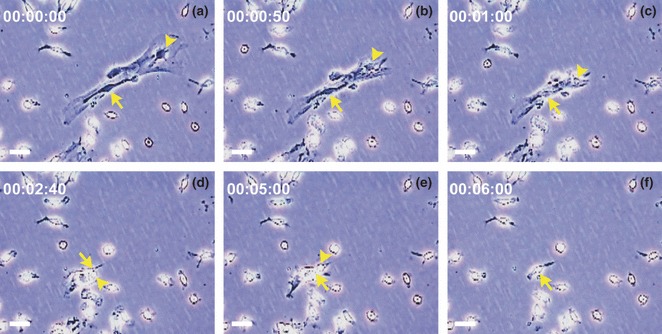
HCs engaged and lifted together with mMSCs in primary bone marrow adherent culture. (a–f) Representative sequential frames from time‐lapse video microscopic recording, showing one HC engaged on top of mMSCs, at the beginning of the recording, which lifted together with mMSCs by trypsin digestion within 6 min (arrowhead). HCs adjacent to but not overlapped with mMSCs (arrow) were not lifted. Scale bars represent 50 μm.
Isolation of mMSCs by tLDA approach
To segregate plastic‐adherent fibroblastic cells efficiently from HC contaminants, first, optimal dilution factors (from 5 × 104 to 1.25 × 104 cells/cm2) that allowed for segregation of bone marrow adherent cells to tissue culture plate surfaces were examined. In normal density culture (5 × 104 cells/cm2), most cells engaged and tightly interacted with each other (Fig. 3a). Below this density, adherent cells were better separated (Fig. 3b–d). When the culture was near confluence, these cells were retrieved by trypsin digestion. Indeed, most spherical cells (characterized as HCs) remained adherent to the plastic surface and were not easily trypsinized (data not shown). FACS analysis revealed that the percentage of HCs gradually declined in accordance with increasing dilutions of normal density cultures (Fig. 3f). Nevertheless, remaining mMSCs expanded poorly with a PDT of 48.97 ± 9.57 h (n = 3; Fig. 3e).
Figure 3.
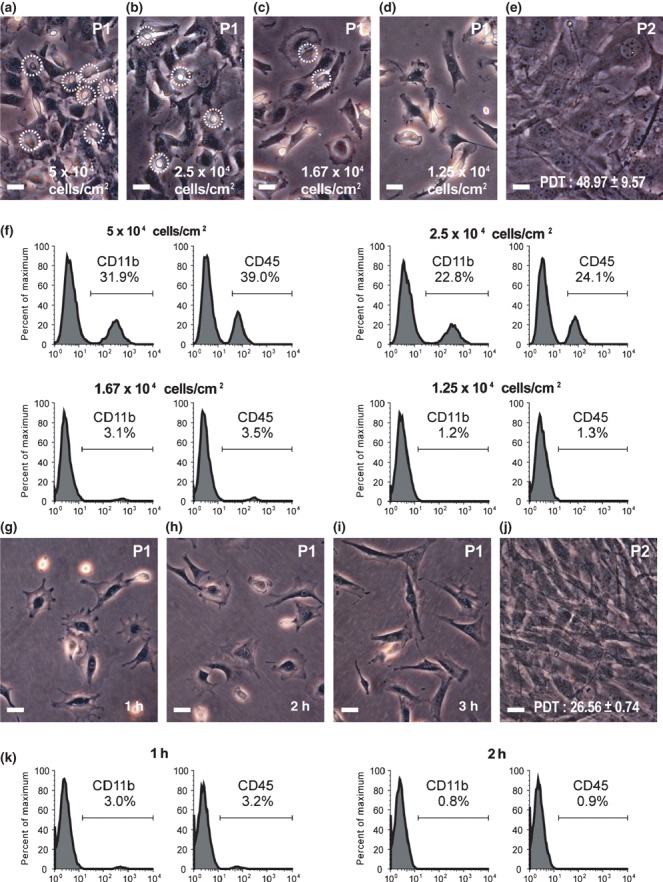
Effects of seeding density and adhering time on purity and proliferative competence of isolated mMSCs. (a–d) Phase‐contrast images of mouse bone marrow adherent cells from different seeding densities demonstrated that engaged HCs (circled) gradually became depleted with increasing dilutions. (e) Replating of confluent cells from lowest density culture (d, 1.25 × 104 cells/cm2) showed PDT of 48.97 ± 9.57 h. (f) After trypsin digestion, percentage of HCs (CD11b+ and CD45+) from different density cultures (a–d) was demonstrated by FACS analysis using anti‐CD11b and anti‐CD45 monoclonal antibodies. (g–i) Phase‐contrast images of mouse bone marrow adherent cells after seeded at density of 1.25 × 104 cells/cm2 for 1–3 h. (j) Replating of cells after 3 h adherence (i) at density of 5 × 104 cells/cm2 had PDT of 26.56 ± 0.74 h. Scale bars represent 50 μm. (k) After trypsin digestion, percentage HCs (CD11b+ and CD45+) from various degrees of adherent culture was demonstrated by FACS analysis using anti‐CD11b and anti‐CD45 monoclonal antibodies.
Next, with a time course experiment, we looked for a condition that would allow for rapid harvesting of mMSCs from the lowest cell density described earlier and simultaneously avoid non‐adherent HCs reappearing in cultures followed by trypsin digestion. By 1 h post‐culture, cells adhered but displayed incomplete morphology, while others floated in the medium, remaining unattached to the tissue culture dish (Fig. 3g). After 2–3 h, most of the cells adhered and showed phenotypes typical of fibroblastic and spherical cells (Fig. 3h,i). Then, these were retrieved by trypsin digestion. Spherical cells remained attached to the plate and were also not easily trypsinized (data not shown). FACS analysis revealed that presence of HCs was barely detectable in the cell population trypsinized after 2–3 h culture (3, 4 respectively). These cells designated ‘tLDA‐mMSCS’ had fibroblast morphology typical of MSCs, with a PDT of 26.56 ± 0.74 h (n = 3; Fig. 3j) compared to PDT of 48.97 ± 9.57 h of cells isolated from confluent lower‐density cultures (n = 3, P < 0.05; Fig. 3e).
Figure 4.
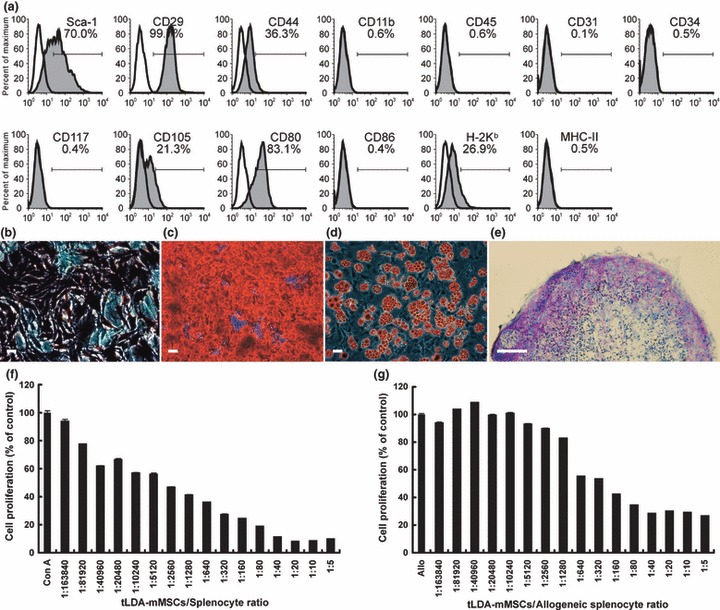
Characterization of tLDA‐mMSCs. (a) FACS analysis of tLDA‐mMSCs. A homogeneous confluent monolayer of tLDA‐mMSCs was trypsinized and analysed by staining with various antibodies. Respective isotype control shown as open histogram. (b–e) Osteogenic, adipogenic and chondrogenic capabilities of tLDA‐mMSCs. Osteogenic capability characterized by ALPase activity (b) and ARS staining (c) after 2 weeks’ and 3 weeks’ induction respectively. Adipogenic capability characterized by oil red O staining (d) after 2 weeks’ induction. Scale bars represent 50 μm (b–d). Chondrogenic capability evaluated by histological section micromass pellet cultures after 3 weeks’ induction. Glycosaminoglycan content of the pellet was revealed by toluidine blue staining (e). Scale bar represents 100 μm. (f–g) Immunosuppressive properties of tLDA‐mMSCs. In mitogen proliferation assays (f), responding splenocytes from C57BL/6 mice were stimulated with ConA in absence or presence of graded numbers of autologous tLDA‐mMSC (n = 6 for each group). In MLR culture (g), responding splenocytes from C57BL/6 mice were cultured with an equal number of mitomycin C‐treated BALB/c splenocytes, with or without graded number of C57BL/6 tLDA‐mMSCs (n = 6 for each group). Results shown as percentage of cell proliferation in comparison to control cell proliferation. Data expressed as mean ± SD of triplicate of two separate experiments. Allo indicates allogeneic splenocyte culture.
For all subsequent assays, we isolated mMSCs in these defined optimal conditions (lower density at 1.25 × 104 cells/cm2; transient adherence of 2–3 h). In general, we could obtain around 2.5 × 106 tLDA‐mMSCs from one mouse in the first passage directly after tLDA procedure. Purified tLDA‐mMSCs had viability of 98 ± 0.95% (n = 12).
Phenotype characterization of tLDA‐mMSCs
tLDA‐mMSCs were analysed for expression of a panel of antigens (Fig. 4a). FACS analysis revealed that these cells uniformly expressed Sca‐1 (Ly‐6A/E; murine haematopoietic and mesenchymal stem/progenitor cell marker), CD29 (β1‐integrin), CD44 (Pgp‐1; receptor for hyaluronate and osteopontin) and variable levels of CD105 (endoglin), but did not express CD11b (integrin αM; monocyte marker), CD34 (mucosialin), CD45 (leucocyte common antigen), CD117 (c‐kit) and CD31 (platelet‐endothelial cell adhesion molecule‐1). In addition, tLDA‐mMSCs did not express MHC II (I‐A) and co‐stimulating molecules for CD86 (B7‐2), but they did express H‐2Kb and CD80 (B7‐1). These data were consistent with mMSCs isolated using magnetic beads coupled with anti‐CD11b and CD45‐specific antibodies (CD11b−/CD45− mMSCs, Fig. S2).
In vitro differentiation of tLDA‐mMSCs
To investigate osteogenic potential, confluent tLDA‐mMSC cultures were grown in osteogenic induction medium. Significantly greater ALPase activity (marker of osteoblastic differentiation), was observed on day 10 (Fig. 4b). By day 14, these cells generated mineral nodules and more than 80% of tLDA‐mMSCs showed mineral accumulation after 21 days. ARS staining confirmed elaborately mineralized matrix brilliant red colour in colour (Fig. 4c).
To determine adipogenic potential, tLDA‐mMSCs were also grown to confluence and cultured in adipogenic induction medium. Neutral lipid droplets were visible within cytoplasm after 7 days and were easily recognizable after 9 days. Lipid droplet‐containing cells accounted for more than 80% of induced tLDA‐mMSC cultures as observed on day 14, as illustrated by oil red O staining (Fig. 4d).
Chondrogenic potential of tLDA‐mMSCs was evaluated by culturing cells using the pelleted micromass system in chondrogenic induction medium. After 3 weeks of differentiation, chondrogenesis was confirmed by toluidine blue staining of sectioned micromasses showing purple stained matrix (Fig. 4e). In all differentiation experiments, undifferentiated mMSCs were used as negative control and were consistently negative for each histochemical staining (data not shown).
Immunosuppressive characteristics of the tLDA‐mMSCs
It has been well documented that multipotent MSCs have immunosuppressive properties. Specifically, they can inhibit T‐cell proliferation when induced by MLR or by non‐specific mitogens. To determine whether tLDA‐mMSCs had similar features, we first co‐cultured mitomycin C‐treated tLDA‐mMSCs from C57BL/6 mice with autologous splenocytes in the presence of ConA. tLDA‐mMSCs significantly inhibited ConA‐induced splenocyte proliferation in a dose‐dependent manner. Notably, even when only around five tLDA‐mMSCs were present in the cultures (tLDA‐mMSCs‐to‐splenocytes ratio: 1/40,960), they could diminish splenocyte proliferation to 61.69 ± 0.41% (n = 6) compared to corresponding negative control (100%) without mMSCs (P < 0.05; Fig. 4f).
We also carried out experiments to determine whether tLDA‐mMSCs could inhibit allogeneic‐induced T‐cell proliferation. To address this, we cultured splenocytes from C57BL/6 mice with allogeneic BALB/c mitomycin C‐treated splenocytes in C57BL/6 tLDA‐mMSCs cultures (one‐way MLR). Likewise, tLDA‐mMSCs exhibited dose‐dependent suppression of allogeneic T‐cell responses. Although presence of up to 40 tLDA‐mMSCs (tLDA‐mMSCs‐to‐allogeneic splenocyte ratio: 1/5,120) they had no impact on T‐cell proliferation and dramatic suppression (>50%) was observed when 1250 tLDA‐mMSCs were present in mixed lymphocyte cultures (tLDA‐mMSCs‐to‐allogeneic splenocyte ratio: 1/160), as compared to allogeneic T‐cell response without mMSCs (42.18 ± 0.26%, n = 6, P < 0.05; Fig. 4g). These results showed that tLDA‐mMSCs were capable of suppressing splenocyte proliferation in vitro, demonstrating their important role in immunosuppression.
Homing and therapeutic effects of tLDA‐mMSCs in a model of osteoporotic disease
We studied function of tLDA‐mMSCs in an animal model of osteoporosis. Osteoporosis was induced by bilateral ovariectomy of 6‐week‐old female mice (C57BL/6). Six months after osteoporosis induction, the mice were injected intravenously with GFP‐labelled tLDA‐mMSCs (GFP‐tLDA‐mMSCs, ICR strain) or PBS. Two months post‐injection, they were killed and GFP‐tLDA‐mMSCs were observed in flushed bone marrow cells, under a fluorescence microscope (Fig. 5a). In addition, the GFP signal was also confirmed by immunohistochemistry (IHC), demonstrating their homing ability to bone marrow (Fig. 5b). Moreover, GFP‐positive cells were densely distributed in cortical and trabecular bone areas (Fig. 5c). Control tissue from osteoporotic mice injected with PBS showed no GFP staining (Fig. 5d). By carrying out histological evaluation, it was demonstrated that tLDA‐mMSC transplantation rescued the hosts, osteoporotic mice (Fig. 5e). Analysis of femoral μCT from mice transplanted with tLDA‐mMSCs also showed increased bone density from distal end of the endocortical compartment when compared to sham controls, without substantial change in cortical bone (Fig. 5f). Moreover, after transplanting GFP‐tLDA‐mMSCs into osteoporotic mice for 2 months, bone volume fraction became indistinguishable when compared to normal mice (P = 0.1485).
Figure 5.
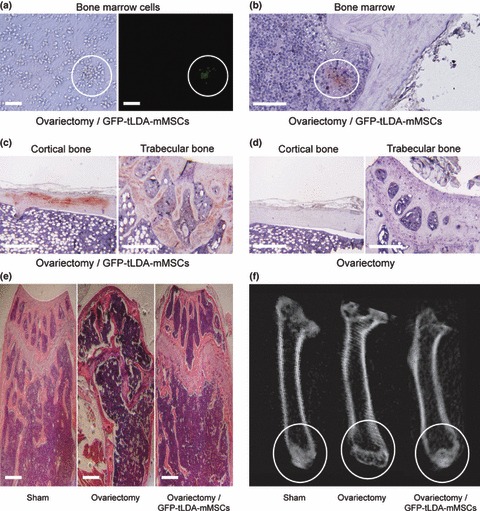
Repair of ovariectomy‐induced bone loss in mice after injection of tLDA‐mMSCs. Mice undergoing osteoporotic injury were injected intravenously with GFP‐tLDA‐mMSCs. (a) Phase‐contrast (left) and fluorescence (right) images of bone marrow cells isolated from osteoporotic mice 2 months after injection of GFP‐tLDA‐mMSCs. (b–c) GFP positive signal was found in bone marrow (b), cortical bone (left) and trabecular bone (right) (c) by IHC staining. (d) Control tissue (left: cortical bone; right: trabecular bone) from osteoporotic mice injected with PBS showed no GFP staining. (e) Histological section of femurs taken from sham‐operated mice (left), OVX mice (middle) and OVX mice receiving GFP‐tLDA‐mMSCs (right). Scale bars represent 50 μm. (f) Images of μCT scans of femurs taken from sham‐operated mice (left), OVX mice (middle) and OVX mice receiving GFP‐tLDA‐mMSCs (right) at 10 μm resolution.
Discussion
There are several advantages in isolating mMSCs using tLDA method. First, the mMSCs can be readily isolated from bone marrow cell adherent cultures within 3 h. Second, mMSCs isolated retain good proliferative and therapeutic potentials. Third, the mMSCs are isolated using their native properties and no prior labelling of the cells is required. Fourth, the tLDA method is extremely straightforward. It can be easily established and readily transferred to other research laboratories. Finally, as the tLDA is a non‐antigenic based method, it can be further performed in combination with immunoselection methods, while the assays are developed for use of specific MSC subtypes.
The mouse is a valuable model animal for human diseases, with unique richness in functional genomics‐related resources and gene‐targeting‐based platforms. In addition, well‐established inbred mouse strains can be used for autotransplantation‐equivalent studies between different individuals of the same genetic background. We therefore would like to use mMSCs for various mechanistic and therapeutic studies. However, the standard unmodified method based on plastic adherence has been confirmed as unsuccessful for mMSCs isolation because of persistent HC contamination within adherent bone marrow cultures. In most cases, the conventional approach to eradicate contaminating HCs from mMSC culture is by repeatedly passaging the adherent bone marrow cells by trypsin digestion (23, 24, 25). Consequently, HC‐free mMSCs are generally obtained after weeks of repeated passaging, with the resulting cells showing poor proliferation and differentiation activities (26, 27, 28). Herein, we have demonstrated the need to remove HCs that engage with mMSCs. These engaged HCs can be lifted together with mMSCs despite their insensitivity to trypsin digestion. Accordingly, we developed the tLDA method that can efficiently and effectively remove HCs from mMSCs within 3 h. The ‘lower density’ of 1.25 × 104 cells/cm2 and ‘transient attachment’ for no more than 3 h are both crucial for viability and differentiation potential of the resulting tLDA‐mMSCs. Seeding density of 1.25 × 104 cells/cm2 is crucial to separate all HCs from mMSCs. This allows all HCs to adhere to the plastic surface directly to avoid being lifted with mMSCs upon subculture. However, if we let these separated cells grow to confluence (which took 4 days in our hands), PDT of resulting mMSCs was significantly higher. For this reason, we would suggest using the transient adherent procedure for no more than 3 h immediately after our lower‐density protocol (Fig. 6). This step helped us to overcome the tedious and time‐consuming mMSC isolation procedures and lowered potential problems caused by long‐term passages in vitro.
Figure 6.
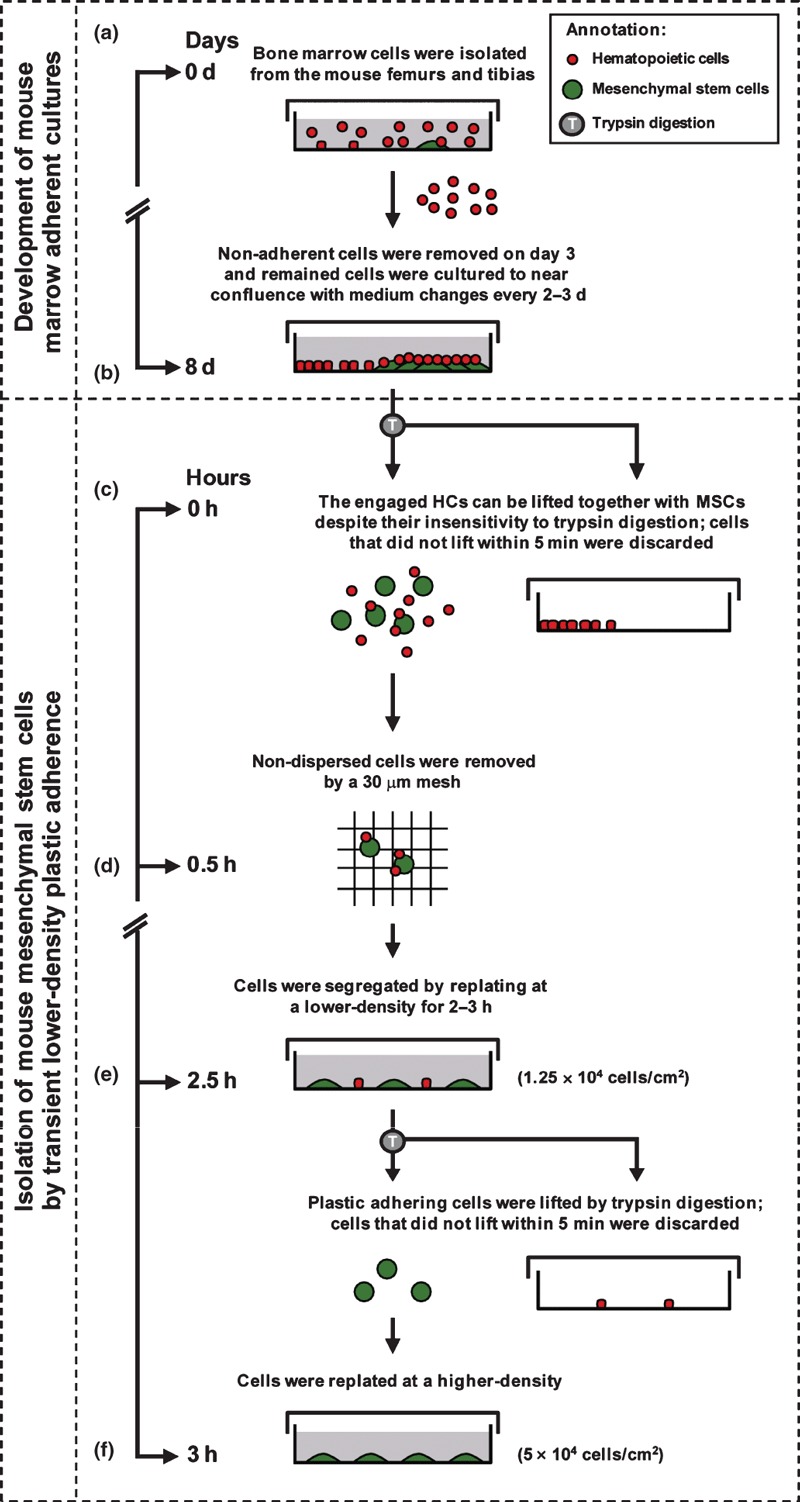
Timeline showing stages required to isolate tLDA‐mMSCs from heterogeneous bone marrow cultures. (a) Bone marrow adherent cell cultures were initiated in a plastic dish. Non‐adherent cell population was removed on day 3 and remaining adherent cells were cultured to near confluence with medium changes twice weekly. (b) By day 8, mMSCs clone developed, with some HCs adhering to the plastic surface of the dish, while others engaged on mMSCs. (c) After trypsin digestion, some engaged HCs were lifted with mMSCs (most HCs were segregated from mMSCs, whereas some of them still adhered to mMSCs). Plastic‐adherent trypsin‐insensitive HCs were depleted. (d) To isolate mMSCs using the tLDA method, non‐dispersed cells were first removed using a 30 μm mesh after trypsin digestion. (e) mMSCs could be segregated from HCs using lower‐density culture (1.25 × 104 cells/cm2) on plastic within 2.5 h. After being lifted by trypsin digestion, mMSCs were retrieved from the cultures, whereas trypsin‐insensitive HCs were depleted from recovered cells as they remained adherent to plastic. (f) tLDA‐mMSCs could be obtained with good proliferative and therapeutic potentials when cells were replated at a density of 5 × 104 cells/cm2.
Several previous studies have introduced that very low‐density plating (50 cells/cm2) can obtain clonally expanded mMSCs from bone marrow adherent cultures (33, 39). However, this method may not be practical enough due to the tedious culture schemes that take more than 3 months. In addition, Miura et al. (46) demonstrated accumulation of chromosomal instability during prolonged cultures/passages of mMSCs. As a result, when they used these mMSCs for therapeutic transplantation, malignant transformation was observed. These studies demonstrated a potential risk in using long‐term repeat cultured mMSCs for therapeutic purposes. With our tLDA protocol, we can dramatically cut down duration of in vitro culture/manipulation, before we obtain HC‐free mMSCs for extensive transplantation or mechanistic studies, thus significantly increasing the accuracy of the experimental outcome.
Although immunodepletion using antibodies against HCs expressing surface markers, CD11b+, CD34+ and CD45+, can also be used to rapidly remove contaminated HCs and greatly shorten mMSCs isolation procedure, this protocol might reduce viability of mMSCs as a result of down‐regulation of genes involved in cell proliferation and cell cycle progression (16). Moreover, obvious disadvantages for implementing such an isolation protocol are cost of magnetic antibody labelling, consumption of many non‐reusable separation columns and need of specific magnet separators. In contrast, our tLDA approach is relatively simple, and no special reagents and devices are required. In addition, this tLDA step does not require labelling of antibodies to cells precluding impairment of cell viability as seen for immunodepletion, in which the magnetic labelling process is performed under cold conditions using pre‐cooled solutions to prevent capping of antibodies on cell surfaces, and non‐specific cell labelling. In our observations, despite tLDA‐mMSCs exhibiting homogeneous surface markers with the CD11b−/CD45− mMSCs, purified tLDA‐mMSCs had viability of 98 ± 0.95% (n = 12), compared to 84 ± 3.27% (n = 12) in the CD11b−/CD45− mMSCs.
Recently, there have been studies to discover specific surface antigens for isolating pure populations of MSCs from mouse bone marrow, by immunoselection (47). In addition, a series of monoclonal antibodies initially prepared for other cell types have also been used to characterize mMSCs (48, 49, 50). Although published markers targeting mMSCs are useful, some surface markers may vary between different mMSC culture conditions and mouse strains (51, 52, 53, 54). Moreover, some immunoselection schemes may introduce genetic and epigenetic changes from the isolated cells (3) Therefore, isolating mMSCs with any specific antibody has not yet provided satisfactory results. This underscored our proposal that use of the non‐antigenic‐based tLDA method might provide a useful approach for isolating mMSCs; tLDA can also be performed in combination with immunoselection or immunodepletion, if identification of a particular subtype of mMSCs is necessary.
The tLDA‐mMSCs possessed most characteristics published for bone marrow‐derived mMSCs. Using flow cytometry, we did not find any substantial differences between tLDA‐mMSCs and bone marrow‐derived mMSCs (16, 25, 39, 55). Moreover, we validated MSC properties by inducing differentiation of tLDA‐mMSCs along the three main mesenchymal lineages: osteoblasts, adipocytes and chondrocytes. This multipotency allowed us to conclude that these cells were MSCs as defined by Pittenger et al. (56). When we transplanted GFP‐labelled tLDA‐mMSCs into mice suffering from osteoporosis, low, but clearly detectable levels of GFP‐positive cells were observed in the allogeneic bone marrow. Assays of histopathology and μCT scans illustrated improved microstructure in newly formed trabecular bone tissue. These results suggested that tLDA‐mMSCs retained their capacity of homing to bone marrow and also that they localized to sites of injured bone and prevented/recovered bone loss in mice suffering from osteoporosis.
In summary, we demonstrated a simple and effective tLDA method to isolate HC‐free mMSCs. The tLDA‐mMSCs are homogeneous with regard to surface epitopes, have multidifferentiation potentials, have immunosuppressive properties and are therapeutically functional as demonstrated in the osteoporosis model. Thus, this innovative method could significantly facilitate mMSC‐based differentiation studies in molecular and genetics/epigenetic levels and MSC‐mediated therapies in mouse models of human diseases.
Supporting information
Fig. S1 Presence of HCs within adherent mouse bone marrow cultures at different passages. (a) HCs were analysed by FACS at different passages using anti‐CD11b and anti‐CD45 antibodies. (b) PDT of adherent mouse bone marrow cells at different passages. Data are expressed as mean ± SD, triplicate of one representative experiment. (c) Phase‐contrast image of serial‐passaged mMSCs (passage 10). Scale bars represent 50 mm. (d–f) Osteogenic, adipogenic and chondrogenic capabilities of serial‐passaged mMSCs (passage 10). Osteogenic capability characterized by ARS staining (d) after 3 weeks’ induction. Adipogenic capability characterized by oil red O staining (e) after 2 weeks’ induction. Scale bars represent 50 mm. Chondrogenic capability evaluated in histological section of a micromass pellet cultures after 3 weeks’ induction. Glycosaminoglycan content of the pellet revealed by toluidine blue staining (f). Scale bar represents 100 mm.
Fig. S2 Immunophenotype of CD11b‐/CD45‐mMSCs. A homogeneous confluent monolayer of CD11b‐/CD45‐ mMSCs was trypsinized and stained with surface antibodies as indicated, and analysed using FACS. Plots show isotype control IgG‐staining profile (open histogram) versus specific antibody staining profile (shaded histogram).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
We thank Dr Shiaw‐Min Hwang, Chuan‐Mu Chen, Shan‐Hui Hsu, Shou‐Cheng Chen and Bernhard Payer for discussions and comments on the manuscript, as well as Dr Masaru Okabe and Jun‐Ichi Miyazaki (Osaka University, Japan) for providing the pCX‐EGFP plasmid. This study was supported by grants from the National Taiwan University (Grants 97R0066‐40 to W.T.‐K.C.; 97R0066‐41 to S.‐C.W.; 97R0066‐46 to S.‐P.L.) and the National Science Council (Grant NSC 94‐2313‐B‐002‐044 to W.T.‐K.C.).
References
- 1. Bianco P, Robey PG, Simmons PJ (2008) Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell 2, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chamberlain G, Fox J, Ashton B, Middleton J (2007) Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 25, 2739–2749. [DOI] [PubMed] [Google Scholar]
- 3. Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair – current views. Stem Cells 25, 2896–2902. [DOI] [PubMed] [Google Scholar]
- 4. Liu H, Kemeny DM, Heng BC, Ouyang HW, Melendez AJ, Cao T (2006) The immunogenicity and immunomodulatory function of osteogenic cells differentiated from mesenchymal stem cells. J. Immunol. 176, 2864–2871. [DOI] [PubMed] [Google Scholar]
- 5. Nauta AJ, Fibbe WE (2007) Immunomodulatory properties of mesenchymal stromal cells. Blood 110, 3499–3506. [DOI] [PubMed] [Google Scholar]
- 6. Kawada H, Fujita J, Kinjo K, Matsuzaki Y, Tsuma M, Miyatake H et al. (2004) Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood 104, 3581–3587. [DOI] [PubMed] [Google Scholar]
- 7. Giordano A, Galderisi U, Marino IR (2007) From the laboratory bench to the patient’s bedside: an update on clinical trials with mesenchymal stem cells. J. Cell. Physiol. 211, 27–35. [DOI] [PubMed] [Google Scholar]
- 8. Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M et al. (1999) Transplantability and therapeutic effects of bone marrow‐derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 5, 309–313. [DOI] [PubMed] [Google Scholar]
- 9. Petite H, Viateau V, Bensaid W, Meunier A, De Pollak C, Bourguignon M et al. (2000) Tissue‐engineered bone regeneration. Nat. Biotechnol. 18, 959–963. [DOI] [PubMed] [Google Scholar]
- 10. Prockop DJ, Olson SD (2007) Clinical trials with adult stem/progenitor cells for tissue repair: let’s not overlook some essential precautions. Blood 109, 3147–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quarto R, Mastrogiacomo M, Cancedda R, Kutepov SM, Mukhachev V, Lavroukov A et al. (2001) Repair of large bone defects with the use of autologous bone marrow stromal cells. N. Engl. J. Med. 344, 385–386. [DOI] [PubMed] [Google Scholar]
- 12. Ringden O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lonnies H et al. (2006) Mesenchymal stem cells for treatment of therapy‐resistant graft‐versus‐host disease. Transplantation 81, 1390–1397. [DOI] [PubMed] [Google Scholar]
- 13. Friedenstein AJ, Chailakhjan RK, Lalykina KS (1970) The development of fibroblast colonies in monolayer cultures of guinea‐pig bone marrow and spleen cells. Cell Tissue Kinet. 3, 393–403. [DOI] [PubMed] [Google Scholar]
- 14. Owen M, Friedenstein AJ (1988) Stromal stem cells: marrow‐derived osteogenic precursors. Ciba Found. Symp. 136, 42–60. [DOI] [PubMed] [Google Scholar]
- 15. Prockop DJ, Gregory CA, Spees JL (2003) One strategy for cell and gene therapy: harnessing the power of adult stem cells to repair tissues. Proc. Natl. Acad. Sci. USA 100(Suppl. 1), 11917–11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baddoo M, Hill K, Wilkinson R, Gaupp D, Hughes C, Kopen GC et al. (2003) Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J. Cell. Biochem. 89, 1235–1249. [DOI] [PubMed] [Google Scholar]
- 17. Bearpark AD, Gordon MY (1989) Adhesive properties distinguish sub‐populations of haemopoietic stem cells with different spleen colony‐forming and marrow repopulating capacities. Bone Marrow Transplant. 4, 625–628. [PubMed] [Google Scholar]
- 18. Deryugina EI, Muller‐Sieburg CE (1993) Stromal cells in long‐term cultures: keys to the elucidation of hematopoietic development? Crit. Rev. Immunol. 13, 115–150. [PubMed] [Google Scholar]
- 19. Kerk DK, Henry EA, Eaves AC, Eaves CJ (1985) Two classes of primitive pluripotent hemopoietic progenitor cells: separation by adherence. J. Cell. Physiol. 125, 127–134. [DOI] [PubMed] [Google Scholar]
- 20. Simmons PJ, Masinovsky B, Longenecker BM, Berenson R, Torok‐Storb B, Gallatin WM (1992) Vascular cell adhesion molecule‐1 expressed by bone marrow stromal cells mediates the binding of hematopoietic progenitor cells. Blood 80, 388–395. [PubMed] [Google Scholar]
- 21. Barda‐Saad M, Rozenszajn LA, Ashush H, Shav‐Tal Y, Ben Nun A, Zipori D (1999) Adhesion molecules involved in the interactions between early T cells and mesenchymal bone marrow stromal cells. Exp. Hematol. 27, 834–844. [DOI] [PubMed] [Google Scholar]
- 22. Deans RJ, Moseley AB (2000) Mesenchymal stem cells: biology and potential clinical uses. Exp. Hematol. 28, 875–884. [DOI] [PubMed] [Google Scholar]
- 23. Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E et al. (2003) Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen‐specific T cells to their cognate peptide. Blood 101, 3722–3729. [DOI] [PubMed] [Google Scholar]
- 24. Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J et al. (1999) Cardiomyocytes can be generated from marrow stromal cells in vitro. J. Clin. Invest. 103, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun S, Guo Z, Xiao X, Liu B, Liu X, Tang PH et al. (2003) Isolation of mouse marrow mesenchymal progenitors by a novel and reliable method. Stem Cells 21, 527–535. [DOI] [PubMed] [Google Scholar]
- 26. Baxter MA, Wynn RF, Jowitt SN, Wraith JE, Fairbairn LJ, Bellantuono I (2004) Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 22, 675–682. [DOI] [PubMed] [Google Scholar]
- 27. Digirolamo CM, Stokes D, Colter D, Phinney DG, Class R, Prockop DJ (1999) Propagation and senescence of human marrow stromal cells in culture: a simple colony‐forming assay identifies samples with the greatest potential to propagate and differentiate. Br. J. Haematol. 107, 275–281. [DOI] [PubMed] [Google Scholar]
- 28. Guo Z, Li H, Li X, Yu X, Wang H, Tang P et al. (2006) In vitro characteristics and in vivo immunosuppressive activity of compact bone‐derived murine mesenchymal progenitor cells. Stem Cells 24, 992–1000. [DOI] [PubMed] [Google Scholar]
- 29. Tropel P, Noel D, Platet N, Legrand P, Benabid AL, Berger F (2004) Isolation and characterisation of mesenchymal stem cells from adult mouse bone marrow. Exp. Cell Res. 295, 395–406. [DOI] [PubMed] [Google Scholar]
- 30. Nadri S, Soleimani M (2007) Isolation murine mesenchymal stem cells by positive selection. In Vitro Cell. Dev. Biol. Anim. 43, 276–282. [DOI] [PubMed] [Google Scholar]
- 31. Van Vlasselaer P, Falla N, Snoeck H, Mathieu E (1994) Characterization and purification of osteogenic cells from murine bone marrow by two‐color cell sorting using anti‐Sca‐1 monoclonal antibody and wheat germ agglutinin. Blood 84, 753–763. [PubMed] [Google Scholar]
- 32. Eslaminejad MB, Nikmahzar A, Taghiyar L, Nadri S, Massumi M (2006) Murine mesenchymal stem cells isolated by low density primary culture system. Dev. Growth Differ. 48, 361–370. [DOI] [PubMed] [Google Scholar]
- 33. Wang QR, Wolf NS (1990) Dissecting the hematopoietic microenvironment. VIII. Clonal isolation and identification of cell types in murine CFU‐F colonies by limiting dilution. Exp. Hematol. 18, 355–359. [PubMed] [Google Scholar]
- 34. Modderman WE, Vrijheid‐Lammers T, Lowik CW, Nijweide PJ (1994) Removal of hematopoietic cells and macrophages from mouse bone marrow cultures: isolation of fibroblastlike stromal cells. Exp. Hematol. 22, 194–201. [PubMed] [Google Scholar]
- 35. Falla N, Van V, Bierkens J, Borremans B, Schoeters G, Van Gorp U (1993) Characterization of a 5‐fluorouracil‐enriched osteoprogenitor population of the murine bone marrow. Blood 82, 3580–3591. [PubMed] [Google Scholar]
- 36. Meirelles Lda S, Nardi NB (2003) Murine marrow‐derived mesenchymal stem cell: isolation, in vitro expansion, and characterization. Br. J. Haematol. 123, 702–711. [DOI] [PubMed] [Google Scholar]
- 37. Soleimani M, Nadri S (2009) A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 4, 102–106. [DOI] [PubMed] [Google Scholar]
- 38. Nadri S, Soleimani M, Hosseni RH, Massumi M, Atashi A, Izadpanah R (2007) An efficient method for isolation of murine bone marrow mesenchymal stem cells. Int. J. Dev. Biol. 51, 723–729. [DOI] [PubMed] [Google Scholar]
- 39. Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ (2004) Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 103, 1662–1668. [DOI] [PubMed] [Google Scholar]
- 40. Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU (1998) In vitro chondrogenesis of bone marrow‐derived mesenchymal progenitor cells. Exp. Cell Res. 238, 265–272. [DOI] [PubMed] [Google Scholar]
- 41. Lee KH, Chuang CK, Wang HW, Stone L, Chen CH, Tu CF (2007) An alternative simple method for mass production of chimeric embryos by coculturing denuded embryos and embryonic stem cells in Eppendorf vials. Theriogenology 67, 228–237. [DOI] [PubMed] [Google Scholar]
- 42. Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 108, 193–199. [DOI] [PubMed] [Google Scholar]
- 43. Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y (1997) ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 407, 313–319. [DOI] [PubMed] [Google Scholar]
- 44. Bellino FL (2000) Nonprimate animal models of menopause: workshop report. Menopause 7, 14–24. [DOI] [PubMed] [Google Scholar]
- 45. Jan M‐L, Ni T‐C, Chen K‐W, Liang H‐C, Chuang K‐S, Fu Y‐K (2006) A combined micro‐PET/CT scanner for small animal imaging. Nucl. Instrum. Methods Phys. Res. A 569, 314–318. [Google Scholar]
- 46. Miura M, Miura Y, Padilla‐Nash HM, Molinolo AA, Fu B, Patel V et al. (2006) Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells 24, 1095–1103. [DOI] [PubMed] [Google Scholar]
- 47. Gindraux F, Selmani Z, Obert L, Davani S, Tiberghien P, Herve P et al. (2007) Human and rodent bone marrow mesenchymal stem cells that express primitive stem cell markers can be directly enriched by using the CD49a molecule. Cell Tissue Res. 327, 471–483. [DOI] [PubMed] [Google Scholar]
- 48. Anjos‐Afonso F, Bonnet D (2007) Nonhematopoietic/endothelial SSEA‐1+ cells define the most primitive progenitors in the adult murine bone marrow mesenchymal compartment. Blood 109, 1298–1306. [DOI] [PubMed] [Google Scholar]
- 49. Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC (2007) SSEA‐4 identifies mesenchymal stem cells from bone marrow. Blood 109, 1743–1751. [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Hisha H, Taketani S, Adachi Y, Li Q, Cui W et al. (2006) Characterization of mesenchymal stem cells isolated from mouse fetal bone marrow. Stem Cells 24, 482–493. [DOI] [PubMed] [Google Scholar]
- 51. Dazzi F, Ramasamy R, Glennie S, Jones SP, Roberts I (2006) The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 20, 161–171. [DOI] [PubMed] [Google Scholar]
- 52. Devine SM (2002) Mesenchymal stem cells: will they have a role in the clinic? J. Cell. Biochem. Suppl. 38, 73–79. [DOI] [PubMed] [Google Scholar]
- 53. Javazon EH, Beggs KJ, Flake AW (2004) Mesenchymal stem cells: paradoxes of passaging. Exp. Hematol. 32, 414–425. [DOI] [PubMed] [Google Scholar]
- 54. Pittenger MF, Martin BJ (2004) Mesenchymal stem cells and their potential as cardiac therapeutics. Circ. Res. 95, 9–20. [DOI] [PubMed] [Google Scholar]
- 55. Stagg J, Galipeau J (2007) Immune plasticity of bone marrow‐derived mesenchymal stromal cells. Handb. Exp. Pharmacol. 180, 45–66. [DOI] [PubMed] [Google Scholar]
- 56. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Presence of HCs within adherent mouse bone marrow cultures at different passages. (a) HCs were analysed by FACS at different passages using anti‐CD11b and anti‐CD45 antibodies. (b) PDT of adherent mouse bone marrow cells at different passages. Data are expressed as mean ± SD, triplicate of one representative experiment. (c) Phase‐contrast image of serial‐passaged mMSCs (passage 10). Scale bars represent 50 mm. (d–f) Osteogenic, adipogenic and chondrogenic capabilities of serial‐passaged mMSCs (passage 10). Osteogenic capability characterized by ARS staining (d) after 3 weeks’ induction. Adipogenic capability characterized by oil red O staining (e) after 2 weeks’ induction. Scale bars represent 50 mm. Chondrogenic capability evaluated in histological section of a micromass pellet cultures after 3 weeks’ induction. Glycosaminoglycan content of the pellet revealed by toluidine blue staining (f). Scale bar represents 100 mm.
Fig. S2 Immunophenotype of CD11b‐/CD45‐mMSCs. A homogeneous confluent monolayer of CD11b‐/CD45‐ mMSCs was trypsinized and stained with surface antibodies as indicated, and analysed using FACS. Plots show isotype control IgG‐staining profile (open histogram) versus specific antibody staining profile (shaded histogram).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item