

Abstract
Abstract. Atypical protein kinase C‐iota (PKC‐ι) protects cells against apoptosis and may play a role in cell proliferation. However, in vivo, the status and function of PKC‐ι in human normal brain tissue, gliomas, benign and malignant meningiomas as well as its in vitro status in proliferating and confluent glioma cells, remains unknown. Objectives: The objectives of our research were to determine whether expression of PKC‐ι is altered either in gliomas or in benign and malignant meningiomas, compared to normal brain. In addition, we wished to establish the expression of PKC‐ι in proliferating plus in cell cycle‐arrested glioma cell lines, as well as the relationship between PKC‐ι siRNA on PKC‐ι protein content and cell proliferation. Materials and Methods: Western blot analyses for PKC‐ι were performed on 12 normal brain biopsies, 15 benign meningiomas, three malignant meningiomas and three gliomas. Results: Results demonstrated no (n = 9) or very weak (n = 3) detection of PKC‐ι in normal brain tissue. In comparison, PKC‐ι was robustly present in the majority of the benign meningiomas. Similarly, PKC‐ι was abundant in all malignant meningiomas and gliomas. Western blotting for PKC‐ι in confluent or proliferating glioma cell lines depicted substantial quantities of PKC‐ι in proliferating T98G and U‐138MG glioma cells. In contrast, confluent cells had either 71% (T98G) or 21% (U‐138MG) less PKC‐ι than proliferating cells. T98 and U‐138 MG glioma cells treated with 100 nm PKC‐ι siRNA had lower levels of cell proliferation compared to control siRNA‐A and complete down‐regulation of PKC‐ι protein content. Conclusion: These results support the concept that presence of PKC‐ι may be required for cell proliferation to take place.
INTRODUCTION
Protein kinase Cs (PKCs) compose a family of 14 known isozymes found in varying ratios in the cytosolic and membrane fractions of cells, depending on the type of tissue and its physiological state (Nishizuka 1992). PKC isozymes can be classified into three groups. Group I includes Ca2+‐dependent isozymes: cPKC‐α, cPKC‐βl cPKC‐βll and cPKC‐γ. Isozymes in group II, nPKC‐ɛ, nPKC‐δ, nPKC‐η and nPKC‐θ are Ca2+‐independent. Group III includes the atypical PKC: aPKC‐ι (Selbie et al. 1993), aPKC‐ζ, aPKC‐ζII (Hirai et al. 2003), aPKC‐µ (protein kinase D) and aPKC‐ν (Hayashi et al. 1999) that are insensitive to both diacylglycerol and calcium and neither bind to nor are activated by phorbol esters. PKC regulates cellular functions, metabolism and proliferation by phosphorylating proteins in response to transmembrane signals from hormones, growth factors, neuro‐transmitters and pharmacological agents.
Literature summarizing PKCs in general in normal brain tissue (Tanaka & Saito 1992), gliomas (da Rocha et al. 2002) and meningiomas (Johnson & Toms 2005) have been published.
Of special interest is atypical PKC‐ι that does not contain a Ca2+‐binding region, has one zinc finger‐like motif and is the human homologue of the mouse PKC‐λ (Diaz‐Meco et al. 1996). PKC‐ι may play a role in development of malignancy at the cellular level, as shown by its association with the transformed phenotype of human melanomas in vivo and in vitro (Selzer et al. 2002) and that PKC‐ι protects cells against drug‐induced apoptosis (Murray & Fields 1997; Xie et al. 2000). In cells of some types of human lung cancer, PKC‐ι is a Bad kinase that can phosphorylate and inactivate the pro‐apoptotic BH3‐region containing protein, leading to enhanced survival and chemoresistance (Jin et al. 2005). In glioblastoma cells, PKC‐ι protects against cisplatin cytotoxicity by attenuation of p38 mitogen‐activated protein kinase signalling (Baldwin et al. 2006). Additionally, in glioma cells a link between PKC‐ι and cyclin‐dependent kinase 7 (Cdk7) activation has been reported previously (Acevedo‐Duncan et al. 2002; Bicaku et al. 2005). PKC‐ι is also required for oncogenic Ras‐ and carcinogen‐mediated colon carcinogenesis in vivo (Murray et al. 2004).
This study investigates the content of PKC‐ι in cells of normal brain tissue, gliomas, benign and malignant meningiomas and in the T98G and U‐138MG cell lines.
MATERIALS AND METHODS
Brain tissue
Human autopsy‐derived brain tissue and meninigiomas were obtained from the Cooperative Human Tissue Network (Southern Division) at the University of Alabama (Birmingham, AL, USA). Tissue specimens were obtained from both males and females of varying ages (23–80 years of age). Normal brain tissue included specimens from the frontal lobe, brain cortex, cerebellum, hippocampus, pons, corpus collosum and basal ganglia. Tumours labelled ‘benign’ were meningiomas. Malignant tumours were either meningiomas or gliomas. Brain tissue protein lysates (50 µg) were subjected to gel electrophoresis and Western blotting was performed using monoclonal antibodies against PKC‐ι (cat. no. 610176, BD Transduction, San Diego, CA, USA) at a 1: 2000 dilution (5 µg). Secondary antibodies were obtained from Accurate (JOM035146, Westbury, NY, USA), and were used at 1.5 : 10 000 dilution (48 µg). Western blots were probed for actin with a goat polyclonal antibody to actin (SC‐1616) at 2.5 : 2000 dilution (10 µg) and secondary antibodies SC‐2350 at a 1 : 2000 dilution (8 µg, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Positive control for PKC‐ι immunoreactivity was U‐373MG cell lysate (81 µg) that is known to contain PKC‐ι.
Passage of T98G and U‐138MG glioma cells
Human glioma U‐138MG (ATCC no. HTB‐16) and T98G (ATCC no. CRL‐1690) cells were obtained from the American Tissue Culture Collection (Rockville, MD, USA). U‐138MG cells were originally isolated and established into a stable cell line by Ponten & MacIntyre (1968), having been derived from an astrocytoma‐glioblastoma (grade III) of a 47‐year‐old Caucasian male. These cells have oligodendroglia histological features and a hyperdiploid to pentaploid karyotype. They grow in semisolid medium but are not tumourigenic in immunosuppressed mice. In our laboratory, U‐138MG cells have a doubling time of 36 h. The T98G cell line was isolated as a spontaneous variant of the parental T98G cells by Stein (1979), derived from a glioblastoma multiforme tumour of a 61‐year‐old Caucasian male. T98G appeared sometime between population doubling level 25–300 and the growth characteristics have been stable. The cells have a hyperpentaploid chromosome count, display anchorage independence and immortality but are not tumourigenic in nude mice. The reported doubling time of T98G cells is 26 ± 2 h (Stein 1979); however, in our conditions T98G cell populations double every 18 h. We are not certain why this happens, because we use ATCC media to grow the cells and they are subcultured according to ATCC procedures. The one parameter that distinguishes T98G cells from fully transformed cells is that they behave similarly to normal cells and can become arrested and stationary in G1 phase (Stein 1979). T98G and U‐138MG glioma cells were grown as adherent cultures. Cells were seeded into 0.22 mm filter 75 cm2 flasks containing 90% minimal essential media (MEM), 10% fetal calf serum, and standard antibiotics. Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 until they became no more than 70–80% confluent. Medium was changed (level of 5 mL medium/25 cm2 growth area) every other day, because these cells metabolize it quickly as judged by its yellow colouration if left for a longer period.
Inhibition of gene expression with siRNA
RNA interference functions by a regulatory mechanism for sequence‐specific gene silencing through double stranded (ds)RNA. Sequence specific RNA 19–25 nucleotides in length were synthesized by Santa Cruz Biotechnology against PKC‐ι. PKC‐ι siRNA were transfected into glioma cells (U‐138MG and T98G) using lipid based siRNA transfection reagent (Santa Cruz Biotechnology). Sequence‐specific PKC siRNA were as follows: PKC‐ι siRNA is a pooled sequence that consists of three combined RNA sequences – mRNA location. Gene accession number for PKC‐ι is NM_002740. PKC‐ι siRNA: 663: 5′‐CAAGCCAAGCGUUUCAACA‐3′ and 5′‐UGUUGAAACGCUUGGCUUG‐3′; 739: 5′‐GGAACGAUUGGGUUGUCAU‐3′ and 5′‐AUGACAACCCAAUCGUUCC‐3′; and 2137: 5′‐CCCAAUAUCUUCUCUUGUA‐3′ and 5′‐UACAAGAGAAGAUAUUGGG‐3′.
In addition to these siRNAs, negative controls containing a scrambled sequence (which does not lead to specific degradation of any known cellular mRNA) was synthesized; this is proprietary and Santa Cruz Biotechnology does not reveal it.
Cell viability assay
Effects of PKC‐ι siRNA were determined in exponentially growing T98G or U‐138MG glioma cells in complete media over 72 h. Cells were plated on 75 cm2 vessels at a density of 3.75 × 105 cells/flask. Twenty‐four hours after plating, cells were incubated with either siRNA‐A or PKC‐ι siRNA (100 µm) according to the manufacture's instructions (Santa Cruz Biotechnology). Following initial exposure to siRNA, no additional siRNA was applied or removed during the 3‐day incubation period. Following treatments, cells were washed with phosphate‐buffered saline (PBS), were trypsinized and re‐suspended in 3 mL of PBS. Cell viability was quantified using the trypan blue exclusion assay. Two hundred microlitres of the cell suspension was added to 50 µL of trypan blue and numbers of unstained and stained cells were counted. For Fig. 3, the same cultures from cell proliferation assays were harvested after counting and were prepared for Western blot analysis. Equal amounts of cellular protein (15 µg) were loaded per well. Anti‐PKC‐ι (P20520; Transduction Laboratories, Lexington, KY, USA) was used at 1 : 12 000 dilution (0.83 µg). Western blots were also performed to verify that protein loading (15 µg) and protein integrity were equal. Western blots were probed with a monoclonal antibody to β‐actin (Santa Cruz Biotechnology; SC‐8432) at 5 : 1000 dilution (40 µg). Secondary antibodies were obtained from Accurate (JGM035146,Westbury) and were used at 1 : 15 000 dilutions (1 µg).
Figure 3.
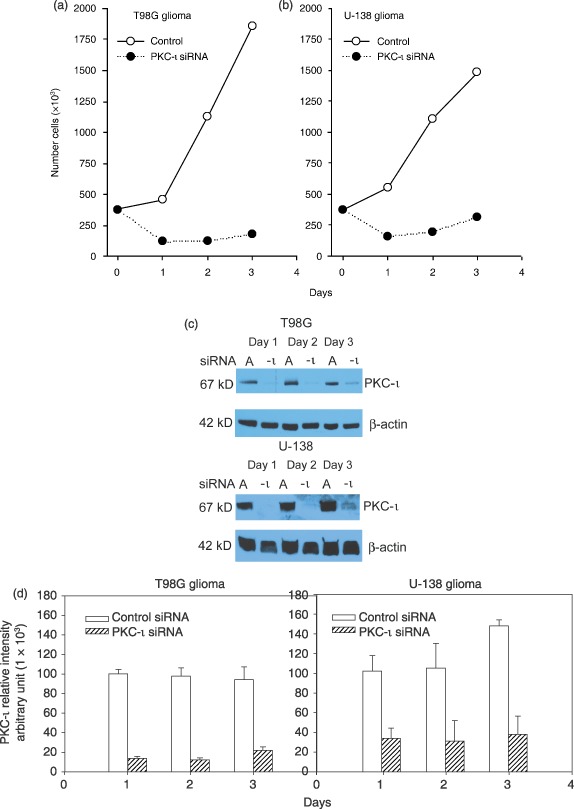
Effects of PKC‐ι siRNA on proliferation of T98G and U‐138MG glioma cells and PKC‐ι protein content. Cells were plated in 75 cm2 flasks at a density of 3.75 × 105 cells/flask. Twenty‐four hours after plating, cells were incubated with either siRNA‐A (100 nm; vehicle–control) or PKC‐ι siRNA (100 nm) for 6 h according to manufacture's instructions (Santa Cruz Biotechnology). During the 3‐day incubation viable cells were quantified by trypan blue dye exclusion assay (a, T98G; b, U‐138 MG). Open symbols ( ) represent control (siRNA‐A) treated cells, solid symbols (
) represent control (siRNA‐A) treated cells, solid symbols ( ) represent cells treated with PKC‐ι siRNA (100 nm). Western blots of PKC‐ι present in U‐138MG and T98G cells, treated with PKC‐ι siRNA (100 nm) for three days (c). Band intensity was quantified by densitometry (d). Data are representative of three experiments.
) represent cells treated with PKC‐ι siRNA (100 nm). Western blots of PKC‐ι present in U‐138MG and T98G cells, treated with PKC‐ι siRNA (100 nm) for three days (c). Band intensity was quantified by densitometry (d). Data are representative of three experiments.
Tissue fractionation
Brain tissue, T98G or U‐138MG glioma cells were re‐suspended and sonicated in 2 mL homogenization buffer [50 mm HEPES (pH 7.5), 150 mm NaCl, 0.1% Tween‐20, 1 mm EDTA (ethylenediaminetetraacetic acid) and 2 mm EGTA (glycol‐bis(2‐aminoethylether)‐N,N,N′,N′‐tetraacetic acid), 0.1 mm orthovanadate, 1 mm NaF, 2 mm PMSF (phenylmethylsulphonly fluoride), 2.5 µg/mL leupeptin, 1 mm DTT (dithiothreitol), 0.15 U/mL aprotinin; Agrawal et al. 1995]. The suspension was sonicated for 3 × 15 s, cycles on ice. Brain tissue suspensions or cell lysates were centrifuged at 100 000 g for 30 min to obtain cell extracts. Protein content was measured according to Bradford (1976).
Cell cycle analysis by flow cytometry
Cell cycle analysis was performed as previously described (Acevedo‐Duncan et al. 1997). Confluent cell cultures were either rapidly proliferating or semisynchronized by contact inhibition and serum starvation for 48 h. Subsequently, cells were collected and were washed twice with PBS and then were trypsinized. Cells in the suspension were centrifuged then the trypsin was decanted. To fix the cells, first, 3 mL of ice‐cold PBS was added and the pellet was re‐suspended. While vortexing gently, 7 mL of ethanol was added drop wise. The day before analysis, the 70% ethanol was decanted off and PBTB (PBS, 0.2% Triton and 1% bovine serum albumin) was added. Cells were counted and diluted to 1 × 106 cells/mL with PBTB. Then, they were filtered and 50 µL of RNase was added. Nuclei were analysed for DNA content using a propidium iodine (10 µL) staining protocol plus flow cytometry (Carlton et al. 1991). Distributions of 40 000 nuclei were quantified using a FAC STARPlus, flow cytometer (Becton Dickinson, San Jose, CA, USA) and ModFitLT cell cycle analysis program (version 2.0; Verity Software House Inc., Topsham, ME, USA). Mean separation was by using Student's t‐test (Minitab Inc., State College, PA, USA).
Western blot analysis
Cell extracts containing equal amounts of protein in each lane were run on sodium dodecyl sulfate‐polyacrylamide gel electrophoresis according to the protocol of Laemmli (1970); proteins were transblotted according to the method of Towbin et al. (1979). Brain tissue or glioma cell lysate PKC‐ι was probed with monoclonal antibodies against PKC‐ι (BD Transduction Laboratories), secondary antibodies were obtained from Accurate (JOM000003, Westbury). Immunoreactive bands were visualized with enhanced chemiluminescence according to the manufacturer's instructions (Amersham, Piscataway, NJ, USA). For Fig. 2, duplicate cultures from the flow cytometry experiment were harvested at the indicated times and were prepared for Western blot analysis. Equal amounts of cellular protein (20–50 µg) were loaded per well. Anti‐PKC‐α, anti‐PKC‐β1, anti‐PKC‐δ, anti‐PKC‐ɛ and anticaspase 7 (SC‐208, SC‐209, SC‐937, SC‐214 and SC‐8510; Santa Cruz Biotechnology) were used at 1 : 1300 dilution (3 µg). Anti‐PKC‐ι (P20520; Transduction Laboratories) was used at 1 : 12 000 dilution (0.83 µg). Western blots were also performed to verify that protein loading (20 µg) and protein integrity are equal; they were then probed with a monoclonal antibody to actin (Santa Cruz Biotechnology; SC‐8432) 5 : 1000 dilution (40 µg). Secondary antibodies were obtained from Accurate (JGM035146 and JG2035744, Westbury) and were used at 1 : 15 000 dilutions (1 µg).
Figure 2.
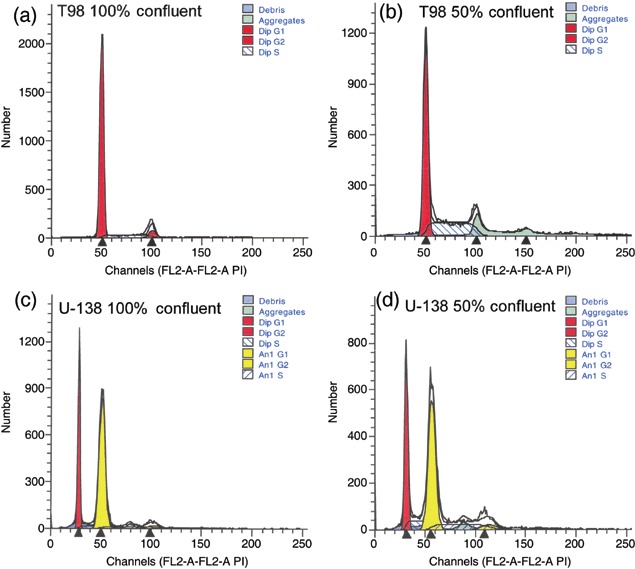
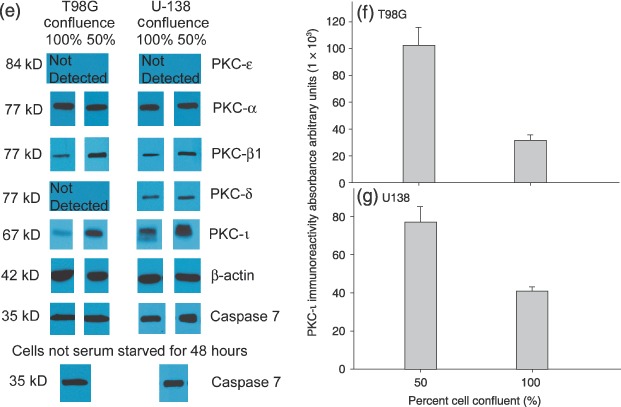
Effects of cell density and cell cycle progression on PKC‐ι concentration in T98G and U‐138MG glioma cells. FACS analysis of DNA content in 100% confluent T98G (a) or U‐138MG cells (c) and 50% confluent T98G (b) or U‐138 MG (d) cells. U‐138 glioma cells are aneuploid; U‐138MG DNA histograms are from one representative experiment and illustrate two cycling populations with a G0/G1 peak at 2 N (first red shaded peak) and another at 4 N (second yellow unshaded peak). Total DNA content for G0/G1, DNA synthetic phase (S), gap 2 and mitosis (G2/M) was quantified by addition of each of the phases in both populations. Forty thousand events were collected per time point and treatment group. Western blots of PKC‐α, PKC‐β1, PKC‐δ, PKC‐ɛ, PKC‐ι, and anticaspase 7 present in T98G or U‐138MG cells that were at different stages of confluence (e). Band intensity of PKC‐ι in T98G (f) and U‐138 MG (g) was quantified by densitometry.
Densitometry
The intensity of each band was measured using Gel Base/Gel Blot‐Pro software (Synoptics Ltd., Cambridge, UK); briefly, background intensity was subtracted from the intensity of each band, to derive the corrected intensity.
RESULTS
The relationship between absence of PKC‐ι in normal brain tissue and its robust presence in either benign/malignant meningiomas or gliomas is summarized in Table 1. Western blots probed for PKC‐ι in 12 normal brain biopsies, 15 benign meningiomas and 6 malignant tumours revealed complete absence (n = 9) or very low detection (n = 3) of PKC‐ι in normal brain tissue (Table 1). By comparison, PKC‐ι was very clearly evident in the majority of benign meningiomas (n = 14) but was only weakly present in one. Similarly, PKC‐ι was abundant in all malignant meningiomas (n = 3) and gliomas (n = 3). Table 2 presents the main characteristics (age, gender and World Health Organization classification of tumours) of the patients and tumour specimens. Western blots corresponding to some of the data present in Table 1 are shown in Fig. 1a. PKC‐ι was identified in Western blots by a band of molecular weight 67 kDa that corresponded to the immunoreactive signal obtained from U‐373MG glioma cells that contain PKC‐ι. Western blot controls for PKC‐βll did not show a pattern of expression specific to either normal brain tissue, or benign or malignant brain tumours (data not shown). Control actin Western blots showed actin immunoreactive bands at a molecular weight of 42 kDa. Actin immunoreactive bands were of equal intensity, indicating that equal amounts of protein were loaded into each lane. Data presented in Table 1 and Fig. 1a are also presented graphically in Fig. 1b that depicts a 38–46‐fold increase in PKC‐ι immunoreactivity in glioma, benign and malignant menigiomas when compared to normal brain tissue. These data were subjected to analysis of variance and mean separation by Tukey's honestly significant difference test (Minitab Inc., State College, PA, USA). The level of PKC‐ι in normal brain tissue differed from that in gliomas, and benign or malignant meningiomas (P < 0.001), while there was no significant difference (P > 0.05) between levels of PKC‐ι immunoreactivity between gliomas, benign and malignant meninigiomas. This study demonstrates that PKC‐ι is overexpressed in gliomas, benign and malignant meningiomas compared to normal brain tissues.
Table 1.
Status of PKC‐ι in brain tissues, meningiomas and gliomas a
| Tissue type | Not present | Weakly present | Positively present |
|---|---|---|---|
| Normal brain | 9 | 3 | 0 |
| Glioma | 0 | 0 | 3 |
| Benign meningioma | 0 | 1 | 14 |
| Malignant meningioma | 0 | 0 | 3 |
Normal brain tissue was obtained from frontal lobe, cortex, cerebellum, hippocampus, pons, corpus collosum or basal ganglia; benign tumours were meningiomas. Malignant tumours were either meningiomas or gliomas.
Table 2.
Patient data and WHO classification of brain tissues, meningiomas and gliomas
| Specimena | Sex | Age | WHO classification |
|---|---|---|---|
| N1 | M | 76 | N/A |
| N2 | M | 54 | N/A |
| N3 | M | 59 | N/A |
| N4 | M | 47 | N/A |
| N5 | M | 44 | N/A |
| N6 | M | 52 | N/A |
| N7 | F | 79 | N/A |
| B1 | M | 53 | Meningioma – N/L |
| B2 | F | 61 | Fibrous meningioma – grade I |
| B3 | F | 46 | Fibrous meningioma – grade I |
| M1 | F | 62 | Glioblastoma multiforme – grade IV |
| M2 | M | 23 | Glioblastoma multiforme – grade IV |
| M3 | M | 57 | Malignant meningioma – N/L |
| M4 | F | 64 | Atypical meningioma – grade II |
| M5 | M | 36 | Astrocytoma – grade IV |
| M6 | M | 56 | Anaplastic meningioma – grade III |
| B4 | F | 46 | Meningioma – N/L |
| B5 | F | 49 | Meningioma – N/L |
| B6 | F | 90 | Meningioma – N/L |
| B7 | M | 73 | Meningioma – N/L |
| B8 | F | 53 | Meningioma – N/L |
| B9 | M | 80 | Meningothliomatous meningioma – grade I |
| B10 | F | 43 | Meningothliomatous meningioma – grade I |
| B11 | F | 66 | Fibrous meningioma – grade I |
F, female; M, male; N/A, not applicable; N/L, not listed.
Figure 1.
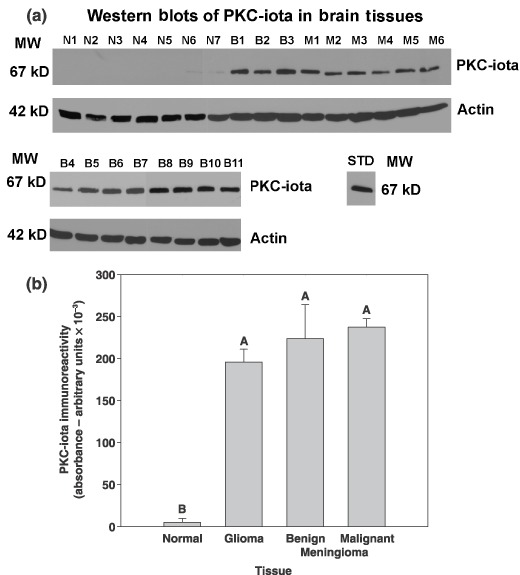
PKC‐iota is present in benign and malignant meningiomas, gliomas but not normal brain tissue. (a) Human autopsy‐derived normal brain tissue (N1, frontal lobe; N2, cortex; N3 and N4, unspecified brain; N5, cortex; N6, cerebellum), benign tumour tissue (B1, B4, B7, B9 and B10, [WHO grade 1] meningothelial meningioma; B5 and B8, meningioma; B6, fibroblastic meningioma; B2, B3 and B11, fibrous meningioma [WHO grade 1], and malignant tumour tissue; M1 and M2, [WHO grade IV] glioblastoma multiforme; M3, right frontal lobe meningioma; M4, atypical meningioma [WHO grade II]; M5, Astrocytoma [WHO grade IV]; M6, anaplastic meningioma [WHO grade III]). Specimens were obtained from the Cooperative Human Tissue Network. (b) Immunoblots from 12 normal brain specimens, 3 gliomas, 15 benign meningiomas and 3 malignant meningiomas were quantified, and mean plus and minus SE value is presented for each tissue type. Treatments indicated by the same letter do not differ, according to Tukey's honestly significant difference test (P = 0.001).
To establish whether PKC‐ι plays a role in cell cycle progression, T98G and U‐138MG glioma cells were plated and samples were taken for flow cytometric analysis or Western blotting, when cells were either serum starved for 48 h and 100% confluent, or when cells were 50% confluent and not serum starved. T98G cells and U‐138MG glioma cells that were 100% confluent had 94% and 74% of the cells in quiescence/Gap 1 (G0/G1), respectively (Fig. 2a,c and Table 3). In contrast, 64% of rapidly dividing 50% confluent T98G cells accumulated in G0/G1 phase and 54% of U‐138MG glioma cells were in G0/G1 phase (Fig. 2b,d). To insure that 48 h serum deprivation did not lead to cell death nor alteration in intracellular cascades, we evaluated caspase 7 activation. Caspase 7 is an executioner/effector caspase that promotes apoptotic morphological alteration by cleaving several death substrates (Araya et al. 2002). In cells undergoing apoptosis, 35 kDa caspase 7 is cleaved into 20 kDa and 10 kDa active subunits. Figure 2e shows that 48 h serum deprivation did not induce degradation of caspase 7 into the 20 kDa subunit, as judged by lack of the cleaved fragment on Western blots and invariant levels of the 35 kDa caspase 7 in T98G and U‐138MG glioma cells (Fig. 2e). Western blotting for PKC‐ι in these cell populations depicted robust quantities of PKC‐ι in proliferating 50% confluent T98G and U‐138MG glioma cells. In contrast, 100% confluent cells had either 71% (T98G) or 47% (U‐138MG) less PKC‐ι than 50% confluent cells (Fig. 2e–g). Differences between PKC‐ι protein content in 100% confluent and 50% confluent rapidly proliferating cells was significant at P < 0.05 (n = 3). To establish that variations in PKC‐ι may be specific for PKC‐ι, we randomly performed Western blots for PKC‐α, PKC‐β1, PKC‐δ and PKC‐ɛ in confluent and proliferating cells. PKC‐β1 is known to play a role in angiogenesis and tumourigenesis (Teicher et al. 2001). Similarly, PKC‐δ (Jane et al. 2006) and PKC‐ɛ (Sharif et al. 2001) are involved in glioma cell proliferation. Westerns blots for PKC‐α showed invariant levels of PKC‐α in confluent and proliferating cells (Fig. 2e). Of interest, Western blots for PKC‐δ did not detect PKC‐δ in T98G glioma cells and invariant levels were detected in U‐138MGs.
Table 3.
Summary of cell cycle phases a
| Cell type | G0/G1 | S | G2/M |
|---|---|---|---|
| 100% Confluent T98G | 94 ± 1 | 4 ± 3 | 2 ± 3 |
| 50% Confluent T98G | 64 ± 3 | 33 ± 3 | 4 ± 4 |
| 100% Confluent U‐138MG | 74 ± 11 | 20 ± 10 | 2 ± 3 |
| 50% Confluent U‐138MG | 54 ± 17 | 39 ± 18 | 7 ± 1 |
n = 3 experiments per cell line.
Western blotting for PKC‐β1 depicted decreased levels (35% and 53% reduction) of PKC‐β1 in 100% confluent T98G and U‐138MG cultures compared to rapidly proliferating 50% confluent cells, respectively (Fig. 2e). Differences between PKC‐β1 protein content in 100% confluent and 50% confluent rapidly proliferating cells were significant at (P < 0.05; n = 3) for both cell lines. In contrast, PKC‐ɛ was not detected in either T98G or U‐138MG cells. Western blots of PKC‐α, PKC‐β1, PKC‐δ and PKC‐ɛ, suggest that the presence or absence of these PKC isozymes may be dependent on cell type. Results depicting a relationship between cell confluence and PKC‐ι protein levels suggest that PKC‐ι along with PKC‐β1, and PKC‐δ may play a role in cell cycle progression and/or proliferation. For subsequent studies, we focused on the role of PKC‐ι in glioma cell proliferation.
Effects of PKC‐ι siRNA on proliferation of T98G and U‐138MG glioma cells and appropriate protein content
The effects of exposure to PKC‐ι siRNA on T98G and U‐138MG glioma cell viability and proliferation were evaluated by trypan blue dye exclusion (Fig. 3a,b). Viable cell number was counted 24–72 h following addition of either control short interfering RNAs (siRNA‐A, vehicle control; 100 nm) or PKC‐ι siRNA (100 nm) according to manufacturer's instructions (Santa Cruz Biotechnology). Exposure of T98G or U‐138MG glioma cells to PKC‐ι siRNA significantly reduced their cell proliferation by 59% (P = 0.002) and 69% (P = 0.03), respectively, at 72 h post‐treatment. Densitometric scanning of Western blots revealed that PKC‐ι siRNA decreased PKC‐ι protein content by 86% to 77% (T98G) and 66% to 74% (U‐138MG) during the 3‐day experimental time course (Fig. 3c,d; n = 3 experiments). Differences between PKC‐ι protein content in control siRNA‐A and PKC‐ι siRNA treated cells were significant at (P < 0.05) for all time points. Control β‐actin Western blots produced β‐actin immunoreactive bands at a molecular weight of 42 kDa. Actin‐immunoreactive bands were of equal intensity, indicating that equal amounts of protein were loaded into each lane. These results indicate that PKC‐ι may be required for cell proliferation to take place.
In this study, we examined the PKC‐ι protein content in normal brain biopsies, gliomas, benign and malignant meningiomas as judged by Western blotting. Of interest were the results depicting increases in PKC‐ι abundance in benign or malignant meningiomas and gliomas compared to normal brain tissue. Results from this study indicate that PKC‐ι may be a characteristic of brain tumourigenesis. We also investigated the effects of PKC‐ι siRNA on T98G and U‐138MG glioma cell lines. Results demonstrated that PKC‐ι siRNA reduced PKC‐ι protein content concomitantly with a decrease in glioma cell proliferation. Taken together, these results suggest that PKC‐ι may play a role in glioma cell proliferation.
DISCUSSION
Typically, high‐grade malignant brain tumours are lethal. Despite rigorous therapy, median survival is less than 1 year for patients with high‐grade tumours (Allalunis‐Turner et al. 1992). While post‐operative radiation therapy clearly delays tumour re‐growth and prolongs survival, total tumour control is rarely achieved. Glioma recurrence and radioresistance may be due to an abundance of hypoxic or tumour stem cells, rapid glioma cell proliferation, low radiosensitivity or the involvement of PKC isozymes in radiation resistance (Baumann et al. 1992; Mitsutake et al. 2001; Tenzer et al. 2001). PKC may be involved in several cell signalling pathways (cell survival including repair of radiation damage) and cell cycle progression (Hallahan et al. 1992), thus its inhibition may result in radiosensitization. Additionally, rapid glioma cell growth rate has been attributed to inherently high levels of PKC (Couldwell et al. 1990; Pollack et al. 1990).
Signal transduction pathways have been studied comprehensively in gliomas, but only lately have they been examined in meningiomas. Roles of growth factor receptors Ras‐Raf‐1‐MEK‐1‐MAPK, PI3K‐Akt/PKB, PLC‐γ1‐PKC, phospholipase A2‐cyclooxygenase, and TGF‐β receptor‐Smad pathways on meningioma growth and apoptosis have been reviewed, and sites that can be targeted along these receptor/kinase pathways have been identified (Johnson & Toms 2005). PKC also appears to be involved in growth regulation of low‐passage number human meningioma cells in vitro, as judged by decreased proliferation of cells following treatment with the PKC inhibitor staurosporine (Todo & Fahlbusch 1994; Huang et al. 2000). Additionally, two PKC‐activating phorbol esters, 12‐O‐tetradecanoyl‐13‐phorbol acetate (TPA) and phorbol‐12,13‐dibutyrate (PDBu) have been shown to demonstrate different effects on proliferation of low‐passage number human meningioma cells in culture (Todo & Fahlbusch 1994). TPA provoked dose‐dependent stimulation of cell proliferation while PDBu produced significant inhibition of cell proliferation, suggesting that PKC‐mediated signalling pathways may play a role in growth regulation of human meningioma cells. However, immunohistochemical studies investigating the presence of PKC in meningiomas depicted variable immunostaining (Reifenberger et al. 1989).
Protein kinase C is the major receptor for tumour‐promoting phorbol esters, but the extent of PKC involvement in cellular malignancy is not clearly defined. Various studies indicate that increased tumourigenicity results from deregulation of PKC activity, or changes in PKC concentration, or both (Kamata et al. 1987; Housey et al. 1988; Mizuguchi et al. 1988; Mishima et al. 1994; Person et al. 1988; Weyman et al. 1988). Two PKC isozymes (PKC‐β11 and PKC‐ι) have been reported to be critical for some forms of cancers, including colon cancer (Murray et al. 1999; Gokmen‐Polar et al. 2001; Murray et al. 2002) and chronic myelogenous leukaemia (Murray et al. 1993; Murray & Fields 1997; Jamieson et al. 1999). PKC‐ι has also been shown to be overexpressed in serous and nonserous ovarian cancers through a mechanism that results in loss of apical‐basal polarity and cyclin E overexpression leading to poor prognosis (Eder et al. 2005).
Although, meningiomas arise from arachnoid cells of the meninges of the brain and spinal cord, and gliomas arise from supportive tissues of the brain glial or neuroglial, this study has demonstrated that PKC‐ι is overexpressed in benign and in malignant meningiomas and in gliomas but not in normal brain tissues. Moreover, we have shown that rapidly proliferating T98G and U‐138MG glioma cell lines have enhanced levels of PKC‐ι compared to confluent cells. Additionally, PKC‐ι siRNA reduced PKC‐ι protein content and decreased cell proliferation, suggesting a role for PKC‐ι in regulating cell proliferation.
ACKNOWLEDGEMENTS
We acknowledge the important contribution of the Flow Cytometry Core, H. Lee Moffitt Cancer Center. This project was supported in part by Inhibition Therapeutics Inc., the Margaret Ewell Dickens Foundation and the Research Service of the Veterans’ Administration.
REFERENCES
- Acevedo‐Duncan M, Zhang R, Cooper DR, Greenberg H (1997) Effects of interferon and PKC modulators on human glioma protein kinase C, cell proliferation and cell cycle. Neurochem. Res. 22, 775–784. [DOI] [PubMed] [Google Scholar]
- Acevedo‐Duncan M, Patel R, Whelan S, Bicaku E (2002) Human glioma PkC‐ι and PKC‐βII phosphorylate cyclin dependent kinase activating kinase during the cell cycle. Cell Prolif. 35, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal D, Dong F, Wang Y‐Z, Kayda D, Pledger WJ (1995) Regulation of cyclin E and p27Kip during mitosis in BALB/c 3T3 cells. Cell Growth Differ. 6, 1199–1205. [PubMed] [Google Scholar]
- Allalunis‐Turner JM, Barron GM, Day RS, Fulton DS, Urtasun RC (1992) Radiosensitivity testing of human primary brain tumor specimens. Int. J. Radiat. Oncol. Biol. Phys. 23, 339–343. [DOI] [PubMed] [Google Scholar]
- Araya R, Takahashi R, Nomura Y (2002) Yeast two‐hybrid screening using constitutive‐active caspase‐7 as bait in the identification of PA28gamma as an effector caspase substrate. Cell Death Differ. 9, 322–328. [DOI] [PubMed] [Google Scholar]
- Baldwin RM, Garratt‐Lalonde M, Parolin DAE, Krzyzanowski PM, Andrade MA, Lorimer IAJ (2006) Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase C‐ι‐mediated attenuation of p38 MAP kinase signaling. Oncogene 25, 2909–2919. [DOI] [PubMed] [Google Scholar]
- Baumann M, Dubois W, Pu A, Freeman J, Suit HD (1992) Response of xenografts of human malignant gliomas and squamous cell carcinoma to fractionated irradiation. Int. J. Radiat. Oncol. Biol. Phys. 23, 803–809. [DOI] [PubMed] [Google Scholar]
- Bicaku E, Patel R, Acevedo‐Duncan M (2005) Cyclin‐dependent kinase activating kinase co‐localizes with protein kinase C‐ι in human glioma cells. Tissue Cell 37, 53–58. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Carlton JC, Terry NHA, White A (1991) Measuring potential doubling times of murine tumors using flow cytometry. Cytometry 12, 645–650. [DOI] [PubMed] [Google Scholar]
- Couldwell W, Antel JP, Apuzzo ML, Yong VW (1990) Inhibition of growth of established human glioma cell lines by modulators of the protein kinase‐C system. J. Neurosurg. 73, 594–600. [DOI] [PubMed] [Google Scholar]
- Diaz‐Meco MT, Municio MM, Sanchez P, Lozano J, Moscat J (1996) Lamda‐interacting protein, a novel protein that specifically interacts with the Zinc finger domain of the atypical Protein Kinase C isotype λ/ι and stimulates its kinase activity in vitro and in vivo . Mol. Cell. Biol. 16, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, Kango‐Singh M, Lu KH, Warneke CL, Atkinson EN, Bedrosian I, Keyomarsi K, Kuo WL, Gray JW, Yin JC, Liu J, Halder G, Mills GB (2005) Atypical PKC‐ι contrubutes to poor prognosis through loss of apical‐basal polarity and cyclin E overexpression in ovarian cancer. Proc. Natl. Acad. Sci. USA 35, 12519–12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokmen‐Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP (2001) Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 61, 1375–1381. [PubMed] [Google Scholar]
- Hallahan DE, Virudachalam S, Grdina D, Weichselbaum RR (1992) The isoquinoline sulfonamide H7 attenuates radiation‐mediated protein kinase C activation and delays the onset of X‐ray induced G2 arrest. J. Radiat. Oncol. Biol. Phys. 24, 687–692. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Seki N, Hattori A, Kozuma S, Saito T (1999) PKCν, a new member of the protein kinase C family, composes a fourth subfamily with PKC‐µ. Biochim. Biophys. Acta 1450, 99–106. [DOI] [PubMed] [Google Scholar]
- Hirai T, Niino Y, Chida K (2003) PKC‐ζII, a small molecule of protein kinase C ζ, specifically expressed in mouse brain. Neurosci. Lett. 348, 151–154. [DOI] [PubMed] [Google Scholar]
- Housey GM, Johnson MD, Hsiao WL, O’Brian CA, Murphy JP, Kirschmeier P, Weinstein IB (1988) Overproduction of protein kinase C causes disordered growth control in rat fibroblasts. Cell 52, 343–354. [DOI] [PubMed] [Google Scholar]
- Huang B, Lei T, Liu K, Zhang L, Li L, Zhang Z, Xue D (2000) The regulatory effects of protein kinase C on the proliferation of cultured human low‐passage number meningioma cells. J. Tongji Med. Univ. 20, 217–219. [DOI] [PubMed] [Google Scholar]
- Jamieson L, Carpenter L, Biden TJ, Fields AP (1999) Protein kinase C iota activity is necessary for Bcr‐Abl‐mediated resistance to drug‐induced apoptosis. J. Biol. Chem. 274, 3927–3930. [DOI] [PubMed] [Google Scholar]
- Jane EP, Premkumar DR, Pollack IF (2006) Coadministration of sorafenib with rottlerin potently inhibits cell proliferation and migration in human malignant glioma cells. J. Pharmacol. Exp. Ther. 319, 1070–1080. [DOI] [PubMed] [Google Scholar]
- Jin Z, Xin M, Deng X (2005) Survival function of protein kinase C‐ι as a novel nitrosamine 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone‐activated bad Kinase. J. Biol. Chem. 280, 16045–16052. [DOI] [PubMed] [Google Scholar]
- Johnson M, Toms S (2005) Mitogenic signal transduction pathways in meningiomas: novel targets for meningioma chemotherapy? J. Neuropathol. Exp. Neurol. 64, 1029–1036. [DOI] [PubMed] [Google Scholar]
- Kamata T, Sullivan NF, Wooten MW (1987) Reduced protein kinase C activity in a ras‐resistant cell line derived from Ki‐MSV transformed cells. Oncogene 1, 37–46. [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 237, 680–685. [DOI] [PubMed] [Google Scholar]
- Mishima K, Ohno S, Shitara N, Yamaoka K, Suzuki K (1994) Opposite effects of the overexpression of protein kinase C gamma and delta on the growth properties of human glioma cell line U251 MG. Biochem. Biophys. Res. Commun. 201, 363–372. [DOI] [PubMed] [Google Scholar]
- Mitsutake N, Namba H, Shklyaev SS, Tsukazaki T, Ohtsuru A, Ohba M, Kuroki T, Ayabe H, Yamashita S (2001) PKC delta mediates ionizing radiation‐induced activation of c‐Jun NH(2)‐terminal kinase through MKK7 in human thyroid cells. Oncogene 20, 989–996. [DOI] [PubMed] [Google Scholar]
- Mizuguchi J, Nakabayashi H, Yoshida Y, Huang KP, Uchida T, Sasaki T, Ohno S, Suzuki K (1988) Increased degradation of protein kinase C without diminution of mRNA level after treatment of WEHI‐231 B lymphoma cells with phorbol esters. Biochem. Biophy. Res. Commun. 155, 1311–1317. [DOI] [PubMed] [Google Scholar]
- Murray NR, Baumgardner GP, Burns DJ, Fields AP (1993) Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that betaII protein kinase C is required for proliferation. J. Biol. Chem. 268, 15847–15853. [PubMed] [Google Scholar]
- Murray NR, Davidson LA, Chapkin RS, Gustafson WC, Schattenberg DG, Fields AP (1999) Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J. Cell Biol. 145, 699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Fields AP (1997) Atypical protein kinase C iota protects human leukemia cells against drug‐induced apoptosis. J. Biol. Chem. 272, 27521–27524. [DOI] [PubMed] [Google Scholar]
- Murray NR, Jamieson L, Yu W, Zhang J, Gokmen‐Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP (2004) Protein kinase Cι is required for Ras transformation and colon carcinogenesis in vivo . J. Cell Biol. 164, 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Weems C, Chen L, Guo H, Ethridge R, Ceci JD, Evers BM, Thompson EA, Fields AP (2002) Protein kinase C betaII and TGFbetaRII in omega‐3 fatty acid‐mediated inhibition of colon carcinogenesis. J. Cell Biol. 157, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y (1992) Intracellular signally by hydrolysis of phospholipids and activation of protein kinase C. Science 258, 607. [DOI] [PubMed] [Google Scholar]
- Persons DA, Wilkison WO, Bell RM, Finn OJI (1988) Altered growth regulation and enhanced tumorigenicity of NIH 3T3 fibroblasts transfected with protein kinase C‐I DNA. Cell 52, 447–458. [DOI] [PubMed] [Google Scholar]
- Pollack IF, Randall MS, Kristofik MP, Kelly RH, Selker RG, Vertosick FT Jr (1990) Response of malignant glioma lines to activation and inhibition of protein kinase C mediated pathways. J. Neurosurg. 73, 98–105. [DOI] [PubMed] [Google Scholar]
- Ponten J, MacIntyre EH (1968) Long term culture of normal and neoplastic human glia. Acta Pathol. Microbiol. Scand. 74, 465–486. [DOI] [PubMed] [Google Scholar]
- Reifenberger G, Deckert M, Wechsler W (1989) Immunohistochemical determination of protein kinase C expression and proliferative activity in human brain tumors. Acta Neuropathol. (Berl). 78, 166–175. [DOI] [PubMed] [Google Scholar]
- Da Rocha AB, Mans DR, Regner A, Schwartsmann G (2002) Targeting protein kinase C: new therapeutic opportunities against high‐grade malignant gliomas? Oncologist 7, 17–33. [DOI] [PubMed] [Google Scholar]
- Selbie LA, Schmitz‐Peiffer C, Sheng Y, Biden T (1993) Molecular cloning and characterization of PKC iota, an atypical isoform of PKC derived from insulin‐secreting cells. J. Biol. Chem. 268, 24296–24302. [PubMed] [Google Scholar]
- Selzer E, Okamoto I, Lucas T, Kodym R, Pehamberger H, Jansen B (2002) Protein Kinase C isoforms in normal and transformed cells of the melanocytic lineage. Melanoma Res. 12, 201–209. [DOI] [PubMed] [Google Scholar]
- Sharif TR, Sasakawa N, Sharif M (2001) Regulated expression of a dominant negative protein kinase C epsilon mutant inhibits the proliferation of U‐373MG human astrocytoma cells. Int. J. Mol. Med. 7, 373–377. [DOI] [PubMed] [Google Scholar]
- Stein GH (1979) T98G: An anchorage‐independent human tumor cell line that exhibits stationary phase G1 arrest in vitro . J. Cell. Physiol. 99, 43–54. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Saito N (1992) Localization of subspecies of protein kinase C in the mammalian central nervous system. Neurochem. Int. 21, 499–512. [DOI] [PubMed] [Google Scholar]
- Teicher BA, Menon K, Alvarez E, Galbreath E, Shih C, Faul M (2001) Antiangiogenic and antitumor effects of a protein kinase C‐beta inhibitor in human T98G glioblastoma multiforme xenografts. Clin. Cancer Res. 7, 634–640. [PubMed] [Google Scholar]
- Tenzer A, Zingg D, Rocha S, Hemmings B, Fabbro D, Glanzmann C, Schubiger PA, Bodis S, Pruschy M (2001) The phosphatidylinositide 3′‐kinase/Akt survival pathway is a target for the anticancer and radiosensitizing agent PKC412, an inhibitor of protein kinase C. Cancer Res. 61, 8203–8210. [PubMed] [Google Scholar]
- Todo T, Fahlbusch R (1994) Involvement of protein kinase C in growth regulation of human meningioma cells. Acta Neurochir. (Wien) 131, 282–288. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon PE (1979) Electrophoretic transfer of proteins form polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyman CM, Taparowsky EJ, Wolfson M, Ashendel CL (1988) Partial down‐regulation of protein kinase C in C3H10tl/2 mouse fibroblasts transfected with the human Ha‐ras oncogene. Cancer Res. 48, 6535. [PubMed] [Google Scholar]
- Xie J, Guo Q, Zhu H, Wooten M, Mattson M (2000) Protein kinase C iota protects neural cells against apoptosis induced by amyloid beta‐peptide. Mol. Brain Res. 82, 107–113. [DOI] [PubMed] [Google Scholar]