

Abstract.
Objectives: Initiation and maintenance of pro‐inflammatory reactions elicited by bacterial lipopolysaccharide and/or cytokines in the macrophage lineage have been reported to play a crucial role in acute and chronic pathogenic effects. Whether pro‐inflammatory responses triggered by lipopolysaccharide in growth arrested cells differ from those in proliferating cells remains unanswered.
Materials and methods: Olomoucine and roscovitine are cyclin‐dependent kinase (CDK) inhibitors that prevent progression through the cell cycle. After treatment with CDK inhibitors, expression of pro‐inflammatory genes was analysed by reverse transcriptase–polymerase chain reaction. Protein levels of inducible nitric oxide synthase (iNOS) and nuclear factor kappaB (NF‐κB) were determined by Western blotting. Promoter activity of iNOS was measured by the luciferase activity assay.
Results: In this study we have demonstrated that both olomoucine and roscovitine inhibit cell proliferation and diminish nitric oxide production and cytokine gene expression, in lipopolysaccharide‐stimulated murine RAW264.7 macrophages. In addition, olomoucine reduces iNOS promoter activity and alleviates NF‐κB transcription activation. After co‐transfection with E2F1 interference RNA, suppression of lipopolysaccharide‐mediated iNOS promoter activity and NF‐κB activation was observed. Furthermore, we demonstrated that olomoucine‐induced growth arrested cells reduce expression of the p65 subunit of NF‐κB.
Conclusions: The findings of this study suggest that inhibition of cell‐cycle progression is capable of reducing pro‐inflammatory responses via down‐regulation of NF‐κB.
Introduction
The family of cyclin‐dependent kinases (CDK) is responsible for orderly progression of cells through various phases of the cell cycle (1). Unlike other protein kinases, CDKs are regulated by binding to their cyclin protein partners to form active heterodimeric complexes. They play a crucial role in progression of cells from G1 to S phase and this occurs by regulation of phosphorylation state of the retinoblastoma gene product (Rb). After Rb phosphorylation by CDKs, E2Fs released from Rb become active and regulate gene expression at the initiation of S phase. These active CDKs are then involved in transcriptional control, mitotic progression, DNA repair, and cell differentiation (2, 3, 4, 5, 6). Dysfunctional molecules controlling the cell cycle have been considered to play an important role in tumour pathogenesis. Thus, CDKs represent very attractive targets for cancer therapy (7, 8).
Olomoucine, a purine‐based CDK inhibitor, is one of the first CDK inhibitors to be developed. Moreover, roscovitine is an olomoucine derivative that shows even more potent inhibitory activity (9, 10). These chemicals exhibit similar selectivity regarding inhibition of CDKs 1, 2, 5 and 7, while this is not the case for CDKs 4 and 6. Olomoucine and roscovitine are able to cause 50% inhibition of growth in the National Cancer Institute panel of 60 tumour cell lines, at average concentrations of 60.3 and 16 µm, respectively. When cells are treated with these drugs, the results typically show them arrested at G1/S and G2/M transitions. In addition to inhibiting cell‐cycle progression, they also cause cell death (7, 11, 12, 13, 14). Paradoxically, they are also reported to rescue cells from apoptosis when it is induced by withdrawal of neurotrophic factors and furthermore, to provide neuroprotection after traumatic brain injury (15, 16, 17, 18).
Cells of the monocyte/macrophage lineage have been reported to play a crucial role in initiation and maintenance of bacterial lipopolysaccharide and/or cytokine‐elicited inflammatory reactions (19, 20). In order to eliminate pathogens, activated macrophages secrete a spectrum of cytokines and release free radicals, such as nitric oxide (NO) (11). However, these changes may also lead to acute and/or chronic pathogenic effects. Relevant clinical manifestations in septic shock, autoimmune diseases, cerebral infarction and diabetes mellitus have been attributed to dysregulated inflammatory reactions (21, 22). Although mechanisms of lipopolysaccharide‐induced effects at the molecular level are not clear, various kinases such as the three subclasses of mitogen‐activated protein kinases, including extracellular signal‐regulated kinases, p38 mitogen‐activated protein kinase, and c‐Jun N‐terminal kinases, are well known to be involved in some of these pathways (19). In addition, nuclear factor kappaB (NF‐κB) transcription factors have also been implicated in lipopolysaccharide‐induced signal transduction. Induction of NO and cytokines, including interleukin‐1 and tumour necrosis factor‐α (TNF‐α), converge on a common activation pathway that leads to phosphorylation of Iκ‐B by the IKK kinases; this, in turn, leads to Iκ‐B protein degradation by ubiquitin‐mediated proteasome activity, that results in release of the active NF‐κB subunit, p65 (RelA) from the cytoplasm for entry into the nucleus (20, 23, 24). When active NF‐κBs enter the nucleus, they activate expression of genes involved in the pro‐inflammatory responses.
Inhibitors olomoucine and roscovitine may prevent CDK activity and in turn, cause cell growth arrest. Whether growth arrested cells may change lipopolysaccharide‐induced pro‐inflammatory responses remains to be solved. In this study, we have demonstrated that CDK inhibitors olomoucine and roscovitine inhibit NO production and cytokine gene expression in lipopolysaccharide‐stimulated RAW264.7 macrophages. Moreover, olomoucine also reduces iNOS promoter activity and alleviates NF‐κB transcription activation. After co‐transfection with E2F1 interference RNA (E2F1i), there is suppression of lipopolysaccharide‐mediated iNOS promoter activity and NF‐κB activation. In addition to the above, olomoucine reduces expression of NF‐κB but promotes expression of CD14. Taken together, these findings suggest that growth arrested cells resulting from treatment with CDK inhibitors, may decline lipopolysaccharide‐induced pro‐inflammatory responses through inhibition of expression of the transcription factor NF‐κB.
Material and methods
Reagents
Olomoucine, roscovitine and iso‐olomoucine were purchased from Calbiochem (San Diego, CA, USA). Lipopolysaccharide (Escherichia coli, serotype 0111:B4) was purchased from Sigma (St. Louis, MO, USA). Cell culture materials and transfection reagents were obtained from Invitrogen Life Science (Carlsbad, CA, USA). Reagents for the luciferase activity assay and 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) were purchased from Promega (Madison, WI, USA). Specific antibodies against iNOS and p65 were the products of Cell Signalling Technology Inc. (Beverly, MA, USA). All other reagents were obtained from Merck KGaA (Darmstadt, Germany).
Cell culture
The mouse macrophage RAW264.7 cell line was obtained from the American Type Culture Collection and cultured as a monolayer in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum, penicillin (100 U/ml) and streptomycin (100 µg/ml) at 5% CO2 and 37 °C. Cells were treated with olomoucine, iso‐olomoucine or roscovitine for 24 h and then lipopolysaccharide was added. Dimethyl sulfoxide was used as solvent control for the CDK inhibitors.
Cytotoxicity assay
Effect of CDK inhibitors on cell population growth inhibition were determined by MTS uptake method as described in CellTiter Aqueous Non‐Radioactive Cell Proliferation assay (Promega). Briefly, 1 × 104 cells were incubated with CDK inhibitor for 24 or 48 h in triplicate in 96‐well plates. Thereafter, 20 µl of MTS was added to each well and incubated for 2 h. Optical density was measured at 490 nm using a 96‐well ELISA reader.
Reverse transcriptase–polymerase chain reaction
The cells were lysed in 1 ml of RNA isolation kit (Sigma), and total RNA was harvested as described in the manufacturer's protocol. Two micrograms of total RNA served as template for the reverse transcriptase–polymerase chain reaction (RT‐PCR) using oligo‐dT and SuperScrip Reverse Transcriptase II (Invitrogen). Following reverse transcription, 1/10 of total RT reaction was used in PCR with the following primers: GADPH (accession number: XM_001473623.1): forward, 5′‐AACTTTGGCATTGTGGAAGG‐3′ (+565–+584), reverse, 5′‐ACACATTGGGGGTAGGAACA‐3′ (+768–+787); IL‐1α (accession number: NM_008361.3): forward, 5′‐TACAGGCTCCGAGATGAACAACAA (+441–+464), reverse, 5′‐TGGGGAAGGCATTAGAAACAGTCC‐3′ (+889–+912); IL‐6 (accession number: NM_031168.1): forward, 5′‐CCACTTCACAAGTCGGAGGCTT‐3′ (+171–+192), reverse, 5′‐CCAGCTTATCTGTTAGGAGA‐3′ (+547–+567); TNF‐α (accession number: NM_013693.2): forward, 5′‐TTCTGTCTACTGAACTTCGGGGTGATCGGTCC‐3′ (+298–+329), reverse, 5′‐GTATGAGATAGCAAATCGGCTGACGGTGTGGG‐3′ (+620–+651); iNOS (accession number: NM_010927.3): forward, 5′‐CATGGCTTGCCCCTGGAAGTTTCTCTTCAAA‐3′ (+183–+214), reverse, 5′‐GCAGCATCCCCTCTGATGGTGCCATCG‐3′ (+984–+1010); Mn‐SOD (accession number: NM_013671.3): forward, 5′‐ATTAACGCGCAGATCATGCAG‐3′ (+331–+351), reverse, 5′‐TTTCAGATAGTCAGGTCTGACGTT‐3′ (+790–+813); and Cu/Zn SOD (accession number: NM_011434.1): forward, 5′‐ATGGCGATGAAAGCGGTGTGCGTG‐3′ (+117–+140), reverse, 5′‐TTAATGGTTTGAGGGTAGCAG‐3′ (+615–+635). PCR products were then separated electrophoretically in a 1.5% agarose DNA gel and stained with ethidium bromide.
Transient transfection and luciferase assay
Mouse iNOS promoter from –1200 to –10 was introduced into pGL2 plasmid (Promega), which contains a luciferase reporter gene. NF‐κB reporter DNA was purchased from Clontech (Mountain View, CA, USA). E2F and Rb reporters were obtained from Panomics (Redwood, CA, USA). E2F1 interfering RNA expression vector and E2Fc control vector were purchased from Panomics. Transient transfection was carried out with a total of 2 µg of DNA mixture using Lipofectamine (Invitrogen, Carlsbad, CA, USA) reagent according to the manufacturer's instructions. Cells were cultured with olomoucine for 24 h and then treated with lipopolysaccharide for 24 h prior to harvesting. Luciferase activity of cell extracts was measured using the luciferase assay system (Promega).
Western blotting
Following treatment, cells were washed with phosphate‐buffered saline and then lysed in 300 µl lysis buffer (50 mm Tris‐HCl pH 7.4, 1% Triton X‐100, 150 mm NaCl, 1% glycersol, 1 mm EDTA, 0.1 mm phenylmethylsulphonyl fluoride, 50 mm NaF, and 1 mm Na3VO4). The supernatant was collected by centrifugation at 14 000 g for 10 min at 4 °C. Protein concentration was determined using a Bio‐Rad Protein Assay Dye Reagent (Hercules, CA, USA). In total, 20 µg of protein per sample was resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) and then electro‐transferred on to nitrocellulose membranes, blotted with each antibody and detected using ECL reagents (Amersham Bioscience). Quantification of Western blot data was performed using densitometery with Image Calculator software.
NO detection
RAW264.7 cells (1 × 104/well) were cultured in flat bottom microtitre plates in triplicate for 24 h. Cells were incubated with CDK inhibitor for 24 h and then stimulated with lipopolysaccharide (1 µg/ml) for 24 h. Amounts of nitrite present (a stable metabolite of NO), was measured using Griess reagent. Briefly, cell‐free culture media (50 µl) was reacted with 1 : 1 mixture (50 µl) of 1% sulphanilamide in 5% H3PO4 and 0.1% N‐(1‐naphthyl) ethylenediamine in distilled water, and then absorbance at 540 nm was measured. Concentration of nitrite (µm) was calculated from a standard curve created from known concentrations of sodium nitrite dissolved in DMEM.
Data analysis
The results are shown as mean ± standard error of the mean (SEM) of three independent experiments. Comparisons were analysed by one‐way analysis of variance followed by the Dunnet test. Values of P < 0.01 were considered significant.
Results
Olomoucine and roscovitine reduce lipopolysaccharide‐induced NO production
CDK inhibitors – olomoucine and roscovitine – have been shown to inhibit CDK activity and block cell‐cycle progression. As shown in Fig. 1(a), RAW264.7 macrophage proliferation was effectively arrested by olomoucine (75 µm) and roscovitine (20 µm) treatment. Since NO induction after lipopolysaccharide treatment is one of the most reliable pro‐inflammatory responses in RAW macrophages, we first examined the effect of CDK inhibitors on lipopolysaccharide‐induced NO production. RAW cells were pre‐incubated for 24 h with CDK inhibitors to cause growth arrest, then they were treated with lipopolysaccharide. Reduction of lipopolysaccharide‐mediated NO production was observed in cells treated with CDK inhibitor (Fig. 1b). Iso‐olomoucine, a structural isomer of olomoucine with no biological activity, was used as negative control. Treatment with 75 µm did not produce any effect on cell proliferation or NO production. Results indicate that effects of CDK inhibitors are attributed to their inhibition of the CDK activity.
Figure 1.
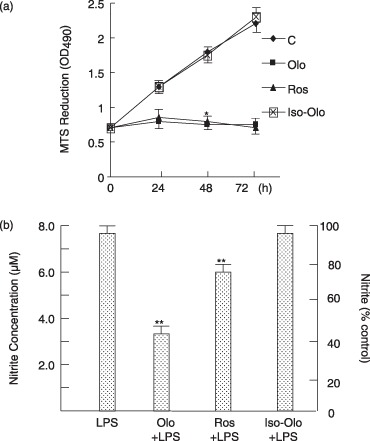
Reduction in lipopolysaccharide‐induced nitric oxide production by CDK inhibitors. (a) RAW cells (1 × 104/well) were incubated with 75 µm olomoucine (Olo), 20 µm roscovitine (Ros) or 100 µm iso‐olomoucine (iso‐Olo) for indicated times. Cell population growth was determined by the MTS reduction method. (b) Cells were pretreated with olomoucine, roscovitine or iso‐plomoucine for 24 h and then stimulated with 1 µg/ml lipopolysaccharide for 24 h. Nitrite concentration (µm) was determined by the value extrapolated from a nitrite standard concentration curve and divided by the value for MTS reduction. Results were expressed as a percentage of nitrite control culture with lipopolysaccharide treatment. Values are mean ± SEM of four independent experiments. *P < 0.001 vs. control cells. **P < 0.001 compared to cells treated with lipopolysaccharide alone.
Expression of lipopolysaccharide‐stimulated pro‐inflammatory genes is diminished by CDK inhibitors
With the exception of NO production, pro‐inflammatory genes are normally induced by lipopolysaccharide in macrophages. Cells were pre‐incubated with the inhibitors for 24 h and then were treated with lipopolysaccharide for 4, 8 or 12 h. RT‐PCR was then performed to analyse mRNA expression of pro‐inflammatory genes (Fig. 2a). In comparison to expression of glyceraldehydes‐3‐phosphate dehydrogenase (GAPDH) as a loading control, levels of IL‐1α, IL‐6, TNFα and iNOS transcripts were significantly reduced by treatment with olomoucine and roscovitine. However, no notable differences in expression of Cu/Zn‐SOD and Mn‐SOD were found. Treatment with 75 µm iso‐olomoucine produced no effect on lipopolysaccharide‐induced pro‐inflammatory gene expression (Fig. 2b). Apparently, expression patterns exhibited by lipopolysaccharide treatment were not changed, but levels of the transcripts were reduced in CDK inhibitor‐treated cells (Fig. 2c). These findings imply that there is a common regulatory mechanism involved in inhibitory effects elicited by CDK inhibitors. Next, we investigated whether decline in iNOS transcripts is reflected at the protein level. As shown in Fig. 2(d), the amount of iNOS protein was significantly lowered after cells were incubated with CDK inhibitors, as compared to cells that were treated with lipopolysaccharide only.
Figure 2.
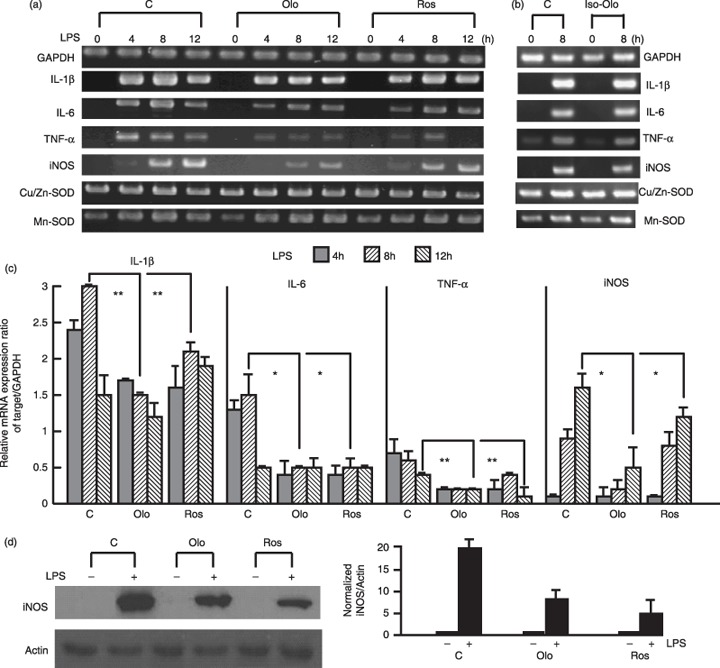
Lipopolysaccharide‐induced pro‐inflammatory gene expression was reduced by pretreatment with CDK inhibitors. (a) Cells were pretreated with olomoucine (Olo), roscovitine (Ros) or iso‐olomoucine (b) for 24 h and then incubated with lipopolysaccharide (1 µg/ml) for the indicated times. Pro‐inflammatory gene expression was detected by RT‐PCR. (c) Bars indicate mean ± SEM of three independent experiments for (a) determined from densitometry relative to GAPDH transcript. **P < 0.01 vs. lipopolysaccharide alone‐treated group. *P < 0.001 compared with lipopolysaccharide alone‐treated group. (d) Cells were pretreated with olomoucine or roscovitine for 24 h; then cells were stimulated with 1 µg/ml lipopolysaccharide for 12 h. Normalized soluble extracts of protein were then prepared and separated by SDS‐PAGE for immunoblot analysis of iNOS induction. Actin was used as loading control. Normalized level of iNOS is shown in the graph and bars indicate mean ± SEM of three independent experiments.
Olomoucine decreases iNOS promoter transcription activity
According to our observations, CDK inhibitors were capable of reducing expression of the pro‐inflammatory genes. To examine whether inhibitory effects occured at the transcription level, we analysed effects of olomoucine on activation of a 1.5‐kb fragment of the iNOS promoter. In a similar way to iNOS mRNA level, the inhibitor was observed to reduce induction level of lipopolysaccharide‐induced activation of the iNOS promoter Fig. 3a). Olomoucine is able to reduce phosphorylation of p38, c‐Jun N‐terminal kinase and IKK (data not shown). This suggests that transcription factors such as NF‐κB may be affected by the inhibitor, which might lead to blocking of their activation from an upstream signal transduction pathway. In Fig. 3(b), decrease in NF‐κB reporter activity is shown after olomoucine treatment at basal as well as after lipopolysaccharide stimulation. These results demonstrate that alleviation of lipopolysaccharide‐induced pro‐inflammatory responses by olomoucine may be due to reduction of NF‐κB transcription at the transcriptional level.
Figure 3.
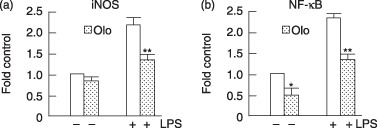
Reduction of lipopolysaccharide‐induced iNOS promoter activity and NF‐κB reporter activity by olomoucine. RAW cells were transiently transfected with 2 µg of iNOS promoter‐luciferase and NF‐κB reporter using the lipofectamine method. Cells were pretreated with olomoucine (close column) at 12 h and then treated 1 µg/ml lipopolysaccharide for 24 h. Extracts were prepared and luciferase activity measured. Results were expressed as fold induction of control luciferase activity in the culture without lipopolysaccharide treatment. Values are mean ± SEM of six independent experiments. *P < 0.01 compared with the media alone‐treated cells. **P < 0.001 compared with lipopolysaccharide alone‐treated group.
Knockdown of E2F1 decreases NF‐κB transcription activity
In the quiescent cells, E2F activity is blocked by hypophosphorylated Rb. Therefore, E2F1 activity is inactivated by CDK inhibitors through prevention of CDK phosphorylation of Rb. To examine this possibility, E2F1i was used to knock down expression of E2F1. E2F1i effectively reduced E2F1 RNA (data not shown) and protein levels (Fig. 4a) in our macrophages. In Fig. 4(b), olomoucine and E2F1i are able to reduce E2F reporter gene activity, indicating that CDK inhibition and E2F1i are able to ablate E2F transcriptional activity. Remarkably, similar to the effects of CDK inhibitors, E2F1i abrogates lipopolysaccharide‐induced iNOS promoter activation as well as NF‐κB reporter activity (Fig. 4c). However, it is thought that the iNOS promoter does not contain an E2F‐binding site and therefore, E2F1 should be unable to bind directly to NF‐κB‐response sequences of the reporter gene. These observations imply that inhibition of cell‐cycle progression by olomoucine may impair pro‐inflammatory responses due to inhibition of activation of E2F1, and this then down‐regulates NF‐κB expression. To examine this hypothesis, RT‐PCR was performed to analyse expression of p65, the NF‐κB subunit (Fig. 5a). As compared with RNA expression of CD14, p65 showed decrease in mRNA expression after olomoucine treatment. At the protein level, there was reduction in level of NF‐κB p65 subunit (Fig. 5b). These findings indicate that growth arrested cells are able to directly decrease expression of the NF‐κB p65 subunit and this in turn, reduces lipopolysaccharide‐stimulated expression of pro‐inflammatory genes.
Figure 4.
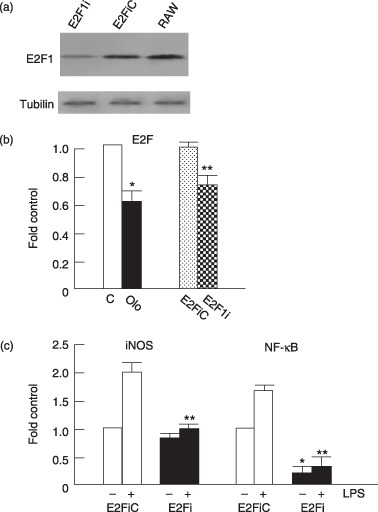
Olomoucine and knockdown E2F1 abrogated lipopolysaccharide‐induced iNOS promoter activity and activation of NF‐κB. (a) RAW264.7 cells were transfected with E2F1 interference RNA (E2Fi) and control vector (E2FiC). Transfected cells were analysed for presence of E2F1 and tubulin using Western blotting. (b) Cells were transfected with 1 µg E2F‐luciferase reporter DNA. Olomoucine was added 12 h later and incubated for 24 h. On co‐transfection, 1 µg E2F1i or E2F1iC (as a control, C) were co‐transfected with E2F‐luciferase reporter DNA and cells were cultured for 36 h. Extracts were prepared and luciferase activity measured. Values are mean ± SEM of three independent experiments. *P < 0.01 compared with the media alone‐treated cells. **P < 0.01 compared with E2F1iC group. (c) RAW cells were transiently cotransfected with 1 µg of the iNOS promoter‐luciferase or the NF‐κB reporter and 1 µg E2F1i or E2F1iC (as a control). The cells were cultured for 24 h and then treated with 1 µg/ml lipopolysaccharide (+) for 24 h. Extracts were prepared and luciferase activity measured. Results were expressed as fold induction of luciferase activity normalized against the control. Values are mean ± SEM of three independent experiments. *P < 0.01 compared with E2F1iC group. **P < 0.001 compared with lipopolysaccharide‐treated E2F1iC group.
Figure 5.
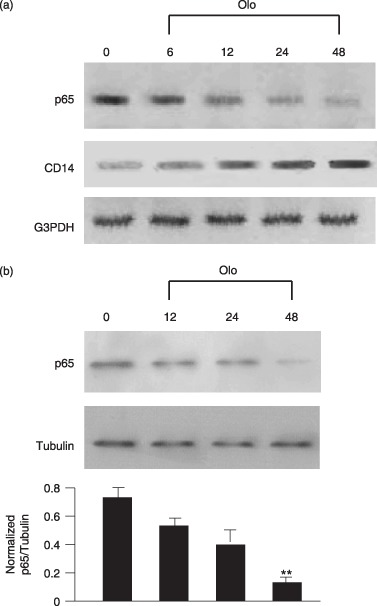
Olomoucine down‐regulates NF‐κB p65 expression. Cells were treated with olomoucine for the indicated intervals. p65 was detected by RT‐PCR (a) and Western blotting (b). Tubulin was used as a loading control. Normalized level of p65 is shown in the graph presented. Values are mean ± SEM of three independent experiments. **P < 0.01 compared with media alone‐treated cells.
Discussion
In this study, we have demonstrated that olomoucine and roscovitine are able to inhibit cell proliferation in murine RAW264.7 macrophages. Compared to proliferating cells, arrested cells showed reduced NO production and cytokine gene expression under lipopolysaccharide stimulation. In addition, we investigated activation of lipopolysaccharide‐induced iNOS promoter and of NF‐κB reporter gene in inhibition of cell‐cycle progression upon olomoucine treatment. We showed that cells reduce iNOS promoter activity and alleviate NF‐κB transcription activation. After co‐transfection with E2F1i, suppression of lipopolysaccharide‐mediated iNOS promoter activity and NF‐κB activation was observed. Moreover, we demonstrated that olomoucine reduces both RNA and protein expression levels of the p65 subunit of NF‐κB. Based on our observation, we proposed a model underlying molecular mechanisms that inhibition of cell‐cycle progression may attenuate lipopolysaccharide‐induced pro‐inflammatory responses through down‐regulation of transcription factor NF‐κB (Fig. 6).
Figure 6.
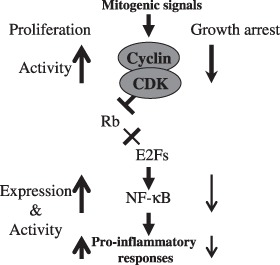
Hypothetical model of growth arrested cells with decreased pro‐inflammatory response in lipopolysaccharide‐stimulated macrophages. Mitogenic signals increase CDK activity (↑), which phosphorylates Rb to promote E2F transcription activity, and induces cell‐cycle progression. E2Fs may be implicated with expression of NF‐κB in the proliferating cells. Therefore, growth arrested cells, which decrease activity of CDKs (↓) and E2F transcription activity, reduce lipopolysaccharide‐stimulated pro‐inflammatory responses by down‐regulation of NF‐κB activation and expression (↓).
We also evaluated effects of nontoxic doses of CDK inhibitors olomoucine and roscovitine, in lipopolysaccharide‐induced pro‐inflammatory cytokine expression of RAW264.7 cells. Remarkably, the inhibitors caused growth arrest along with reduced expression of inflammatory cytokines, but not SODs (Fig. 2a). Furthermore, we demonstrated that inhibition of NO production was concomitant with suppression of iNOS expression at both mRNA and protein levels, as shown by RT‐PCR and Western blotting (Fig. 2d). Based on these observations, we suggest that declines of pro‐inflammatory responses in cells inhibited from cell‐cycle progression may be due to blockade of a common activation pathway. Lipopolysaccharide induces production of pro‐inflammatory cytokines and NO through the CD14‐TLR4‐MD2 complex (21). NF‐κB activation in lipopolysaccharide‐stimulated macrophages is via MyD88 that activates downstream molecules IRAK/TRAF6 followed by activation of TAK/TAB to phosphorylate IKKs, or via TRIF that activates downstream molecules RIP/TBK1. Both MyD88 and TRIF pathways can lead to NF‐κB activation (19, 25). However, olomoucine enhances expression of CD14 at both the RNA (Fig. 5) and protein levels (data not shown), indicating that its inhibitory effect would seem to be independent of blockade of lipopolysaccharide binding to its receptor. NF‐κB has been shown to play a major role in lipopolysaccharide‐induced expression of pro‐inflammatory cytokines and iNOS (26, 27). Thus, anti‐inflammatory activity of the CDK inhibitors can occur by inhibiting NF‐κB activation and expression.
E2F1 activation and hyperphosphorylation of Rb have been associated with cell proliferation (28) and observed in mouse and human ulcerative colitis, chronic inflammation regions (29). Systemic leucocyte‐directed siRNA silencing cyclin D1 declined experimentally in induced colitis in mice, by suppressing leucocyte proliferation and T helper cell cytokine expression, demonstrating that cyclin D1 may be involved in inflammatory processes (30). In our results, inhibition of cell‐cycle progression by CDK inhibitor treatment may suppress E2F transcription activity, thereby decreasing Rb phosphorylation by inhibiting CDK activity in cells. To distinguish inhibitory effects on E2F1 transcription activity from effects on upstream kinase pathways, expression of E2F1 was knocked down by RNAi. Our findings show that E2F1i is capable of directly attenuating lipopolysaccharide‐stimulated promoter activity of iNOS as well as NF‐κB activation (Fig. 4). In addition, RAW macrophages treated with olomoucine are able to down‐regulate expression of p65 subunit of NF‐κB (Fig. 5). Therefore, inhibition of cell‐cycle progression would seem to partially decrease pro‐inflammatory responses by abolishing activation of E2Fs, which may affect expression of NF‐κB. However, it remains to be elucidated whether E2Fs are directly involved in NF‐κB expression through binding its promoter region and induction of RNA expression.
E2F1 is required for transcription activation of lipopolysaccharide‐induced pro‐inflammatory genes, such as IL‐1 and TNF‐α (31). Moreover, NF‐κB activity may be regulated by its interaction with E2F1 (32). It has been shown that E2F1 is able to repress human immunodeficiency virus type‐1 promoter through its binding to the NF‐κB enhancer region and interaction with NF‐κB. In murine embryonic fibroblasts, E2F1 released from Rb has been found to inhibit NF‐κB activity in a cell‐cycle‐dependent manner (33). Instead of repressing gene expression, we demonstrated that CDK inhibitor treatment or interference of E2F1 expression may reduce lipopolysaccharide‐stimulated pro‐inflammatory responses. Our finding implies that E2F1 is implicated in activation of NF‐κB to mediate expression of pro‐inflammatory genes in response to lipopolysaccharide.
In the nervous system, CDK inhibitors have been shown to promote neuroprotection, reduce astroglial scar formation and microglial activation, and to attenuate reactive astrogliosis and microglia‐induced inflammatory responses (16, 34, 35, 36). Theoretically, neuroprotective effects provided by CDK inhibitors may be a result of inhibition of cyclin/CDKs that are involved in transition through the cell cycle. However, based on to our observations, neuroprotective effects resulting from treatment with CDK inhibitors may partially be attributable to down‐regulated expression of pro‐inflammatory gene products, such as TNF‐α, that could elicit cell death.
In conclusion, cell‐cycle arrest induced by treatment with CDK inhibitors, olomoucine and roscovitine, in RAW 264.7 macrophages may reduce lipopolysaccharide‐stimulated pro‐inflammatory responses via down‐regulation of NF‐κB activation and transcription. Our results reveal that cell‐cycle regulatory genes may participate in regulating pro‐inflammatory responses by cross‐talk between NF‐κB and E2F1 signalling pathways. This study suggests that targeting the cell‐cycle pathway of Cyclin‐CDK, may be feasible for anti‐inflammatory therapies.
Acknowledgements
This work was supported in part by grants from the National Science Council (NSC‐95‐2313‐B‐019‐004), VTY Joint Research Program, and the Cheng‐Hsin Rehabilitation Medical Center (Taipei, Taiwan).
References
- 1. Vermeulen K, Van Bockstaele DR, Berneman ZN (2003a) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36, 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Souza SJ, Vespa A, Murkherjee S, Maher A, Pajak A, Dagnino L (2002) E2F‐1 is essential for normal epidermal wound repair. J. Biol. Chem. 277, 10626–10632. [DOI] [PubMed] [Google Scholar]
- 3. Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15, 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stevaux O, Dyson NJ (2002) A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 14, 684–691. [DOI] [PubMed] [Google Scholar]
- 5. Stevens C, La Thangue NB (2003) E2F and cell cycle control: a double‐edged sword. Arch. Biochem. Biophys. 412, 157–169. [DOI] [PubMed] [Google Scholar]
- 6. Vermeulen K, Berneman ZN, Van Bockstaele DR (2003b) Cell cycle and apoptosis. Cell Prolif. 36, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dai Y, Grant S (2003) Cyclin‐dependent kinase inhibitors. Curr. Opin. Pharmacol. 3, 362–370. [DOI] [PubMed] [Google Scholar]
- 8. Haesslein JL, Jullian N (2002) Recent advances in cyclin‐dependent kinase inhibition. Purine‐based derivatives as anti‐cancer agents. Roles and perspectives for the future. Curr. Top. Med. Chem. 2, 1037–1050. [DOI] [PubMed] [Google Scholar]
- 9. Alessi F, Quarta S, Savio M, Riva F, Rossi L, Stivala LA, Scovassi AI, Meijer L, Prosperi E (1998) The cyclin‐dependent kinase inhibitors olomoucine and roscovitine arrest human fibroblasts in G1 phase by specific inhibition of CDK2 kinase activity. Exp. Cell Res. 245, 8–18. [DOI] [PubMed] [Google Scholar]
- 10. Hardcastle IR, Golding BT, Griffin RJ (2002) Designing inhibitors of cyclin‐dependent kinases. Annu. Rev. Pharmacol. Toxicol. 42, 325–348. [DOI] [PubMed] [Google Scholar]
- 11. Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boutis A, Papazisis K, Pistevou‐Gompaki K, Lambropoulos A, Sofroniadis I, Papageorgiou A, Destouni E, Kortsaris A (2006) Cyclin‐dependent kinase (CDK) inhibitor olomoucine enhances gamma‐irradiation‐induced apoptosis and cell cycle arrest in Raji cells. Anticancer Res. 26, 3493–3498. [PubMed] [Google Scholar]
- 13. Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP (2008) R‐Roscovitine simultaneously targets both the p53 and NF‐κB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ. 15, 263–273. [DOI] [PubMed] [Google Scholar]
- 14. Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez‐Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C (2006) Cyclin‐dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064. [DOI] [PubMed] [Google Scholar]
- 15. Calegari F, Huttner WB (2003) An inhibition of cyclin‐dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J. Cell Sci. 116, 4947–4955. [DOI] [PubMed] [Google Scholar]
- 16. Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 102, 8333–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Markus MA, Kahle PJ, Winkler A, Horstmann S, Anneser JM, Borasio GD (1997) Survival‐promoting activity of inhibitors of cyclin‐dependent kinases on primary neurons correlates with inhibition of c‐Jun kinase‐1. Neurobiol. Dis. 4, 122–133. [DOI] [PubMed] [Google Scholar]
- 18. Monaco EA III, Beaman‐Hall CM, Mathur A, Vallano ML (2004) Roscovitine, olomoucine, purvalanol: inducers of apoptosis in maturing cerebellar granule neurons. Biochem. Pharmacol. 67, 1947–1964. [DOI] [PubMed] [Google Scholar]
- 19. Akira S, Takeda K (2004) Toll‐like receptor signaling. Nat. Rev. Immunol. 4, 499–511. [DOI] [PubMed] [Google Scholar]
- 20. Raetz CR, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bochud PY, Calandra T (2003) Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ 326, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ulevitch RJ (2004) Therapeutics targeting the innate immune system. Nat. Rev. Immunol. 4, 512–520. [DOI] [PubMed] [Google Scholar]
- 23. Karin M, Greten FR (2005) NF‐κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759. [DOI] [PubMed] [Google Scholar]
- 24. Smith PD, Ochsenbauer‐Jambor C, Smythies LE (2005) Intestinal macrophages: unique effector cells of the innate immune system. Immunol. Rev. 206, 149–159. [DOI] [PubMed] [Google Scholar]
- 25. Kawai T, Akira S (2006) TLR signaling. Cell Death Diff. 13, 816–825. [DOI] [PubMed] [Google Scholar]
- 26. Guha M, Mackman N (2001) LPS induction of gene expression in human monocytes. Cell Signal 13, 85–94. [DOI] [PubMed] [Google Scholar]
- 27. Tian B, Brasier AR (2003) Identification of a nuclear factor kappa B‐dependent gene network. Recent Prog. Horm. Res. 58, 95–130. [DOI] [PubMed] [Google Scholar]
- 28. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ying L, Marino J, Hussain SP, Khan MA, You S, Hofseth AB, Trivers GE, Dixon DA, Harris CC, Hofseth LJ (2005) Chronic inflammation promotes retinoblastoma protein hyperphosphorylation and E2F1 activation. Cancer Res. 65, 9132–9136. [DOI] [PubMed] [Google Scholar]
- 30. Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M (2008) Systemic leukocyte‐directed siRNA delivery revealing cyclin D1 as an anti‐inflammatory target. Science 319, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim C‐A, Yao F, Wong j J‐Y, George J, Xu Han Chiu KP (2007) Genome‐wide mapping of RELA (p65) binding identifies E2F1 as a transcriptional activator recruited by NF‐κB upon TLR4 activation. Mol. Cell 27, 622–635. [DOI] [PubMed] [Google Scholar]
- 32. Kundu M, Guermah M, Roeder RG, Amini S, Khalili K (1997) Interaction between cell cycle regulator, E2F‐1, and NF‐κB mediates repression of HIV‐1 gene transcription. J. Biol. Chem. 272, 29468–29474. [DOI] [PubMed] [Google Scholar]
- 33. Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, Albanese C, Machii T, Testell RG, Kanakura Y (2002) E2F1 and c‐Myc potentiate apoptosis through inhibition of NF‐κB activity that facilitates MnSOD‐mediated ROS elimination. Mol. Cell 9, 1017–1029. [DOI] [PubMed] [Google Scholar]
- 34. Tian DS, Yu ZY, Xie MJ, Bu BT, Witte OW, Wang W (2006) Suppression of astroglial scar formation and enhanced axonal regeneration associated with functional recovery in a spinal cord injury rat model by the cell cycle inhibitor olomoucine. J. Neurosci. Res. 85, 1053–1063. [DOI] [PubMed] [Google Scholar]
- 35. Tian DS, Xie MJ, Yu ZY, Zhang Q, Wang YH, Chen B, Chen C, Wang W (2007) Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 1135, 177–185. [DOI] [PubMed] [Google Scholar]
- 36. Zhu Z, Zhang Q, Yu Z, Zhang L, Tian D, Zhu S, Bu B, Xie M, Wang W (2007) Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo . Glia 55, 546–558. [DOI] [PubMed] [Google Scholar]