

Abstract
Objective
Haem oxygenase‐1 (HO‐1) plays important roles in cytoprotection and tumour growth. Cholangiocarcinoma (CCA) is a deadly malignancy with very poor prognosis. The role of HO‐1 in tumour progression in CCA up to now has been relatively unexplored, thus, its possible therapeutic implications in CCA have been investigated here.
Materials and methods
HO‐1 expression in tumour tissues from 50 CCA patients was determined by immunohistochemical analysis and its association with survival time was evaluated using the Kaplan–Meier method. Its role in CCA cells in vitro was evaluated by transwell and wound healing assays and suppression of HO‐1 expression by siRNA. Effects of HO‐1 inhibition on gemicitabine (GEM)‐mediated tumour suppression was evaluated in nude mice xenografted with CCA cells.
Results
HO‐1 expression was inversely associated with median overall survival time. Hazard ratio of patients with high HO‐1 expression was 2.42 (95% CI: 1.16–5.08) with reference to low expression and HO‐1 knock‐down expression inhibited transwell cell migration. Suppression of HO‐1 by Zn‐protoporphyrin (ZnPP) enhanced cytotoxicity to GEM in CCA cells, validated in CCA xenografts. Treatment with GEM and ZnPP almost completely arrested tumour growth, whereas treatment with only a single reagent, retarded it. Tumour inhibition was associated with reduction in expression of Ki‐67 and microvascular density, and enhanced p53 and p21 immunohistochemical staining.
Conclusion
High HO‐1 expression was associated with poor prognosis of CCA. Synergistic role of HO‐1 inhibition in chemotherapy of CCA is a promising insight for treatment of this tumour and warrants further investigation.
Introduction
Cholangiocarcinoma (CCA) is a particularly devastating malignancy as the majority of cases develop without any clinical symptoms, thus initially its diagnosis difficult 1. Its prognosis is typically extremely poor; treatment outcomes and patient survival have improved only slightly over the past number of decades 1. Currently, complete tumour resection is the only effective treatment to prolong survival, tumour recurrence is common and occurs in up to 63% of patients within 2 years 2, 3. However, the majority of patients are already at advanced stages of the disease at diagnosis, thus chemotherapy is the only remaining therapeutic option for most patients with unresectable tumours. The current chemotherapeutic regimen, gemcitabine‐based or fluoropyrimidine‐based, results in average overall survival of less than 1 year, and 5‐year survival is almost nil 3, 4. There is therefore great need for a more effective treatment to improve survival rates and quality of life for these patients 5, 6. Targeting prosurvival mechanisms in cancer cells is one strategy to overcome resistance to chemotherapy; HO‐1, NQO1 and Nrf2 signalling systems are involved in cytoprotective mechanisms and are therefore possible therapeutic targets 7, 8, 9, 10.
Haem oxygenase‐1 (HO‐1) is a microsomal enzyme which catalyzes the first rate‐limiting step in degradation of haem, leading to equimolar production of biliverdin, carbon monoxide and free iron. HO‐1 is induced in response to cell stress, oxidative stimuli, hypoxia and also during tumour growth 11. More importantly, clinically HO‐1 is found to be over‐expressed in some cancers including those of the prostate, bladder, colon and pancreas 12, 13, 14. In these, tumours express high levels of HO‐1 and this over‐expression seems to provide cell proliferation advantages and possibly drug resistance 15, 16, 17; HO‐1 also has roles in growth and neoangiogenesis. It is also involved in cytoprotection in tumours against oxidative insult in the tumour microenvironment, resulting from chemotherapy and radiotherapy 15, 18, probably due to its anti‐oxidative and anti‐apoptotic activities 15, 19. Expression of HO‐1 has been found to be associated with clinicopathologic features, including tumour growth, invasiveness and overall survival in oesophageal, bladder and gallbladder cancers 13, 20, 21, however, whether its expression is associated with prognosis of cancer of the bile duct has still been unexplored. Inhibition of HO‐1 activity may suppress growth and metastasis of CCA, and thus might prove to be a successful target for increasing chemosensitivity of these tumours. We have recently shown that inhibition of HO‐1 in CCA cell lines, by HO‐1 siRNA or by pharmacological inhibitor zinc protoporphyrin IX (ZnPP), resulted in their remarkably enhanced sensitivity to GEM 16, although the mechanism of the effect has remained unknown.
In the present study, we evaluated relationships between expression of HO‐1 in tumour tissue from CCA patients and the patients' subsequent prognosis. Effects of HO‐1 suppression on tumour growth, migration and sensitivity to chemotherapeutic agents were examined. We further evaluated effects of combined therapy of GEM and ZnPP using an in vivo model with xenografted nude mice.
Materials and methods
Subjects and tissue samples
Fifty patients admitted to Srinagarind Hospital, Faculty of Medicine, Khon Kaen University for treatment of CCA, were recruited into the study; the protocol was approved by the Khon Kaen University Ethics Committee for Human Research (HE541033) and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients. They had been diagnosed with CCA confirmed by histopathological examination, and had undergone surgical resection of tumours with partial hepatectomy. Thirty‐four (68%) received adjuvant chemotherapy, either 5‐fluorouracil‐based (20%) or oral fluoropyrimidine (48%); sixteen (32%) had no adjuvant chemotherapy. Also, CCA tissue sections from the specimen bank of the Liver Fluke and Cholangiocarcinoma Research Center were used for immunohistochemical (IHC) study.
Cell lines and cell cultures
Human CCA cell lines KKU‐100, KKU‐M213 and KKU‐M055 were kindly provided by Prof. Banchob Sripa, Department of Pathology, Faculty of Medicine, Khon Kaen University. These had been derived from CCA tissues and had previously been characterized 22, 23. Samples of KKU‐100 and M213 cell lines were deposited in the National Institute of Biomedical Innovation, JCRB Cell Bank, Japan. All cell lines were routinely cultured in complete media consisting of Ham's F12 medium, supplemented with 10% foetal calf serum, 12.5 mm HEPES, pH 7.3, 100 U/ml penicillin G and 100 μg/ml gentamicin 24.
KKU‐100 cells were seeded on to 96‐well plates, 7500 cells/well, and left overnight. They were then treated with a selection of concentrations of ZnPP and GEM from 1 to 1000 nm, for 24 h. For combination treatment, KKU‐100 cells were pre‐treated with ZnPP for 4 h prior to GEM, and incubation was continued for 24 h. Cytotoxicity was measured using the sulphorhodamine B assay, as previously described 24.
Cell motility and wound healing assays
For cell motility and wound healing assays, KKU‐100 and M213 cells with HO‐1 knocked down by HO‐1 siRNA, were used. KKU‐100 and M213 cells were transfected with HO‐1 siRNA as described below for 24 h, then seeded on to transwell inserts in 300 μl medium. After 6 h incubation, filter membranes were cut and placed on glass slides, covered with mounting medium containing DAPI, and then covered with cover‐slips. Cell migration across membranes was measured using a Nikon Eclipse Ti fluorescence microscope (Nikon Singapore, Capital Square, Singapore).
For the wound healing assay, after transfection with HO‐1 siRNA for 24 h, KKU‐100 and M213, cells were seeded on to 24‐well plates, 150 000 cells per well, in full culture medium, to obtain 80–90% confluence on the day of the experiment. Next day, dishes of cultured cells were each scratched in a straight line with a sterile 200 μl pipette tip, to make wounds. Wound width outlines were measured and recorded and images were captured using a phase‐contrast microscope, 0, 6, 12 and 24 h after scratching.
HO‐1 small interfering RNA transfection
Transfection of HO‐1 siRNA was performed using siGENOME SMARTpool of four sequences of siRNA (M‐006372‐02‐0005: Dharmacon (Lafayette, CO, USA) at final concentration of 200 nm, and lipofectamine 2000 (Invitrogen, Calsbad, CA, USA) according to the manufacturer's instructions. As negative control, siGENOME non‐targeting siRNA (D‐001210‐02‐05) was introduced into the cells using the protocol as previously described 16.
Reverse transcription real‐time polymerase chain reaction
The three CCA cell lines, KKU‐100, M213 and M055, were seeded at 1.5 × 104 cells/well in six‐well plates and allowed to grow for 24 h. Total RNA was isolated and PCR products were generated using a previously described method 25. Primer sequences were as follows: HO‐1: forward, 5′‐CTG ACC CAT GAC ACC AAG GAC‐3′ and HO‐1 reverse: 5′‐AAA GCC CTA CAG CAA CTG TCG‐3′, GenBank accession number NM_002133.2; β‐actin: forward 5′‐AGT GTA GCC CAG GAT GCC CTT‐3′ and β‐actin: reverse, 5′‐GCC AAG GTC ATC CAT GAC AAC‐3′, Gen Bank accession number NM_00246.5. To quantify relative expression of genes, relative quantification was performed using the standard curve method. Amount of HO‐1 mRNA was expressed as a ratio to β‐actin mRNA.
Anti‐tumour activity in the animal model
All experimental procedures were performed according to standards for experimental animal handling, and the study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) (application #2011/SHS/650) SingHealth, Singapore. KKU‐100 cells (2 × 106) in 200 μl Ham‐F12 medium were injected subcutaneously beneath the dorsal skin of 6‐week‐old 28 female Balb/c nude mice (Animal Resource Center, Western Australia), housed under specific pathogen‐free conditions. When tumours grew to volumes of around 200 mm3, mice were randomly divided into four groups; control, GEM (Gemzar®; Eli Lilly, Indianapolis,IN, USA), ZnPP (Enzo Life Sciences, Farmingdale, NY, USA) and combination of both GEM and ZnPP. ZnPP was dissolved in DMSO at 100 mg/ml, and diluted further in PBS immediately before use. GEM was dissolved in PBS, as previously described, to a concentration of 10 mg/ml and diluted further in PBS. ZnPP (12.5 mg/kg) and GEM (60 mg/kg) were administered intraperitoneally once a week for 3 weeks (day 0, 7 and 14). In the combined drug‐treated group, ZnPP was injected 1 h before GEM injection. The control group was treated with vehicle solution following the same schedule. Xenograft tumour volume was measured twice a week using electronic callipers, and tumour volumes were calculated using the equation [L × W 2]/2 (mm3), where L = length and W = width; weight of the mice was measured twice a week. Animals were euthanised on day 17, and tumour masses were measured for further analysis.
Immunohistochemical analysis of human CCA tissues and mouse xenografted tumours
Three micron paraffin wax‐embedded human CCA tissue sections were deparaffinized, followed by antigen retrieval using microwave treatment, for 30 min in 0.01 M citrate buffer (pH 6.0). After rehydratration, tissue sections were incubated with primary antibody against HO‐1, ADI‐SPA‐895 (Enzo Life Sciences) at 4 °C overnight. The following day, tissue sections were incubated at room temperature for 1 h with secondary antibody, using the Dako EnVision® HRP‐labelled polymer anti‐rabbit system (K4003; Dako, Kyoto, Japan) according to the manufacturer's instruction. Tissue sections were then counterstained with haematoxylin and mounted in DPX Mounting Medium (UN 1866; CellPath, Newtown, UK). Areas of HO‐1 protein staining were captured using a Zeiss Axio ScopeA.1 microscope, (ZEISS Singapore, Singapore) original magnification 200×. Quantitative HO‐1 (Q) staining levels were measured as number of positive cells (P) and stain intensity (I), using the formula: Q = P × I. Percentage of positive cell numbers were graded as 0–25% = 1, 26–50% = 2, 51–75% = 3 and 76–100% = 4 and levels of intensity were graded on a scale of 1–4 (1+, no detectable to very weak staining; 2+, weak positive staining; 3+, moderate positive staining; 4+, strong positive staining).
For tumour tissues from xenografted mice, tissues were resected, weighted and fixed in 10% formaldehyde solution and embedded in paraffin wax. Cut sections were then deparaffinized, followed by antigen retrieval, using autoclave treatment for 10 min in 0.01 m citrate buffer (pH 6.0). IHC staining for Ki‐67, CD31, mdm2, p53 and p21 was performed using the streptavidin–biotin complex method and R.T.U. VECTORSTAIN® Universal Quick Kit (PK‐7800; Vector, Burlingame, CA, USA) according to the manufacturer's instructions. Tissue sections were incubated with primary antibodies, anti‐Ki‐67 (clone MIB‐1; Dako) at room temperature for 30 min, anti‐CD31 (ab 28364: Abcam, Cambridge, UK) anti‐mdm2 (sc‐965: Santa Cruz, CA, USA) anti‐p53 (sc‐98) and anti‐p21 (2946; Cell Signaling Technology, Beverly, MA, USA) at 4 °C overnight. Labelled sections were then counterstained with haematoxylin and then mounted in DPX Mounting Medium (UN 1866; CellPath). CD31‐stained vessels were detected under a microscope with original magnification 200×. Mean value of vessel numbers in tumours was calculated from 10 fields in the three most vascularized areas. Areas of mdm2‐ and p21‐positive cell staining were captured using a Nikon DS‐Ri1 microscope with original magnification at 400× and using Image Pro‐plus program Bethesda, MD, USA to count positive cells. Staining levels of p53 were calculated using grading of number of positive cells multiplied by levels of intensity (positive cell numbers graded as 0–30 = 1, 31–60 = 2, 61–90 = 3 and 91–120 = 4, and levels of intensity ranges by 0–4)
Statistical analysis
Data were expressed as mean ± SD. Analysis of variance was used to determine significant differences between experimental groups with the Student–Newman–Keuls post hoc test. Cross‐tabulation was analysed using chi‐square or Fisher's exact test for association of HO‐1 IHC staining, and pathological features of CCA patient tissues. Distribution of HO‐1 expression levels in CCA patients was analysed using the Shapiro–Wilk test to determine normal distribution. Overall survival was analysed with the Kaplan–Meier method and Breslow–Gehan test, to compare survival time among subgroups of predictor variables including HO‐1 expression, histological type and metastasis status. Cox proportional hazards model was used to estimate hazard ratios after adjusting confounders including age, gender, gross pathology, histopathology, metastasis and adjuvant chemotherapy. Level of significance was set at P < 0.05; all analyses were performed using Stata version 10 software (StataCorp, College Station, TX, USA).
Results
HO‐1 expression in CCA tissue
Characteristics of patients are shown in Table 1. HO‐1 is ubiquitously expressed in liver parenchymal tissue, however expression in CCAs, apparent in the cytosol, here varied widely from very low to high levels (Fig. 1b,c). Levels of HO‐1 staining in CCA specimens did not follow normal distribution (P < 0.01), value of antimode in the distribution being used as cut‐off to classify levels of HO‐1 protein expression. Eighteen subjects were classified as low expressers and 32 as high expressers. No significant association was found between HO‐1 expression and other variables including, age (P = 0.42), gender (P = 0.77), pathological gross type (P = 0.79), histological type (P = 0.87), metastatic status (P = 0.067) or adjuvant chemotherapy (P = 0.30).
Table 1.
Characteristics of CCA patients
| Age: year ± SD (min–max) | 56.2 ± 8.8 (33–75) |
| Gender: male:female | 32:18 |
| Tumour gross type | |
| Mass forming | 22 |
| Periductal infiltrating | 23 |
| Intraductal growth type | 5 |
| Histological type | |
| Papillary | 23 |
| Non‐papillary | |
| Well differentiated | 20 |
| Moderately differentiated | 4 |
| Poorly differentiated | 3 |
| Metastasis (no/yes) | 22/28 |
Figure 1.
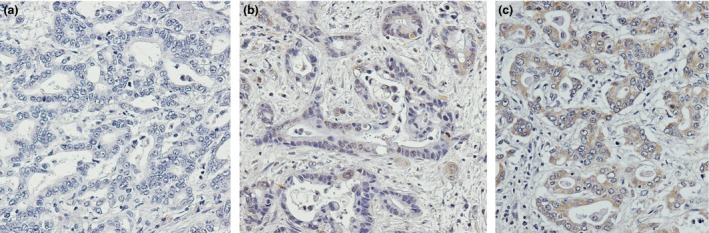
Immunohistochemical staining of HO ‐1 in cholangiocarcinoma tissue from CCA patients. (a) Negative HO‐1 staining control. (b) Tumour tissues with low HO‐1 staining and (c) tumour tissue with high HO‐1 staining (original magnification of 200×)
Survival analysis and various predictor variables
Survival analysis using the Kaplan–Meier method indicated the median overall survival time of subjects in this series as being 257 days (95% CI: 157–386 days). Median survival times of CCA patients stratified by levels of HO‐1 expression, i.e. low or high expression, were 414 days (95% CI: 297–844 days) and 165 days (95% CI: 134–267 days) respectively. Median survival time of patients having histological types of papillary adenocarcinoma or non‐papillary type was 441 days (95% CI: 267–1029 days) and 157 days (95% CI: 113–257 days) respectively. That of patients with no evidence of presence of metastasis was 587 (95% CI: 208–1029 days) and 163 days (95% CI: 134–267 days) respectively. Patients receiving adjuvant chemotherapy with parenteral 5‐FU‐base or oral fluoropyrimidine had median survival times of 1262 days (95% CI: 113–2240 days) or 208 days (95% CI: 141–344 days), respectively, when compared to patients without adjuvant chemotherapy with median survival time of 165 days (95% CI: 65–386 days).
Survival function at mean of covariates is shown in Fig. 2a. Survival function stratified by levels of HO‐1 protein staining, histological type and presence of metastasis are shown in Fig. 2b–d respectively. Multivariate analysis by Cox proportional hazards was performed to explore the impact of various potential predictors. The parameters in the final model of analysis included HO‐1 expression, age, gender, gross types, histological type, presence of metastasis and adjuvant chemotherapy. HO‐1 staining level, age, histological type and metastasis were significant predictors of relative risk to survival of CCA patients. Patients with high HO‐1 expression had poor overall survival levels compared to patients with low HO‐1 tumour expression, with hazard ratio of 2.42 (95% CI: 1.16–5.38), P < 0.05 (Table 2). Patients aged ≥57 years with histological type other than papillary adenocarcinoma and presence of metastasis were at higher risk of short survival time after adjusting for other variables (Table 2). Patients with multiple poor predictor variables were at higher risk than ones with only a single poor predictor. Hazard ratios for patients with the combination of high‐risk predictors included high HO‐1 in combination with histological type of non‐papillary tumours, high HO‐1 with metastasis and combination of all three poor predictors, were 7.59 (95% CI: 2.37–24.29), 6.73 (95% CI: 2.40–19.38) and 21.08 (95% CI: 5.50–80.88) respectively. This result suggests HO‐1 to be an independent prognostic predictor of CCA.
Figure 2.
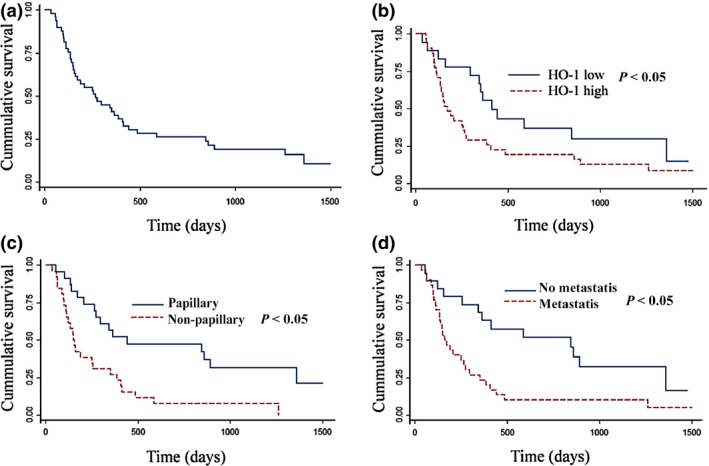
Cumulative survival curves of cholangiocarcinoma patients. (a) Survival function at mean of covariates. (b) Survival function stratified by levels of HO‐1 IHC staining. (c) Survival function stratified by histological types of papillary and non‐papillary adenocarcinoma. (d) Survival function stratified by presence of metastasis.
Table 2.
Mutivariate analysis by Cox proportional hazards
| Variable | Category | Hazard ratio | 95% CI | P‐value |
|---|---|---|---|---|
| Age (year) | <57 | 1 | 1.08–7.24 | 0.03 |
| ≥57 | 2.80 | |||
| Gender | Female | 1 | 0.49–2.06 | 0.99 |
| Male | 1.00 | |||
| Histological types | Papillary | 1 | 1.52–6.44 | 0.002 |
| Non‐papillary | 3.13 | |||
| Metastasis | No | 1 | 1.23–6.29 | 0.014 |
| Presence | 2.78 | |||
| HO‐1 staining | Low | 1 | 1.16–5.08 | 0.019 |
| High | 2.42 | |||
| Adjuvant chemotherapy | No | 1 | ||
| 5‐FU‐based | 0.49 | 0.17–1.38 | 0.176 | |
| Oral fluoropyrimidine | 1.73 | 0.82–3.65 | 0.149 |
HO‐1 expression in CCA cells and contribution to cell migration
Expression levels of HO‐1 were determined in the three CCA cell lines using reverse transcription real‐time‐PCR. KKU‐100 cells had the highest level of HO‐1 mRNA expression, while the other two, M213 and M055, had much lower expression levels (Fig. 3a). KKU‐100 and M213 cells – representative of high and low HO‐1‐expressing cell lines, respectively, were used for further study. To investigate the functional role of HO‐1 in CCA, cells in which HO‐1 was knocked down, cells using siRNA, were subjected to transwell and wound healing assays. HO‐1 siRNA effectively reduced HO‐1 transcripts in KKU‐100 and M213 cells by around 70% and 60%, respectively, compared to control treatment with non‐target siRNA (Fig. 3b). Transwell inserts cultured with CCA cells in which HO‐1 had been knocked down, had reduced numbers of cells migrating through the membrane inserts compared to control cells (Fig. 3c,d). Suppression of cell motility was further supported by results of the wound healing assay. HO‐1 knock‐down cells migrated much more slowly than control cells, resulting in almost no closure of wounds after 24‐h incubation (Fig. 3e). This result suggested that HO‐1 played a role in cell migration in CCA cells, regardless of basal HO‐1 expression.
Figure 3.
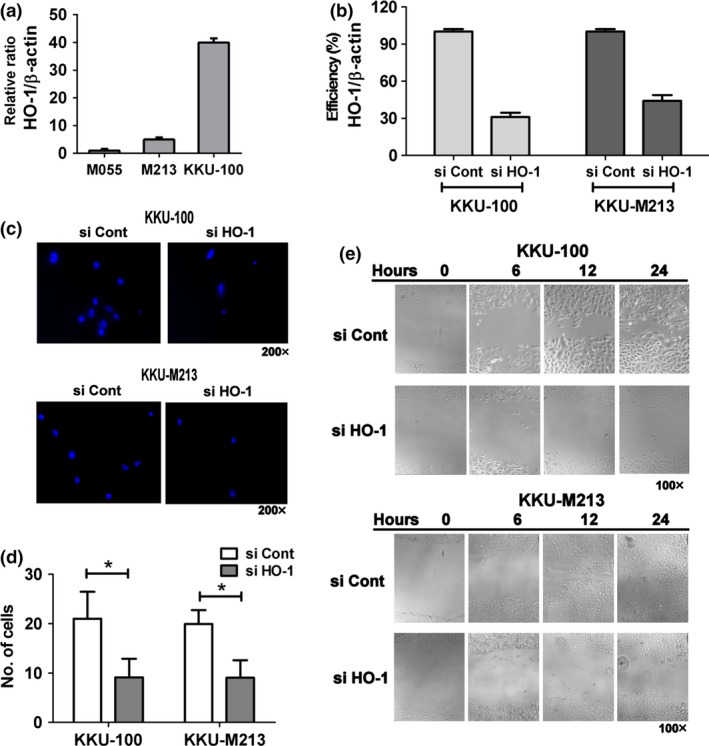
Expression of HO ‐1 in CCA cells and effect of HO ‐1 knock‐down, on cell migration. (a) Basal HO‐1 mRNA expression in CCA cells KKU‐100, M213 and M055, analysed by real‐time PCR. Each bar represents relative expression of HO‐1 normalized with β‐actin expression. (b) KKU‐100 and M213 had knocked down HO‐1 expression by HO‐1 siRNA (si HO‐1) or control transfection (si Cont). Efficiency (%) of knock‐down relative to si Cont was validated using real‐time PCR. (c and d) Cell migration of KKU‐100 and M213 cells was performed using transwell inserts. Representatives of DAPI‐staining images of migrated cells were captured by fluorescence microscopy (original magnification 200 × ). (d) Number of colonies of KKU‐100 and M213 cells was counted over four fields from each insert (original magnification 100 × ). Each bar represents mean ± SD of 12 fields from at least three independent experiments. *P < 0.05 versus controls. (e) Wound healing assay in KKU‐100 and M213 cells was performed. Images of four fields from each well were captured (original magnification 100×) and width of the closing wounds were measured.
Suppression of HO‐1 enhanced anti‐tumour response to GEM in vitro and in vivo
As HO‐1 functions as protective enzyme in various types of cell, it is plausible that its inhibition would increase sensitivity of the cancer to chemotherapeutic agents. The effect of HO‐1 inhibition in relation to GEM‐induced cytotoxicity was evaluated in vitro and in vivo. In the in vitro study, toxicity of ZnPP, an HO‐1 inhibitor, and GEM alone in KKU‐100 cells, was shown in a dose‐dependent fashion (Fig. 4a). ZnPP at 10 nm induced cytotoxicity of less than 10%, and was then used in combination with GEM. Drug combination enhanced cytotoxicity, particularly at low concentrations of GEM (Fig. 4b).
Figure 4.
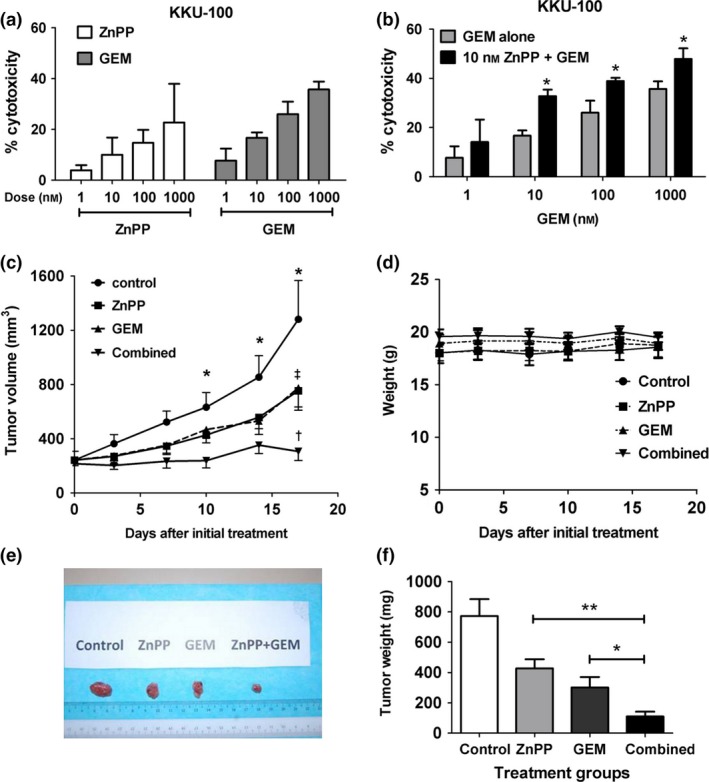
Inhibition of HO ‐1 by Zn PP enhanced anti‐tumour effect of GEM in vitro and in vivo . (a) Dose–response cytotoxicity of ZnPP and GEM in KKU‐100 cells after incubation for 24 h. (b) Enhanced cytotoxicity of GEM by pre‐treatment with ZnPP for 4 h in KKU‐100 cells after incubation with GEM for 24 h. Each bar represents mean ± SD, each from three independent experiment. *P < 0.05 versus GEM alone. (c) Time‐course of tumour growth in nude mice transplanted with KKU‐100 cells. ZnPP, GEM, combination of ZnPP and GEM and saline as vehicle control were administered intraperitoneally on days 0, 7 and 14, when tumour masses had grown to at least 200 mm3. Each value represents mean±SEM, each from seven mice per group. *P < 0.05 combination group versus control group; †P < 0.05 combination versus ZnPP alone, and ‡P < 0.05 combination versus GEM alone. (d) Time‐course of body weights of mice during the drug treatments. (e) Representative tumours resected from mice on day 17. (f) Tumour weights from each treatment group on day 17. Each bar represents mean±SEM each from seven mice in the group. *P < 0.05 drug combination versus GEM alone, and **P < 0.05 drug combination versus ZnPP alone.
Enhanced cytotoxic effects of GEM by ZnPP were further evaluated in nude mice xenografted with KKU‐100 cells. Animals were treated with ZnPP, or GEM, or both in combination, when tumour masses were larger than 200 mm3. Tumour mass in control mice grew exponentially during the study period (Fig. 4c). Treatment with GEM or ZnPP alone for three cycles resulted in partial suppression of tumour growth, compared to non‐treated controls. In contrast, combination of ZnPP and GEM caused almost complete arrest of tumour growth (Fig. 4c). Treatment with individual agents, or in combination, for three cycles did not cause overt toxicity, as indicated by absence of significant changes in body weight (Fig. 4d). The animals showed no signs nor symptoms of distress, and there were no observed mucous membrane lesions. However, the study was limited to only 17 days. Mean tumour weights (mg) on day 17, 3 days after the last treatment cycle, were 772, 427, 300 and 110 mg, for control, ZnPP, GEM and the combination groups respectively (Fig. 4e,f).
Combined GEM and ZnPP treatment suppressed tumour growth by activation of the p53 pathway
We further investigated whether suppression of tumour growth was associated with inhibition of cell proliferation and angiogenesis in tumour tissues. Tumour tissues extracted from four groups of animals were analysed for expression of Ki‐67 and CD31, by immunohistochemistry. Monotherapy with GEM or ZnPP significantly suppressed nuclear expression of Ki‐67 compared to the control group (Fig. 5A). Drug combination further reduced expression of Ki‐67 more than monotherapy with either drug alone. Tumour tissue from the control group had the highest density of blood vessels as measured by CD31 staining, followed by ZnPP‐alone and GEM‐alone groups. Tumours from the drug combination group had the lowest density of blood vessels (Fig. 5B). These results indicate that the drug combination enhanced anti‐proliferation and anti‐angiogenesis in CCA tissues tested.
Figure 5.
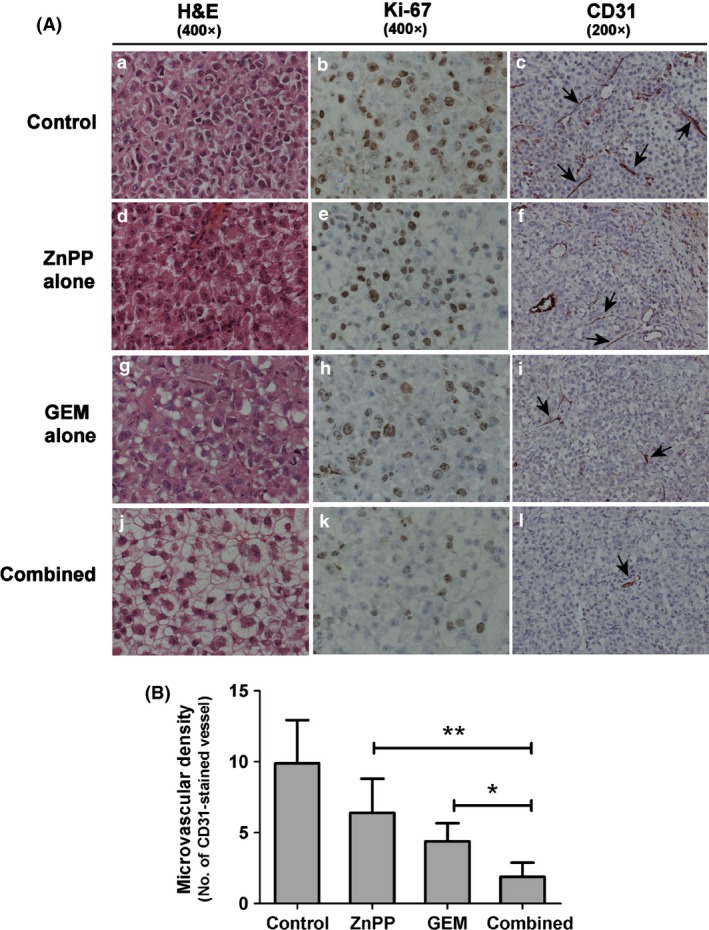
Immunohistochemistry of Ki‐67 and CD 31 staining in tumour tissues. (A) Tumour tissues from mice treated with the drug regimens analysed with H&E and IHC staining for Ki‐67 and CD31. Panels a–c: control; d–f: ZnPP alone, g–i: GEM alone; j–l: combination of GEM and ZnPP. Panels a, d, g, j for H&E staining; b, e, h, k for Ki‐67 staining; c, f, i, l for CD31 staining (the arrows indicate microvessels). (B) Microvascular density as assessed by CD31 staining (original magnification 200×). Each bar represents mean±SD, each from seven mice *P < 0.05 drug combination versus GEM alone and **P < 0.05 drug combination versus ZnPP alone.
As suppression of HO‐1 activity by ZnPP‐enhanced GEM‐induced anti‐proliferation and inhibition of neoangiogenesis, the underlying mechanism was explored. Expression of p53, and its negative regulator, mdm2, and p21 proteins was evaluated. IHC staining indicated that the drug combination induced expression of p53 at higher levels than after single drug treatment (Fig. 6A,C). Conversely, mdm2 was highly expressed in control tissues and was low in GEM or ZnPP groups and remarkably low in the drug combination group (Fig. 6B). Expression of p21 or cyclin‐dependent kinase inhibitor 1 was highest in the combination treatment group, followed by the single drug treatment groups, and the lowest in the controls (Fig. 6D). These results demonstrate that ZnPP‐enhanced GEM‐induced tumour suppression was associated with p53 and downstream protein expression.
Figure 6.
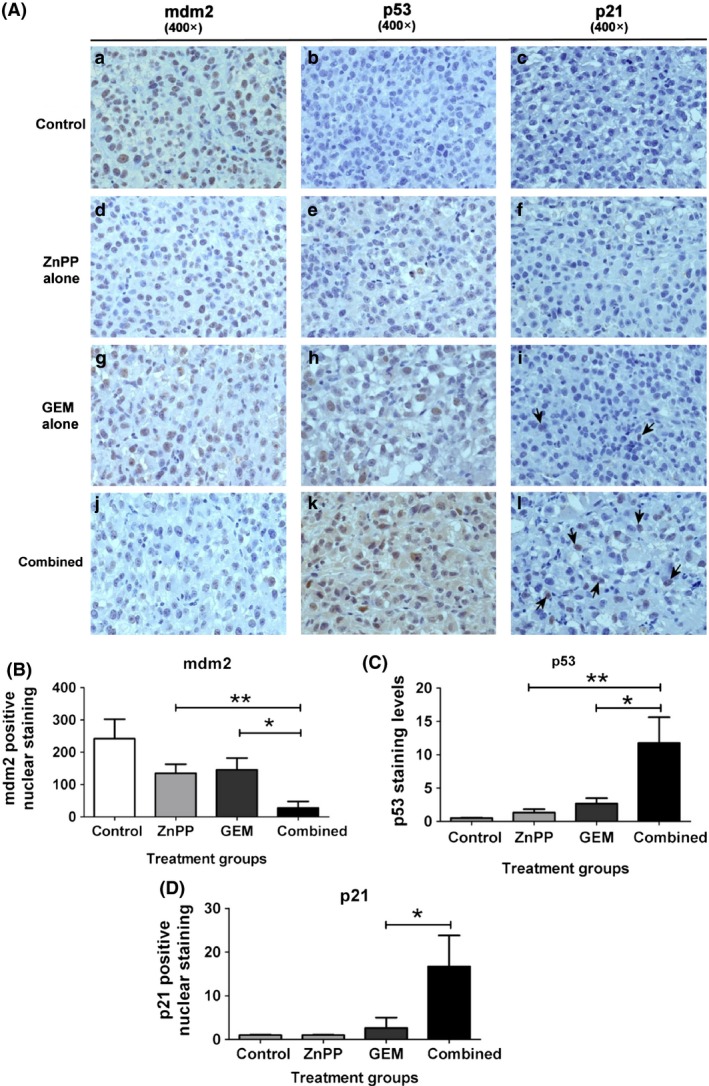
Immunohistochemistry of mdm2, p53 and p21 staining in tumour tissues. (A) Tumour tissues from mice treated with the drug regimens analysed by IHC staining for mdm2, p53 and p21. Panels a–c: control; d–f: ZnPP alone; g–i: GEM alone; j–l: combination of GEM and ZnPP. Panels a, d, g, j for mdm2 staining; b, e, h, k for p53 staining; c, f, i, l for p21 staining (original magnification 400×). (B) Numbers of mdm2‐positive nuclear staining, (C) levels of p53 staining, assessed from intensity and area of staining and (D) Numbers of p21‐positive nuclear staining are shown. Each bar represents mean ± SD, each from seven mice *P < 0.05 drug combination versus GEM alone, and **P < 0.05 drug combination versus ZnPP alone.
Discussion
HO‐1 confers a powerful cytoprotective effect against various insults in normal tissues 26. Several lines of evidence have previously demonstrated that HO‐1 plays an important role in tumour growth, anti‐apoptosis, angiogenesis, metastasis and DNA repair responses following chemotherapeutic or radiotherapeutic treatment 27. In this study, we showed HO‐1 expression in tumour tissue to be an independent predictor associated with patient prognosis. Suppression of HO‐1 sensitized CCA cells to chemotherapeutic agents, where suppression was associated with induction of p53 and p21, and inhibition of angiogenesis and tumour growth.
In some cancers, HO‐1 expression is elevated and is also correlated with clinicopathologic features 13, 20, 21. We demonstrated here that high HO‐1 expression in tumours of the bile duct was associated with short overall survival time of patients. Apart from HO‐1 expression, histological papillary adenocarcinoma and absence of metastasis are associated with long‐term overall survival, compared to other histological types and presence of metastasis. Our results are consistent with previous reports on CCA patients 10, 28. Although patients treated with 5‐FU‐based chemotherapy appeared to have longer median survival time than patients without treatment, or patients treated with oral chemotherapy, it is not possible to propose conclusions on effectiveness of these treatments from the present study. Accepted study design to analyse efficacy of intervention is a prospective randomized control trial study, with intention‐to‐treat analysis. Moreover, histopathology of the tumour is not associated with HO‐1 expression, suggesting that HO‐1 and histological type of tumours, are independent predictors of CCA prognosis. Although association between HO‐1 expression and presence of metastasis was not statistically significant (P = 0.067), probably due to the rather small sample size, there was a tendency for high expression of HO‐1 to be associated with presence of metastasis. Possible association of HO‐1 expression and metastasis is supported by the finding that HO‐1 knock‐down suppressed growth of CCA cells in transwell and wound healing assays.
Other independent prognostic markers of CCA include NQO110, STAT3 28, CD133/Oct3/4 29 and mucin 6 30. Among these markers, NQO1 and STAT3, have been suggested to be possible targets for therapeutic invention in CCA 7, 9, 28, 31. As HO‐1 plays a critical role in cytoprotection, angiogenesis and tumour growth, suppression of HO‐1 is also a logical target for intervention.
A previous study of ours demonstrated HO‐1 to be strongly induced by chemotherapeutic agents, in both high or low HO‐1‐expressing cells, and inhibition of HO‐1 by siRNA, or pharmacological inhibitor ZnPP, renders CCA cells more sensitive to these agents 16. The present investigation further shows HO‐1 to be critically important for cell growth and migration/invasion, as HO‐1 knock‐down in both high and low HO‐1‐expressing cells, KKU‐100 and M213 cells, resulted in diminished migration in transwell and wound healing assays.
Here, we demonstrated that inhibition of HO‐1 activity by ZnPP significantly potentiated the chemotherapeutic effect of GEM in vivo using the CCA xenograft model. This result is consistent with recent studies using xenografted urothelial tumours, LL/2 lung cancer and pancreatic tumours in mice, where treatment with GEM plus ZnPP resulted in inhibition of tumour growth 17, 32. Previous studies have shown that ZnPP inhibited cancer proliferation by suppressing levels of vascular endothelial growth factor, thereby interrupting neovascularization in tumours 33. In contrast, a further report revealed that xenografted animals with over‐expression of HO‐1 in PC3 cells were found to show neovascularization and microvessel density in tumour masses 18. This suggests an intricate role for HO‐1 in modulation of tumour angiogenesis. Our in vivo mouse data demonstrate inhibition of HO‐1 to be effective chemosensitization in cancer chemotherapy. ZnPP or GEM alone suppressed tumour growth and caused reduction in Ki‐67 (a cell cyle regulatory protein) and CD31 (PECAM‐1), a marker of microvascular density. Combination of HO‐1 inhibitor and GEM strongly reduced Ki‐67 and CD31 expression compared to monotherapy. This suggests enhanced tumour suppression may cause inhibition of cell proliferation and neovascularization.
The mechanism of chemosensitization to CCA growth suppression by combination of ZnPP and GEM, was probably associated with the marked up‐regulation of p53 and p21 expression in CCA tumour tissues, whereas monotherapy of ZnPP or GEM weakly affected expression levels. p21 is a p53‐dependent downstream transcriptional control, and a potent cyclin‐dependent kinase inhibitor, that inhibits cell cycle progression and ptoliferation 34. It is notable that GEM or ZnPP alone only slowed tumour growth, and that neither treatment significantly induced p53 or p21 levels. These present results are consistent with our previous in vitro studies, which found increased p21 and cytochrome c expression after treatment with a drug combination compared to single drug therapy 16. p53 can also cause cell death via Bcl‐2 proteins, which can induce the intrinsic mitochondrial apoptotic pathway 35. mdm2 protein is a negative regulator of p53 by catalysing its ubiquitination with subsequent proteasomal degradation retaining low p53 levels. p53 is stabilized and activated during stress by a complex network, including its phosphorylation and interaction via mdm2‐dependent and mdm2‐independent degradation pathways. The inverse relationship of mdm2 and p53 suggests increased p53 expression could have been mediated by mdm2‐dependence 36. Our previous studies showed that combination of GEM and ZnPP strongly induced massive formation of reactive oxygen species, associated with mitochondrial dysfunction and cell death, in CCA cells 16.
It should be noted that animals in the ZnPP group or combination group did not show any overt signs of toxicity ‐ body weight loss, generally a sensitive index of toxicity, was unchanged in all animals. Dosage regimen could be optimized and possibly increased to maximize anti‐tumour activity and completely eradicate tumours. As drug resistance is the main cause of failure in cancer chemotherapy, the strongly sensitizing effect of HO‐1 inhibition to chemotherapeutic agents is relevant in the clinical setting.
In conclusion, expression of HO‐1 in our tumour tissues was associated with prognosis of CCA patients, with higher HO‐1 expression associated with poor overall survival time. Inhibition of HO‐1 resulted in reduced proliferation and suppression of cytoprotection when given in combination with an anti‐cancer agent (GEM). Animal experiments showed strong tumour suppression effect of using a combination of ZnPP and GEM. This study may provide a novel treatment strategy for CCA by combining HO‐1 inhibitor with chemotherapy.
Competing interest
The authors declare that they have no competing interests.
Authors' contributions
VK, BTT, CKO, UK designed the study; SK, WCO, EYS performed animal experiments; SK, AnP, VK, LS analysed IHC study; NK, AnP provided surgical samples and data; SK, CKO, VK, AP, LS, UK analysed the data and interpreted the results; and VK, SK, CKO wrote the manuscript.
Acknowledgements
This study was supported by National Medical Research Council of Singapore, Thailand Research Fund (BRG5480011), the National Research University Project and Office of the Higher Education Commission through SHeP‐GMS of Khon Kaen University, Grant‐in‐aid from Khon Kaen University, and the Royal Golden Jubilee Ph.D. Programme (to S.K.). We thank Dr. Justin Reese of Publication Clinic, Khon Kaen University for English language assistance.
References
- 1. Khan SA, Davidson BR, Goldin RD, Heaton N, Karani J, Pereira SP et al (2012) Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut 61, 1657–1669. [DOI] [PubMed] [Google Scholar]
- 2. Skipworth JR, Olde Damink SW, Imber C, Bridgewater J, Pereira SP, Malago M (2011) Review article: surgical, neo‐adjuvant and adjuvant management strategies in biliary tract cancer. Aliment. Pharmacol. Ther. 34, 1063–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Farges O, Fuks D, Boleslawski E, Le Treut YP, Castaing D, Laurent A et al (2011). Influence of surgical margins on outcome in patients with intrahepatic cholangiocarcinoma: a multicenter study by the AFC‐IHCC‐2009 study group. Ann. Surg. 254, 824–829; discussion 830. [DOI] [PubMed] [Google Scholar]
- 4. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A et al (2010) Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281. [DOI] [PubMed] [Google Scholar]
- 5. Butthongkomvong K, Sirachainan E, Jhankumpha S, Kumdang S, Sukhontharot OU (2013) Treatment outcome of palliative chemotherapy in inoperable cholangiocarcinoma in Thailand. Asian Pac. J. Cancer Prev. 14, 3565–3568. [DOI] [PubMed] [Google Scholar]
- 6. Patel T (2011) Cholangiocarcinoma – controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 8, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeekpudsa P, Kukongviriyapan V, Senggunprai L, Sripa B, Prawan A (2014) Suppression of NAD(P)H‐quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J. Exp. Clin. Cancer Res. 33, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samatiwat P, Prawan A, Senggunprai L, Kukongviriyapan V (2015) Repression of Nrf2 enhances antitumor effect of 5‐fluorouracil and gemcitabine on cholangiocarcinoma cells. Naunyn Schmiedebergs Arch. Pharmacol. 388, 601–612. [DOI] [PubMed] [Google Scholar]
- 9. Buranrat B, Prawan A, Kukongviriyapan U, Kongpetch S, Kukongviriyapan V (2010) Dicoumarol enhances gemcitabine‐induced cytotoxicity in high NQO1‐expressing cholangiocarcinoma cells. World J. Gastroenterol. 16, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buranrat B, Chau‐in S, Prawan A, Puapairoj A, Zeekpudsa P, Kukongviriyapan V (2012) NQO1 expression correlates with cholangiocarcinoma prognosis. Asian Pac. J. Cancer Prev. 13(Suppl), 131–136. [PubMed] [Google Scholar]
- 11. Prawan A, Kundu JK, Surh YJ (2005) Molecular basis of heme oxygenase‐1 induction: implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 7, 1688–1703. [DOI] [PubMed] [Google Scholar]
- 12. Maines MD, Abrahamsson PA (1996) Expression of heme oxygenase‐1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology 47, 727–733. [DOI] [PubMed] [Google Scholar]
- 13. Miyake M, Fujimoto K, Anai S, Ohnishi S, Nakai Y, Inoue T et al (2010) Clinical significance of heme oxygenase‐1 expression in non‐muscle‐invasive bladder cancer. Urol. Int. 85, 355–363. [DOI] [PubMed] [Google Scholar]
- 14. Yin H, Fang J, Liao L, Maeda H, Su Q (2014) Upregulation of heme oxygenase‐1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 14, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim DH, Kim JH, Kim EH, Na HK, Cha YN, Chung JH et al (2009) 15‐Deoxy‐Delta 12,14‐prostaglandin J2 upregulates the expression of heme oxygenase‐1 and subsequently matrix metalloproteinase‐1 in human breast cancer cells: possible roles of iron and ROS. Carcinogenesis 30, 645–654. [DOI] [PubMed] [Google Scholar]
- 16. Kongpetch S, Kukongviriyapan V, Prawan A, Senggunprai L, Kukongviriyapan U, Buranrat B (2012) Crucial role of heme oxygenase‐1 on the sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. PLoS ONE 7, e34994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nuhn P, Kunzli BM, Hennig R, Mitkus T, Ramanauskas T, Nobiling R et al (2009) Heme oxygenase‐1 and its metabolites affect pancreatic tumor growth in vivo. Mol. Cancer. 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferrando M, Gueron G, Elguero B, Giudice J, Salles A, Leskow FC et al (2011) Heme oxygenase 1 (HO‐1) challenges the angiogenic switch in prostate cancer. Angiogenesis 14, 467–479. [DOI] [PubMed] [Google Scholar]
- 19. Fang J, Sawa T, Akaike T, Greish K, Maeda H (2004) Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int. J. Cancer 109, 1–8. [DOI] [PubMed] [Google Scholar]
- 20. Wang J, Zhang M, Zhang L, Cai H, Zhou S, Zhang J et al (2010) Correlation of Nrf2, HO‐1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J. Surg. Res. 164, e99–e105. [DOI] [PubMed] [Google Scholar]
- 21. Yokoyama S, Mita S, Okabe A, Abe M, Ogawa M (2001) Prediction of radiosensitivity in human esophageal squamous cell carcinomas with heme oxygenase‐1: a clinicopathological and immunohistochemical study. Oncol. Rep. 8, 355–358. [DOI] [PubMed] [Google Scholar]
- 22. Sripa B, Leungwattanawanit S, Nitta T, Wongkham C, Bhudhisawasdi V, Puapairoj A et al (2005) Establishment and characterization of an opisthorchiasis‐associated cholangiocarcinoma cell line (KKU‐100). World J. Gastroenterol. 11, 3392–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yonglitthipagon P, Pairojkul C, Chamgramol Y, Loukas A, Mulvenna J, Bethony J et al (2012) Prognostic significance of peroxiredoxin 1 and ezrin‐radixin‐moesin‐binding phosphoprotein 50 in cholangiocarcinoma. Hum. Pathol. 43, 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tusskorn O, Prawan A, Senggunprai L, Kukongviriyapan U, Kukongviriyapan V (2013) Phenethyl isothiocyanate induces apoptosis of cholangiocarcinoma cells through interruption of glutathione and mitochondrial pathway. Naunyn Schmiedebergs Arch. Pharmacol. 386, 1009–1016. [DOI] [PubMed] [Google Scholar]
- 25. Buranrat B, Prawan A, Sripa B, Kukongviriyapan V (2007) Inflammatory cytokines suppress arylamine N‐acetyltransferase 1 in cholangiocarcinoma cells. World J. Gastroenterol. 16, 6219–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase‐1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354. [DOI] [PubMed] [Google Scholar]
- 27. Jozkowicz A, Was H, Dulak J (2007) Heme oxygenase‐1 in tumors: is it a false friend? Antioxid. Redox Signal. 9, 2099–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dokduang H, Techasen A, Namwat N, Khuntikeo N, Pairojkul C, Murakami Y et al (2014) STATs profiling reveals predominantly‐activated STAT3 in cholangiocarcinoma genesis and progression. J. Hepatobiliary Pancreat. Sci. 21, 767–776. [DOI] [PubMed] [Google Scholar]
- 29. Thanan R, Pairojkul C, Pinlaor S, Khuntikeo N, Wongkham C, Sripa B et al (2013) Inflammation‐related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free Radic. Biol. Med. 65, 1464–1472. [DOI] [PubMed] [Google Scholar]
- 30. Thuwajit P, Chawengrattanachot W, Thuwajit C, Sripa B, Paupairoj A, Chau‐In S (2008) Enhanced expression of mucin 6 glycoprotein in cholangiocarcinoma tissue from patients in Thailand as a prognostic marker for survival. J. Gastroenterol. Hepatol. 23, 771–778. [DOI] [PubMed] [Google Scholar]
- 31. Senggunprai L, Kukongviriyapan V, Prawan A, Kukongviriyapan U (2014) Quercetin and EGCG exhibit chemopreventive effects in cholangiocarcinoma cells via suppression of JAK/STAT signaling pathway. Phytother. Res. 28, 841–848. [DOI] [PubMed] [Google Scholar]
- 32. Miyake M, Fujimoto K, Anai S, Ohnishi S, Nakai Y, Inoue T et al (2010) Inhibition of heme oxygenase‐1 enhances the cytotoxic effect of gemcitabine in urothelial cancer cells. Anticancer Res. 30, 2145–2152. [PubMed] [Google Scholar]
- 33. Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H (2007) Inhibition of heme oxygenase‐1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 120, 500–505. [DOI] [PubMed] [Google Scholar]
- 34. Li CH, Tzeng SL, Cheng YW, Kang JJ (2005) Chloramphenicol‐induced mitochondrial stress increases p21 expression and prevents cell apoptosis through a p21‐dependent pathway. J. Biol. Chem. 280, 26193–26199. [DOI] [PubMed] [Google Scholar]
- 35. Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 10, 481–494. [DOI] [PubMed] [Google Scholar]
- 36. Kruse JP, Gu W (2009) Modes of p53 regulation. Cell 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]