

Abstract
Objective: Alterations in plasma lipid profile and in intracellular cholesterol homoeostasis have been described in various malignancies; however, significance of these alterations, if any, in cancer biology is not clear. The aim of the present study was to investigate a possible correlation between alterations in cholesterol metabolism and expansion of leukaemia cell numbers.
Materials and methods: Lipid profiles in plasma and in primary leukaemia cells isolated from patients with acute or chronic lymphocytic leukaemia (ALL and CLL) were studied.
Results and conclusions: Decreased levels of HDL‐C were observed in plasma of leukaemic patients, levels of total cholesterol, LDL‐C, triglycerides and phospholipids were unchanged or only slightly increased. As compared to normal lymphocytes, freshly isolated leukaemic cells showed increased levels of cholesterol esters and reduction in free cholesterol. Growth stimulation of ALL and CLL cells with phytohemagglutinin led to further increase in levels of cholesterol esters. Conversely, treatment with an inhibitor of cell proliferation such as the mTOR inhibitor, RAD, caused decline in population growth rate of leukaemia cells, which was preceded by sharp reduction in rate of cholesterol esterification. On the other hand, exposure of leukaemic cells to two inhibitors of cholesterol esterification, progesterone and SaH 58‐035, caused 60% reduction in their proliferation rate. In addition to demonstrating tight correlation between cell number expansion and cholesterol esterification in leukaemic cells, these results suggest that pathways that control cholesterol esterification might represent a promising targets for novel anticancer strategies.
Introduction
Cholesterol content in tumour tissues has been the topic of a range of investigations since the beginning of 1900s. Starting from 1916, on the basis of an extensive series of experiments indicating that cholesterol is greatly increased in neoplastic tissue, Roffo concluded that cholesterol must play a predominant part in formation of tumours (1, 2, 3). In 1932, Yasuda and Bloor (4) reported that highly malignant tumours contained a much higher percentage of neutral lipids, mainly phospholipids and cholesterol esters (CEs) compared to less malignant ones. These authors emphasized that increase in CEs was extremely interesting and concluded with the question: ‘is the increase in CEs a predominant factor and a cause for the development of the tumor?’ Many years have passed since they posed this question, but the answer is still missing. It is now widely accepted that cholesterol levels in both plasma and tumour tissues are altered in cancer patients (5); therefore, it is realistic to think that pathways that regulate cell population growth and intracellular cholesterol metabolism might be intertwined and hence that mechanisms that modulate cholesterol esterification may be involved in cell number regulation. It has currently become clear that cholesterol‐enriched membrane microdomains, generally referred to as lipid rafts (which exist within the lipid bilayer of all mammalian cells), play an important role in signalling from the cell surface to various subcellular compartment (6, 7, 8). Cholesterol levels in these rafts affect both abundance and function of raft‐resident proteins including Src family kinases, G proteins, growth factor receptors (such as EGFR), mitogen‐activated protein kinase (MAPK) and protein kinase C, most of which control proliferation of leukaemic cells (9, 10). However, molecular bases by which cholesterol levels modulate cell signalling are still largely unknown. It has been proposed that progressive increases in membrane cholesterol contribute to expansion of rafts, which may potentiate oncogenic pathways of cell signalling (11, 12) and therefore that therapies that are able to decrease cell membrane cholesterol content would be a possible cancer chemotherapy (13, 14). In contrast, several independent investigations have demonstrated that depletion of membrane cholesterol activates EGFR and that it also stimulates MAPK pathways including ERK, p38, JNK and Src (15, 16, 17). Among the mechanism(s) evoked to explain why cholesterol depletion increases EGFR activation, the hypothesis that cholesterol depletion causes removal of EGFR from lipid rafts appears to be the most suggestive (18, 19). It has been proposed that EGFR migration out of rafts allows changes in structural conformation of the receptor, thereby promoting its binding to EGF and phosphorylation (20, 21, 22, 23). This is consistent with the finding that kinase activity of EGFR is suppressed when it is associated with lipid rafts (24, 25). The most common form of cholesterol within cell membranes is free cholesterol (FC); CEs are not readily associated with the plasma membrane (5). Numerous studies have indicated that in tumour cells, FC, whether arising from neosynthesis or from uptake, is preferentially channelled into its storage form, CE via acyl CoA cholesterol acyltransferase (ACAT), rather than being transported to the plasma membrane (26, 27, 28). Impairment of this intracellular transport mechanism may have, as consequence, abnormally low levels of high‐density lipoprotein cholesterol (HDL‐C). Consistent with these observations, reduced plasma HDL‐C levels are commonly observed in cancer patients (29, 30, 31, 32, 33, 34). Thus, intracellular accumulation of CEs observed in tumour cells, rather than being a mere consequence of changed metabolic needs, could indirectly contribute to potentiate mitogenic signalling pathways by limiting amounts of FC that can be transported to raft membranes. To build up evidence in support of this hypothesis, in the present study we have investigated lipoprotein profiles in plasma of patients newly diagnosed with acute and chronic lymphocytic leukaemias (ALL, CLL), and intracellular cholesterol metabolism in primary leukaemic cells from the same patients. We also studied effects of inhibitors of cholesterol esterification on rate of proliferation and size of intracellular pools of FC and CEs in the same cells. We chose to use these haematological neoplasms as they represent a readily accessible ‘ex vivo’ model system to investigate parallel alterations of cholesterol metabolism in plasma and tumour cells, in the same patient.
Materials and methods
Drugs and reagents
Acyl amide ACAT inhibitor SaH 58‐035 (SaH) and 40‐O‐(2‐hydroxyethyl) rapamycin (RAD) were kindly provided by Novartis Pharma AG, Basel, Switzerland. Silica gel 60 thin‐layer chromatography plates were purchased from Merck (Darmstadt, Germany). Unless otherwise stated, all other drugs and reagents were purchased from Sigma Chemical (St Louis, MO, USA).
Patient selection
Intracellular lipid content and plasma lipid profiles were initially evaluated in eighteen patients with (CLL) (aged 45–65 years) and twelve patients with acute lymphocytic leukaemia (ALL) (aged 40–60 years) recruited at diagnosis in local hospitals. Fifteen healthy, age‐matched subjects were also recruited as controls. None of the subjects was dieting and there was no significant difference in BMI or waist circumference between the groups. Ten patients (seven with CLL and three with ALL) were randomly chosen on whom to perform kinetic and molecular analyses. Informed written consent was obtained from all patients and healthy controls before initiating the study, according to the policies of the hospital’s Institutional Review Boards.
Cell types and culture conditions
Normal lymphocytes (NL) and leukaemic cells (LC) were obtained by centrifuging blood samples at 600 g for 15 min. After centrifugation, plasma was removed, transferred to centrifuge tubes and utilized for plasma lipid profile determination. The buffy coat was collected, and LC and NL separated by Ficoll‐Hypaque density gradient. Cells were then resuspended (1 × 106 cells/ml) in RPMI‐1640 supplemented with 10% FCS and incubated overnight at 37 °C. These freshly isolated (ex vivo) cells were utilized for the experiments. Where indicated, non‐adherent cells were seeded on 24‐well plates at 2.0 × 105/ml and incubated in RPMI‐1640‐10% FCS supplemented with PHA (10 μg/ml) for 48 h. Trypan blue exclusion test was performed to assess cell viability.
For inhibition experiments, 2.0 × 105/ml non‐adherent cells were incubated at 37 °C in RPMI‐1640 supplemented with 10% FCS and PHA (10 μg/ml) in the presence and absence of SaH, progesterone (PG) or RAD. Preliminary experiments were carried out to determine drug dosages exerting minimal cell toxicity effects (PG, 10 μm, SaH, 4 μm and RAD, 20 nm, respectively).
Lipid testing
Total cholesterol (TC), triglyceride (TG) and phospholipid (PL) levels were determined enzymatically (Boehringer Mannheim Diagnostics, Indianapolis, IN, USA). High‐density lipoprotein cholesterol (HDL‐C) levels were determined after precipitation of apolipoprotein B (Apo‐B)‐containing particles, by magnesium chloride and dextran sulphate (35).
Intracellular lipid content
For cell lipid content determinations, neutral lipids were extracted from freshly isolated cells with cold acetone and evaporated under nitrogen. Dry lipids were re‐suspended with 50 μl of chloroform, spotted on kiesegel plates and run in solvent system containing n‐heptane/isopropyl ether/formic acid (60:40:2, v/v/v). Plates were stained with iodine vapor and spots corresponding to FC, CEs, TG and PL, identified by comparison with reference standards run simultaneously side‐by‐side. Spots were cut out and lipids were eluted in chloroform. After elution, masses of different lipid subclasses were measured using a standard enzymatic method (Boehringer Mannheim Diagnostics).
Lipid staining
NL and LC were cultured as described above. After indicated incubations, cells were washed three times in PBS and fixed by soaking in 10% formalin. Cells were then treated with isopropyl alcohol (60%), washed, stained in oil red O (ORO) for NL and counterstained with Mayer’s haematoxylin. Stained cells were examined by light microscopy. Cytoplasmic red‐stain intensity indicating neutral lipid accumulation was quantified using Image J software (National Institutes of Health, United States). ORO intensity was expressed as mean pixels ± SD/cell obtained by manually selecting three regions of interest (ROIs).
[3H]‐thymidine incorporation
Proliferating cells were identified after [3H]‐thymidine incorporation. Freshly isolated NL and LC cultured as described above were growth‐stimulated by phytohemagglutinin (PHA) treatment in presence or in absence of SaH or RAD. Cells had been labelled with [3H] thymidine (2.5 μCi/ml) during the last 6 h of culture and harvested at 6 and 48 h. Then they were rinsed twice in ice‐cold PBS, washed with 5% cold TCA and lysed with 1 m NaOH. Aliquots of cell lysate were processed for protein content. Amounts of radioactivity were measured using a Beckman LS‐250 β‐counter (Palo Alto, CA, USA). Any drug toxicity effect was excluded by examination of cells after trypan blue uptake.
Cholesterol esterification
Cholesterol esterification was evaluated by incubating cells for 6 h in medium containing [1‐14C] oleic acid (Dupont, NEN 55 mCi/mmol), bound to bovine serum albumin (BSA). After incubation, cells were washed in PBS. Neutral lipids, extracted in cold acetone, dried and re‐suspended with 50 μl of chloroform, were submitted to characterization by thin layer chromatography as described above. For scintillation counting, spots corresponding to CEs were excised and added directly to counting vials containing 10 ml of liquid scintillation fluid. Radioactivity was determined using a Beckman LS‐250 liquid scintillation counter.
RT‐PCR and Southern blotting
mRNA levels for low density lipoprotein receptor (LDL‐R), hydroxy‐methyl‐glutaryl coenzyme A reductase (HMGCoA‐R), sterol regulatory element‐binding protein‐2 (SREBP‐2), ATP‐binding cassette A (ABCA1), acyl CoA‐cholesterol acyltransferase (ACAT‐1), neutral cholesterol ester hydrolase (nCEH), cyclin D1 (Cyc‐D1) and caveolin‐1 (cav‐1), were evaluated by reverse transcription polymerase chain reaction (RT‐PCR) using appropriate primer sets as previously described (36). Total RNA was extracted from approximately 106 cells using TRIZOL reagent (Invitrogen Corporation, Carlsbad, CA, USA). Equal amounts of total RNA (1 μg) were reverse transcribed into cDNA using the random hexamer method and amplified by PCR in presence of specific primers, according to the manufacturer’s instructions (GeneAmp RNA PCR Kit; Perkin‐Elmer Cetus, Norwalk, CT, USA). Amplicons were labelled during PCR, with digoxigenin‐11‐dUTP (Roche Applied Science, Mannheim, Germany), immunodetected with anti‐digoxigenin antibodies conjugated to alkaline phosphatase (Roche Applied Science) and visualized with chemiluminescent substrate CSPD. Intensity of autoradiographic bands was measured after exposure to X‐ray film using Kodak Digital Science Band Scanner Image Analysis System (Kodak, Rochester, NY, USA). Specific bands were detected and analysed by NIH Image 1.63 program (Scion Image, Frederick, MD, USA). Amounts of PCR products for each target mRNA was normalized by using β‐actin as housekeeping gene.
Western blotting
Harvested NL and LC were extracted with radio immuno‐precipitation assay (RIPA) buffer (R0278, Sigma, 0.05 ml/1 000 000 cells) and protein concentration was assessed using Bicinchoninic Acid Protein determination kit (Sigma). For Cyc.‐D1, Cav.‐1, ACAT‐1 and ABCA‐1 determinations, aliquots (30 μg) of cell extracts were separated by 10% SDS–PAGE and blotted on nitrocellulose membranes (Millipore, Bedford, MA, USA), which were then probed with anti‐ Cyc.‐D1, Cav.‐1, ACAT‐1 and ABCA‐1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After incubation with suitable HRP‐conjugated secondary antibodies, specific bands corresponding to target proteins were detected by the ECL procedure (Amersham, Freiburg, Germany), and analysed by NIH Image 1.63 Analysis Software program (Scion Image). Anti β‐actin antibodies (Cell Signalling, Beverly, MA, USA) were used for detection of housekeeping protein.
Statistical analysis
Data are reported as mean ± standard deviation (SD). Statistical calculations were performed using statistical analysis software Origin 8.0 version (Microcal Inc, Northampton, MA, USA). Data analysis was performed using Student’s t‐test and Pearson test; P < 0.05 was considered statistically significant.
Results
Lipid profile of patients with CLL and ALL
We first determined cell lipid mass and fasting plasma lipid profiles in patients with newly diagnosed lymphocytic leukaemia (18 CLL, ages 45–65 years and 12 ALL ages 40–60 years), and in 15 healthy, age‐matched controls. As shown in Table 1, we found that basal levels of CEs were significantly higher in LC compared with NL. In contrast, FC was significantly lower in LC. No significant differences were found in levels of other cell lipid parameters, such as content of TC, TG and PL. Levels of plasma HDL‐C were significantly lower in leukaemic patients compared to controls. Plasma TC, TG, and PL levels were not significantly different between control subjects and tumour‐bearing hosts, although a trend towards hypocholesterolaemia and hypertriglyceridaemia was observed in leukaemia patients.
Table 1.
Lipid profile in cells and plasma from normal subjects and leukaemic patients
| Normal lymphocytes (n = 15) | Leukaemic cells (n = 30) | t‐test (P‐value) | |
|---|---|---|---|
| TC (CE + FC) (μg/106 cells) | 2.6 ± 0.77 | 3.0 ± 1.37 | 0.3 |
| FC (μg/106 cells) | 2.4 ± 0.74 | 1.8 ± 0.66 | 0.008 |
| FC/TC (%) | 92 | 60 | |
| CE (μg/106 cells) | 0.2 ± 0.04 | 1.2 ± 0.55 | 0.000 |
| CE/TC (%) | 8 | 40 | |
| Triglycerides (μg/106 cells) | 5.9 ± 1.9 | 7.0 ± 3.3 | 0.24 |
| Phospholipids (μg/106 cells) | 4.9 ± 1.5 | 4.2 ± 2.2 | 0.27 |
| Normal plasma (n = 15) | Leukaemic plasma (n = 30) | t‐test | |
|---|---|---|---|
| Total cholesterol (mg/dl) | 172 ± 58 | 150 ± 71 | 0.30 |
| HDL‐cholesterol (mg/dl) | 52 ± 15 | 30 ± 11 | 0.000 |
| Triglycerides (mg/dl) | 88 ± 27 | 110 ± 55 | 0.15 |
| Phospholipids (mg/dl) | 150 ± 54 | 162 ± 82 | 0.61 |
TC, total cholesterol; CE, cholesterol ester; FC, free cholesterol.
Accumulation of neutral lipids in LC was confirmed by directly staining then with ORO, a stain able to provide evidence of presence of neutral lipid, but not FC. As shown in Fig. 1, neutral lipids were readily detected in cytoplasm of unstimulated LC, but remained undetectable in resting NL (Fig. 1, time 0). When cells were growth‐stimulated (48 h) with PHA, cytoplasmic staining became apparent in NL (Fig. 1); however, extent of lipid accumulation in response to PHA was significantly higher in LC than in normal ones (Fig. 1b). Interestingly, ALL cells that grow and divide more quickly than CLL cells accumulate even greater amounts of neutral lipids (Fig. 1b). Accordingly, positive correlation (Pearson’s correlation coefficient r = 0.933, P = 0.006) between growth rate as determined by[3H]‐thymidine incorporation into DNA and cholesterol esterification measured by [14C]‐oleate incorporation into CE was also found (Fig. 2a–c).
Figure 1.
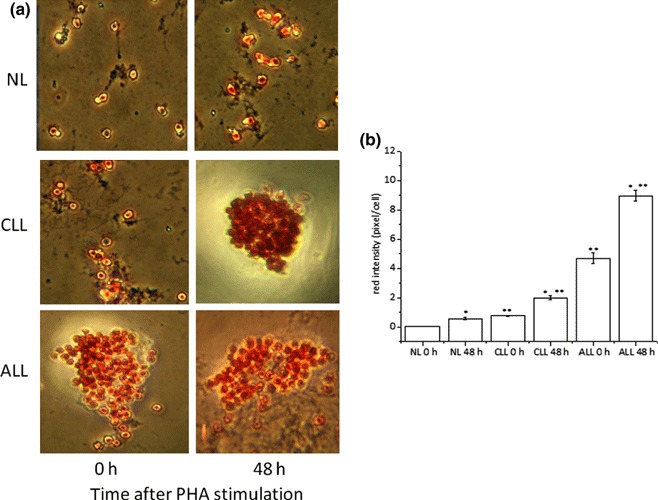
ORO staining of cytoplasmic neutral lipids. NL and LC were incubated for 0–48 h with PHA and then stained with ORO to demonstrate neutral lipids and counterstained with haematoxylin for nuclei. Cells were then examined by light microscopy and two different fields per sample were imaged. Red ORO intensity was measured in these two fields using NIH Image J software. Panel (a) shows representative images of ORO stained NL and LC cultures. Panel (b) shows red intensity expressed as mean pixels ± SD/cell. *P < 0.05 versus corresponding 0 h **P < 0.05 versus corresponding NL.
Figure 2.
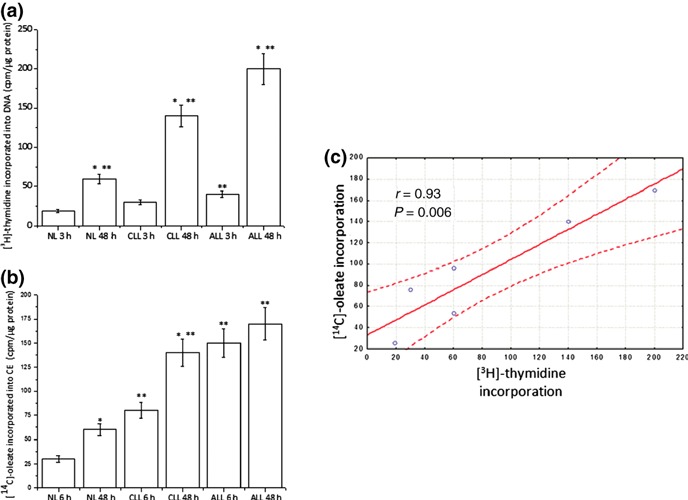
[3H]thymidine and [14C]‐oleate incorporation of leukaemic cells. NL and LC isolated from patients affected by CLL and ALL were incubated at 37 °C in RPMI‐1640‐10% FCS supplemented with PHA (10 μg/ml) for 48 h. Cells were incubated with [3H]thymidine (panel a) and [14C]‐oleate (panel b) 3 and 6 h before harvesting respectively. Data are means ± SD of triplicate determinations from a single normal subject and a single leukaemic patient and are representative of three separate subjects for each group. Panel b: Pearson’s correlation ([3H]thymidine incorporation versus [14C]‐oleate incorporation). *P < 0.05 versus corresponding 3 or 6 h **P < 0.05 versus corresponding NL.
To determine whether changes in CL and CE content in LC may reflect specific alterations in cholesterol homeostasis, in NL and LC we next determined mRNA levels of proteins regulating key steps of cholesterol metabolism, namely cholesterol: (i) uptake, LDL‐R; (ii) neosynthesis, HMGCoA‐R; (iii) homoeostasis, SREBP‐2; (iv) efflux, ABCA‐1; (v) esterification, ACAT‐1 and (vi) CE hydrolysis, nCEH. In addition, we also determined mRNA levels of Cyc‐D1 (responsible for cell cycle transition from G1 to S phase) (37), and of Cav‐1, a negative regulator of Ras‐p42/44 MAP kinase cascade (38) (also involved in transport of cholesterol from endoplasmic reticulum (ER)) and internal stores to lipid rafts (39).
Semiquantitative RT‐PCR analysis showed that Cyc‐D1 mRNA levels was higher and Cav‐1 mRNA was lower in LC at baseline (0 h). This was associated with higher mRNA levels of HMGCoA‐R and ACAT‐1 and with lower mRNA of nCEH, ABCA‐1 and SREBP‐2 in LC compared to NL. Similar differences between NL and LC were observed when cells were growth‐stimulated with PHA for 48 h. No change in baseline mRNA levels of LDL‐R was observed; however, LDL‐R increased in LC after 48 h of PHA stimulation (Fig. 3a,b). All these findings confirm that cholesterol homeostatic mechanisms are generally altered in LC. In particular, accumulation of CE and increased ACAT‐1 mRNA levels associated with decrease in Cav‐1, nCEH and ABCA‐1 mRNAs seems to indicate that during population growth of leukaemic cells, transfer of FC from its site of synthesis, or its uptake into the plasma membrane (PM), is reduced.
Figure 3.
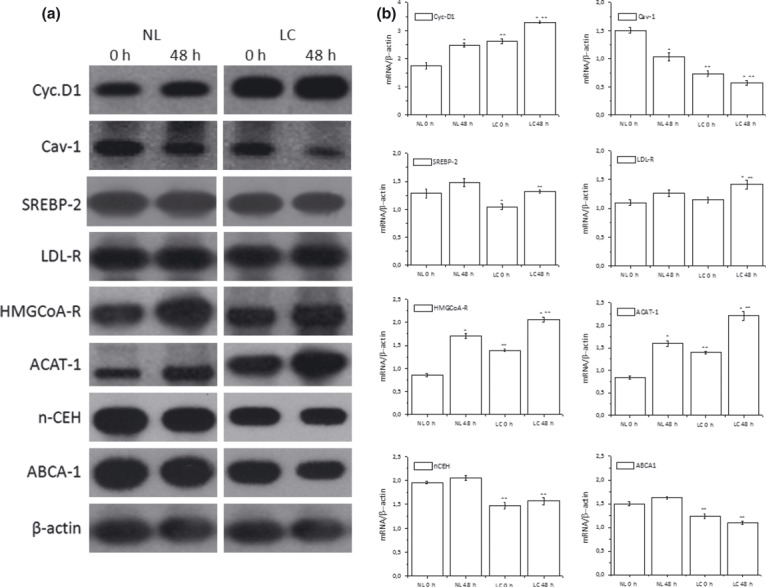
RT‐PCR analyses in NL and LC. Total mRNA was extracted from normal lymphocytes (NL) and leukaemic cells (LC). mRNA levels of indicated genes were determined by RT‐PCR using appropriate primer sets. Specific bands were detected after addition of a chemiluminescent substrate, and analysed using NIH Image 1.63 program (Scion Image). Panel a: blots of target genes, representative of NL and LC isolated from three different subjects for each group. Panel b: densitometric analysis of mRNA levels of NL and LC normalized for endogenous β‐actin mRNA. Histograms represent means ± SD of densitometric scans of triplicate determinations expressed as target gene/β‐actin. *P < 0.05 versus corresponding 0 h **P < 0.05 versus corresponding NL.
Effect of inhibitors of cholesterol esterification on ALL and CLL‐cell expansion
To evaluate further the possible relationship between cholesterol esterification and population growth of LC, we determined incorporation of [3H]‐thymidine and [14C]‐oleate in ALL and CLL stimulated to proliferate by PHA, treated with or without two well‐known inhibitors of cholesterol esterification, SaH and PG. Inhibition of cholesterol esterification, well evident as early as 6 h after exposure to the drugs, in both ALL and CLL (Fig. 4c,d), was followed by reduction in [3H]‐thymidine incorporation (Fig. 4a,b) that became apparent only 48 h after treatment. No obvious cell toxicity was observed, both by visual inspection and by trypan blue exclusion, under experimental conditions that inhibited cell replication. Inhibition of DNA synthesis was independent of extracellular cholesterol concentration as all cells were cultured in 10% foetal calf serum under cholesterol‐clamped conditions.
Figure 4.
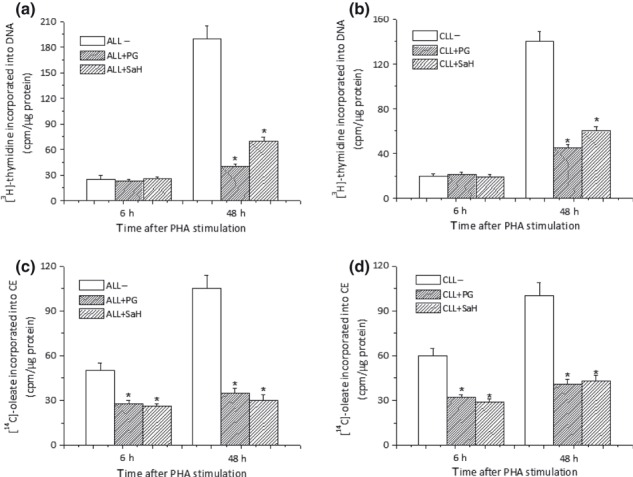
SaH and Pg cause inhibition of cell growth. Freshly isolated ALL and CLL cells were stimulated with PHA in presence or in absence of either PG (10 μm) or SaH (4 μm) and harvested 6 or 48 h later. Cultures were incubated with [3H]‐thymidine (a and b) and [14C]‐oleate (c and d) 6 h before harvest. Histograms represent mean ± SD of triplicate determinations and are representative of 10 different patients (7 CLL and 3 ALL). *P < 0.05 versus untreated cells.
Reduction in ACAT‐1 and Cyc‐D1 mRNA levels associated with an up‐regulation of Cav‐1, nCEH and ABCA‐1 mRNAs was also observed in LC 48 h after SaH and PG treatment (Fig. 5a,b).
Figure 5.
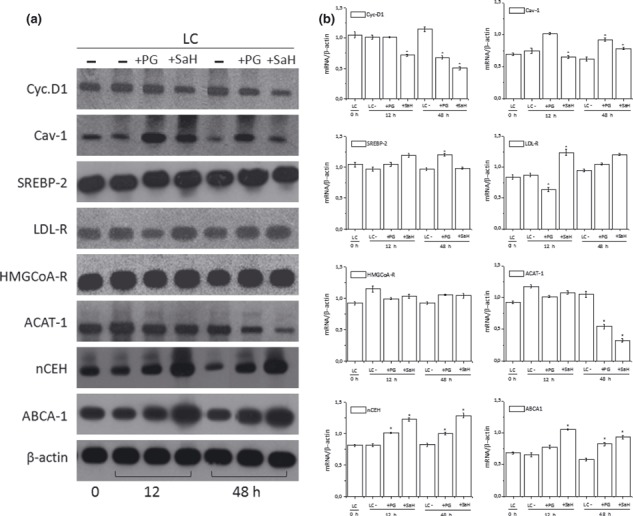
RT‐PCR analyses in LC treated with inhibitors of cholesterol esterification. Total mRNA was extracted from LC and mRNA levels of indicated genes were determined by RT‐PCR using appropriate primer sets. Specific bands were detected after addition of a chemiluminescent substrate, and analysed using NIH Image 1.63 program (Scion Image).). Panel a: blots of target genes, representative of LC treated with or without PG (10 μm) or SaH (4 μm) isolated from three different patients. Panel b: densitometric analysis of mRNA levels of LC normalized for endogenous β‐actin mRNA. Histograms represent mean ± SD of densitometric scans of triplicate determinations expressed as target gene/β‐actin. *P < 0.05 versus corresponding untreated LC.
Effect of RAD on intracellular cholesterol esterification
The above results showing that two different inhibitors of cholesterol esterification suppress proliferation of primary ALL and CLL cells in culture strongly support the hypothesis that cholesterol esterification plays an important role in population growth of LC. This concept was also corroborated by the result that when LC were treated with RAD [an immunosuppressant drug able to induce G1 arrest in cycling B‐CLL cells (40)], inhibition of [3H]‐thymidine incorporation (Fig. 6a) was preceded by significant decrease in rate of [14C]‐oleate incorporation into CE (Fig. 6b).
Figure 6.
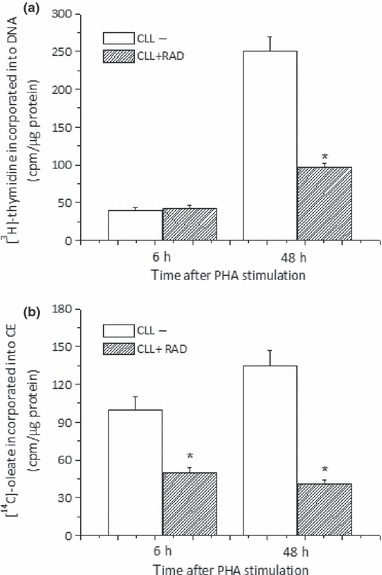
RAD induces inhibition of cholesterol esterification. Freshly isolated CLL cells were stimulated with PHA in presence or in absence of RAD (20 nm) and harvested 6 or 48 h later. [3H]‐thymidine (a) and [14C]‐oleate (b) were added 6 h before harvest. Histograms represent mean ± SD of triplicate determinations and are representative of six different patients *P < 0.05 versus untreated cells.
Inhibitory effects of RAD on cholesterol esterification were also evident by direct detection of cytoplasmic neutral lipids stained with oil‐red O. As shown in Fig. 7, RAD inhibits neutral lipid accumulation in CLL cells to the same extent as the inhibitor of cholesterol esterification PG.
Figure 7.
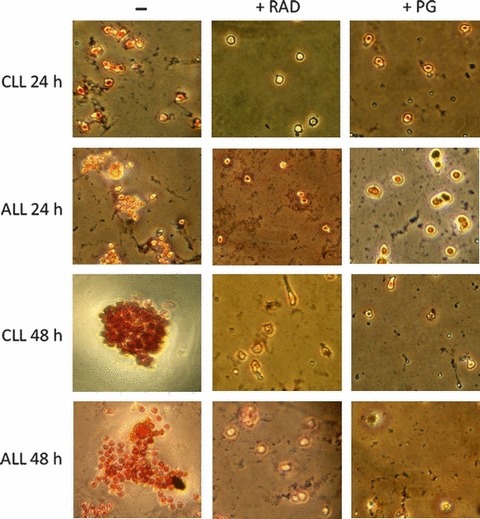
Neutral lipids in RAD and PG‐treated LC. Freshly isolated ALL and CLL cells were stimulated with PHA in presence or in absence of either RAD (20 nm) or PG (10 μm) and harvested 24 or 48 h later. Then, cells were fixed, stained with ORO for neutral lipids and counterstained with haematoxylin for nuclei.
When protein expression of Cyc.‐D1, ACAT‐1, Cav‐1 and ABCA‐1 was examined by western blotting, we found that Cyc‐D1 and ACAT‐1 expression levels were higher in LC compared to NL at 0 and 48h after PHA stimulation; in contrast, Cav‐1 and ABCA‐1 were lower. These data are consistent with the mRNA results. Interestingly in LC, RAD inhibited protein expression of Cyc‐D1 and ACAT‐1, whereas it increased that of Cav‐1 and ABCA‐1 (Fig. 8). These results suggest that RAD, by inhibiting cell proliferation, restores intracellular cholesterol transport mechanisms found altered in LC.
Figure 8.
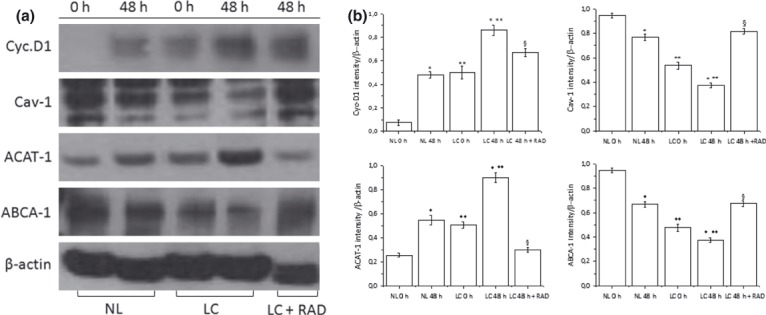
Effect of RAD on protein expression of Cyc.‐D1, Cav.‐1, ACAT‐1 and ABCA‐1 in LC. Freshly isolated LC were stimulated with PHA for 48 h in presence or in absence of RAD (20 nm). β‐actin served as housekeeper gene. (a) Protein levels of Cyc.‐D1, Cav.‐1, ACAT‐1 and ABCA‐1 examined by western blotting (b). Histograms represent means ± SD of the densitometric scans of protein bands from triplicate determinations, representative of six different subjects (three normal and three leukaemic) expressed as intensity of target gene/β‐actin. *P < 0.05 versus corresponding 0 h **P < 0.05 versus corresponding NL. §P < 0.05 versus 48 h untreated LC.
Discussion
It has long been known that cholesterol metabolism is deregulated in most human malignancies (41, 42, 43, 44); however, fewer studies have tried to dissect roles of intracellular pools of FC versus CEs in cancer cells. The current study reveals that in freshly isolated leukaemic cells, multiple anomalies in intracellular cholesterol metabolism, mainly increase in cholesterol esterification potential, abnormal accumulation of CEs and low FC content. An analogous reduction in FC content has been previously observed in the PM of human lymphocytes from leukaemic patients compared to lymphocytes from normal donors, by Inbar and M. Shinitzky (45), FC deficiency in ALL cells being greater than in CLL cells. It is well known that under physiological conditions, FC in the PM is not static, but is steadily delivered to the cell interior and then efficiently returned to the cell surface. It is estimated that cholesterol cycles from PM to the ER and back in about 40 min (46). Intracellular FC can also be esterified by ACAT‐1, and released from the ester storage pool by action of nCEH (5). In most non‐specialized cells, the CE storage pool is quite small (5). In our study, 90% of the cholesterol was free and 8% in esterified form in NL, while in LC, 60% of the cholesterol was free and 40% was esterified. In addition, we show that in LC, increase in cholesterol esterification closely paralleled a two‐fold increase in ACAT‐1 gene expression and that expressions of Cav‐1, nCEH and ABCA‐1 were down‐regulated during proliferation of LC. Thus, it seems that LC possess an inherent defect in PM cholesterol pool cycles, that is, increased ability to entrap cholesterol, in the form of cytoplasmic lipid droplets (LDs). This defect, by reducing transport of cholesterol to the PM, potentially decreases its efflux from the cells. Accordingly, abnormally low HDL‐C levels were found in plasma of ALL and CLL patients. These results highlight the hypothesis that increase in levels of CEs in LC and decrease of HDL‐C in plasma could represent a common and easily detectable biomarker for these types of disease.
Here, ALL and CLL cells responded to PHA mitogenic stimulus by further increasing rate of cholesterol esterification and of accumulation of cytoplasmic neutral lipids; increase in CE synthesis preceded onset of DNA synthesis. Treatment with two different agents, SaH and PG, which are able to inhibit cholesterol esterification by distinct mechanisms (the first by inhibiting ACAT activity, the second one possibly by blocking transport of FC from the PM to the ER), caused growth arrest of LC. On the other hand, inhibition of cell proliferation by RAD, a known antiproliferative agent (40), was associated with very early reduction in cholesterol esterification, which precedes inhibition of LC‐DNA synthesis by several hours. Treatment of LC with these three drugs also resulted in down‐regulation of Cyc‐D1, and in increase in expressions of Cav‐1, nCEH and ABCA‐1 to a value characteristic of NL. In light of these results, it may be argued that in LC, FC reduction, probably secondary to decreased expression of Cav‐1, nCEH and ABCA‐1, may activate signalling pathways involved in cell division, thereby contributing to increased proliferative rate of the tumour cells (15, 16, 17).
These conclusions fit well with recent observations that liver X receptor (LXRs) activation in freshly isolated CLL cells, by synthetic agonists, inhibited cytokine‐induced cell proliferation and cell cycle progression without affecting cell viability (47). LXRs, through their ability to regulate expression of ABCA‐1, have been involved in the process of reverse cholesterol transport (48). LXR activation results in increased levels of plasma HDL and increase in cholesterol excretion. ABCA‐1 expression is virtually absent in untreated CLL cells suggesting that endogenous LXR agonists are largely absent in these cells (47). Therefore, lack of LXR activation in leukaemic cells, by reducing cholesterol efflux, may contribute to cell population growth and cell cycle progression.
Although our findings do not represent final proof of a direct causative involvement of CEs in tumour growth, they add support to the hypothesis that intracellular pathways that regulate cholesterol esterification may contribute to abnormal growth regulation of leukaemic cells. It is well documented that levels of FC in cell membranes regulate a number of critical processes: from de novo cholesterol biosynthesis to signal transduction. FC is particularly abundant in specialized membrane domains called rafts, which house many proteins involved in the transduction of extracellular mitogenic signals (6, 7, 8). Several studies have demonstrated that FC levels in rafts have a profound effect on function of raft‐associated proteins, and that depletion of membrane cholesterol is capable of activating the EGF receptor or the MAP kinase cascade in absence of any extracellular mitogen (15, 16, 17). Cholesterol trafficking across different cell compartments plays a fundamental role in maintenance of FC levels both in rafts and other membrane compartments and, as such, it is a highly regulated process (8). As opposed to the FC pool, intracellular pool of CEs is usually viewed as a mere storage form of cell cholesterol and the process of intracellular cholesterol esterification a simple means to avoid toxic effects of excess FC (5).
In light of our previous studies (5, 29, 32, 33) and findings presented in this paper, a new role for cholesterol esterification in cell proliferation would be suggested. It is tempting to hypothesize that cell machinery that controls cholesterol esterification might be an integral part of cellular apparatus that regulates FC abundance in critical membrane domains such as rafts. In this view, cholesterol esterification could represent a key step along pathways that modulate membrane levels of FC in response to extracellular stimuli, and could directly take part in mechanisms of population growth regulation. In summary here, we show for the first time that ALL and CLL cells display alterations in intracellular cholesterol homoeostasis consisting of increased rate of cholesterol esterification and accumulation of CEs. Although further research is needed to better define exact mechanisms and significance of these alterations, our results pave the way for development of new pharmacological approaches for treatment of this important group of neoplasms.
Acknowledgements
This study was supported by grants from Ministero dell’Università e Ricerca Scientifica (ex 60%). We appreciate Drs Mario Pani and Rosa Manconi for their help in recruitment of controls.
References
- 1. Roffo AH (1916) Montevideo: An. Facult. Med.. [Google Scholar]
- 2. Roffo AH (1933) Heliotropism of cholesterol in relation to skin cancer. Am. J. Cancer. 17, 42–57. [Google Scholar]
- 3. Roffo AH, Pilar F (1930) Bol. Inst. Med.Eexp. Estud. y Trat. Cancer 7, 22 [Google Scholar]
- 4. Yasuda M, Bloor WR (1932) Lipid content of tumors. J. Clin. Invest. 11, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pani A, Dessi S (2003) Cell growth and cholesterol ester. Springer Publication 1–11, 1–150. [Google Scholar]
- 6. Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387, 569–572. [DOI] [PubMed] [Google Scholar]
- 7. Kurzchalia TV, Parton RG (1999) Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11, 424–431. [DOI] [PubMed] [Google Scholar]
- 8. Fielding CJ, Fielding PE (2000) Cholesterol and caveolae: structural and functional relationships. Biochim. Biophys. Acta 1529, 210–222. [DOI] [PubMed] [Google Scholar]
- 9. Meinhardt G, Wendtner CM, Hallek M (1999) Molecular pathogenesis of chronic lymphocytic leukemia: factors and signaling pathways regulating cell growth and survival. J. Mol. Med. 77, 282–293. [DOI] [PubMed] [Google Scholar]
- 10. Stam RW, Schneider P, Hagelstein JA, van der Linden MH, Stumpel DJ, de Menezes RX et al. (2010) Gene expression profiling‐based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 115, 2835–2844. [DOI] [PubMed] [Google Scholar]
- 11. Freeman MR, Solomon KR (2004) Cholesterol and prostate cancer. J. Cell. Biochem. 91, 54–69. [DOI] [PubMed] [Google Scholar]
- 12. Li YC, Park MJ, Ye SK, Kim CW, Kim YN (2006) Elevated levels of cholesterol‐rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol depleting agents. Am. J. Pathol. 168, 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan KK, Oza AM, Siu LL (2003) The statins as anticancer agents. Clin. Cancer Res. 9, 10–19. [PubMed] [Google Scholar]
- 14. Seeger H, Wallwiener D, Mueck AO (2003) Statins can inhibit proliferation of human breast cancer cells in vitro. Exp. Clin. Endocrinol. Diabetes 111, 47–48. [DOI] [PubMed] [Google Scholar]
- 15. Furuchi T, Anderson RGW (1998) Cholesterol depletion of caveolae causes hyperactivation of extracellular signal‐related kinase (ERK). J. Biol. Chem. 273, 21099–21104. [DOI] [PubMed] [Google Scholar]
- 16. Chen X, Resh MD (2002) Cholesterol depletion from the plasma membrane triggers ligand‐independent activation of the epidermal growth factor receptor. J. Biol. Chem. 277, 49631–49637. [DOI] [PubMed] [Google Scholar]
- 17. Westover EJ, Covey DF, Brockman HL, Brown RE, Pike LJ (2003) Cholesterol depletion results in site‐specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects: studies with cholesterol enantiomers. J. Biol. Chem. 278, 51125–51133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ness GC, Chambers CM (2000) Feedback and hormonal regulation of hepatic 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase: the concept of cholesterol buffering capacity. Proc. Soc. Exp. Biol. Med. 224, 8–11. [DOI] [PubMed] [Google Scholar]
- 19. Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ (1999) c‐Src‐mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274, 8335–8343. [DOI] [PubMed] [Google Scholar]
- 20. Mineo C, Gill GN, Anderson RGW (1999) Regulated migration of epidermal growth factor receptor from caveolae. J. Biol. Chem. 274, 30636–30643. [DOI] [PubMed] [Google Scholar]
- 21. Ringerike T, Glystad FD, Levy FO, Madshus IH, Stang E (2002) Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J. Cell Sci. 115, 1331–1340. [DOI] [PubMed] [Google Scholar]
- 22. Warren CM, Landgraf R (2006) Signaling through ERBB receptors: multiple layers of diversity and control. Cell. Signal. 18, 923–933. [DOI] [PubMed] [Google Scholar]
- 23. Grewal T, Enrich C (2009) Annexins – Modulators of EGF receptor signalling and trafficking. Cell. Signal. 21, 847–858. [DOI] [PubMed] [Google Scholar]
- 24. Lambert S, Gniadecki R, Poumay Y (2009) Cholesterol and lipid rafts as regulators of signaling through the EGF receptor in keratinocytes. Open Derm. J. 3, 151–158. [Google Scholar]
- 25. Macdonald‐Obermann JL, Pike LJ (2009) Palmitoylation of the EGF receptor impairs signal transduction and abolishes high‐affinity ligand binding. Biochemistry 48, 2505–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C et al. (1987) Abnormal cholesterol metabolism in renal clear cell carcinoma. J. Lipid Res. 28, 1117–1124. [PubMed] [Google Scholar]
- 27. Pittman RC, Knecht TP, Resenbaum MS, Taylor CA Jr (1987) A non‐endocytotic mechanism for the selective uptake of high density lipoprotein‐associated cholesterol esters. J. Biol. Chem. 262, 2443–2450. [PubMed] [Google Scholar]
- 28. Oram JF, Johnson C, Brown TA (1987) Interaction of high density lipoprotein with its receptor on cultured fibroblasts and macrophages. J. Biol. Chem. 262, 2405–2410. [PubMed] [Google Scholar]
- 29. Dessì S, Batetta B, Pulisci D, Spano O, Cherchi R, Lanfranco G et al. (1992) Altered pattern of lipid metabolism in patients with lung cancer. Oncology 49, 436–441. [DOI] [PubMed] [Google Scholar]
- 30. Umeki S (1993) Decreases in serum cholesterol levels in advanced lung cancer. Respiration 60, 178–181. [DOI] [PubMed] [Google Scholar]
- 31. Siemianowicz K, Gminski J, Stajszczyk M, Wojakowski W, Goss M, Machalski M et al. (2000) Serum HDL cholesterol concentration in patients with squamous cell and small cell lung cancer. Int. J. Mol. Med. 6, 307–311. [DOI] [PubMed] [Google Scholar]
- 32. Dessì S, Batetta B, Pulisci D, Spano O, Anchisi C, Tessitore L et al. (1994) Cholesterol content in tumor tissues is inversely associated with high‐density lipoprotein cholesterol in serum in patients with gastrointestinal cancer. Cancer 73, 253–258. [DOI] [PubMed] [Google Scholar]
- 33. Dessì S, Batetta B, Spano O, Sanna F, Tonello M, Giacchino M et al. (1995) Clinical remission is associated with restoration of normal high‐density lipoprotein cholesterol levels in children with malignancies. Clin. Sci. 89, 505–510. [DOI] [PubMed] [Google Scholar]
- 34. Anchisi C, Batetta B, Sanna F, Fadda AM, Maccioni AM, Dessi S (1995) HDL subfractions as altered in cancer patients. J. Pharm. Biomed. Anal. 13, 65–71. [DOI] [PubMed] [Google Scholar]
- 35. Warnick GR, Benderson J, Albers JJ (1982) Dextran sulfate‐Mg21 precipitation procedure for quantification of high‐density lipoprotein cholesterol. Clin. Chem. 28, 1379–1388. [PubMed] [Google Scholar]
- 36. Pani A, Dessì S, Diaz G, La Colla P, Abete C, Mulas C et al. (2009) Altered cholesterol ester cycle in skin fibroblasts from patients with Alzheimer’s disease. J. Alzheimers Dis. 18, 829–841. [DOI] [PubMed] [Google Scholar]
- 37. Musgrove EA, Lee CS, Buckley MF, Sutherland RL (1994) Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc. Natl. Acad. Sci. USA 91, 8022–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engelman JA, Zhang XL, Razani B, Pestell RG, Lisanti MP (1999) p42/44 MAP kinase‐dependent and ‐independent signaling pathways regulate caveolin‐1 gene expression. Activation of Ras‐MAP kinase and protein kinase a signaling cascades transcriptionally down‐regulates caveolin‐1 promoter activity. J. Biol. Chem. 274, 32333–32341. [DOI] [PubMed] [Google Scholar]
- 39. Uittenbogaard A, Smart E (2000) Palmitoylation of caveolin‐1 is required for cholesterol binding, chaperone complex formation, and rapid transport of cholesterol to caveolae. J. Biol. Chem. 275, 25595–25599. [DOI] [PubMed] [Google Scholar]
- 40. Majewski M, Korecka M, Kossev P, Li S, Goldman J, Moore J et al. (2000) The immunosuppressive macrolide RAD inhibits growth of human Epstein‐Barr virus‐transformed B lymphocytes in vitro and in vivo: a potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc. Natl. Acad. Sci. USA 97, 4285–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen HW, Kandutsch AA, Heininger HJ (1978) The role of cholesterol in malignancy. Prog. Exp. Tumor Res. 22, 275–316. [DOI] [PubMed] [Google Scholar]
- 42. Ho YK, Smith RG, Brown MS, Goldstein JL (1978) Low‐density lipoprotein (LDL) receptor activity in human acute myelogenous leukemia cells. Blood 52, 1099–1114. [PubMed] [Google Scholar]
- 43. Rudling M, Gafvels M, Parini P, Gahrton G, Angelin B (1998) Lipoprotein receptors in acute myelogenous leukemia: failure to detect increased low‐density lipoprotein (LDL) receptor numbers in cell membranes despite increased cellular LDL degradation. Am. J. Pathol. 153, 1923–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harwood HJ, Alvarez IM, Noyes WD, Stacpoole PW (1991) In vivo regulation of human leukocyte 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase: increased enzyme protein concentration and catalytic efficiency in human leukemia and lymphoma. J.Lipid Res. 32, 1237–1252. [PubMed] [Google Scholar]
- 45. Imbar M, Shinitzky M (1974) Cholesterol as a bioregulator in the development and inhibition of leukemia. Proc. Natl. Acad. Sci. USA 71, 4229–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liscum L, Munn NJ (1999) Intracellular cholesterol transport. Biochim. Biophys. Acta 1438, 19–37. [DOI] [PubMed] [Google Scholar]
- 47. Geyeregger R, Shehata M, Zeyda M, Kiefer FW, Stuhlmeier KM, Porpaczy E et al. (2009) Liver X receptors interfere with cytokine‐induced proliferation and cell survival in normal and leukemic lymphocytes. J. Leukoc. Biol. 86, 1039–1048. [DOI] [PubMed] [Google Scholar]
- 48. Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K et al. (2000) Regulation of absorption and ABC1‐mediated efflux of cholesterol by RXR heterodimers. Science 289, 1524–1529. [DOI] [PubMed] [Google Scholar]