

Abstract
Abstract. Objectives/Background: In biological terms, progression means that malignancy increases as genetic mutations accumulate leading to increased proliferation and invasion capacity. By verifying the proliferation capacity, human telomerase reverse transcriptase (hTERT) expression and in vitro invasion, in a group of highly malignant glioblastomas, benign meningiomas and astrocytomas, at the initial stage of progression, we have analysed putative progression in vitro for proliferation and telomerase expression. Materials and Methods: The relative proliferation status (visualized with Ki‐67 antibodies) and presence of hTERT protein was analysed in 27 intracranial tumours (6 astrocytomas, 8 glioblastomas and 13 meningiomas) by immunohistochemistry on paraffin‐embedded biopsy tissue, as well as on primary tumour‐derived cell cultures. A confrontation model was used to analyse invasiveness in vitro. Results: The mean proliferation indices were 22.3 (SD = 18.1) for glioblastomas and 2.1 (SD = 1.9) for low‐grade (LG) astrocytomas. The group of benign meningiomas had a labelling index of 2.2 (SD = 2.7). Mean percentages of staining for hTERT varied between 36.5 (SD = 28.4) for glioblastomas and 10.2 (SD = 8.6) for LG astrocytomas. The group of benign meningiomas had a labelling index of 12.4 (SD = 19.2) for hTERT. A significant difference was seen for Ki‐67 (P < 0.05) and hTERT (P < 0.001) in vivo versus in vitro. No difference was seen between the group of invasive and non‐invasive tumour‐derived cell cultures for the histopathological markers Ki‐67 and hTERT (P > 0.05) in vitro. Conclusions: The elevated expression of hTERT and Ki‐67 in vitro provides a potential prognostic tool for early detection of the progression of brain tumours. As tumour cells require telomerase for continued proliferation, the expression of hTERT may mark immortality, leading to indefinite life span. On the other hand, hTERT expression and cell proliferation are not linked directly to invasion in vitro.
INTRODUCTION
Increase in cell population through uncontrolled cell reproduction is one of the hallmarks of the progression of cancer processes. ‘Tumour progression’ means that the tendency towards malignancy increases as genetic mutations accumulate, leading to increased proliferation and invasion capacity. Expanding cell populations are observed in normal controlled growth, such as embryonic tissues, but uncontrolled cell populations results in tumours (Cavalla & Schiffer 1997).
The proliferation process is linked to telomerase expression; this is a ribonucleoprotein enzyme complex, and is seen in stem cells and in cancer cells. In cancer cells, the presence of telomerase is linked to indefinite lifespan – immortality (Counter et al. 1992; Lustig 1999). One of the major components of telomerase is the human telomerase reverse transcriptase (hTERT), a reverse transcriptase (Kim et al. 1994; Weinrich et al. 1997). In this study, we investigated the proliferation capacity of cells and the presence of hTERT in brain tumours (primary tumour biopsies) and tumour cell cultures derived directly from brain tumour biopsies. The invasiveness of these tumours was analysed in vitro. Three groups of brain tumour were evaluated: on the one hand, a number of astrocytomas at the initial stage of progression and a number of highly malignant glioblastoma multiforme tumours, at the end stage of ‘tumour progression’, and, on the other hand, a group of benign meningiomas considered as non‐progressing tumours. However, amongst the meningiomas, a limited number of examples did progress towards morbidity and mortality (Jaaskelainen et al. 1986; Carroll et al. 1999). All tumour‐derived cells were tested in an invasion model in vitro as a control for the stages of malignancy determined from the in vivo tissue samples (de Ridder & Calliauw 1992; de Ridder 2003).
MATERIALS AND METHODS
Surgical specimens
Biopsies of 14 glial (6 astrocytomas, low grade, and 8 glioblastoma multiforme) and 13 benign meningiomal intracranial tumours were collected from the University Hospital, St. Lucas Hospital and Maria‐Middelares, Ghent, Belgium. The group was comprised of 16 women and 11 men with the patients’ median age at 47.4 (range 8–79 years). Classification and grading of the tumours was based on World Health Organization (WHO) criteria (Kleihues & Cavenee 2000). Immediately after surgery, each tumour fragment was divided for histopathology and to be the bases of primary tumour‐derived cell cultures.
Primary tumour‐derived cell cultures
Explants in vitro from brain tumours give rise to primary monolayers. For this purpose after removal, each half‐biopsy was transferred immediately, aseptically, into a glass Petri dish, containing minimum essential medium (MEM) culture medium (Invitrogen, Merelbeke, Belgium) with Hank's salts, supplemented with 10% foetal calf serum, 0.05% glutamine (Sigma, Bornem, Belgium), 2.5 µg/ml fungizone (Invitrogen, Merelbeke, Belgium) and pen/strep (50 U/ml and 50 µg/ml) (Invitrogen, Merelbeke, Belgium). The fragment was cut into small 1 × 1 × 1 mm cubes and was pipetted into two plastic culture flasks containing the same medium. The slices were allowed to adhere to the bottom of the flasks and viable cells moved out of the biopsies and formed a monolayer of tumour‐derived cells. The cultured cells of one flask were trypsinized and spliced into two parts. One part was explanted on Thermanox (Nunc, Roskilde, Denmark) for immunocytochemical analysis and the other part was placed into a cryotube containing 90% foetal calf serum (FCS) with 10% dimethylsulfoxide (DMSO; Eurolab, Leuven, Belgium) and was stored in liquid nitrogen for later polymerase chain reaction (PCR) analysis. From the second culture flask, flaps of cell clusters were scraped off with a rubber policeman and were transferred into small 5‐ml Erlenmeyer flasks to form spheroids (see succeeding discussions, ‘Invasion model in vitro’).
Immunohistochemical analysis
Preparation of cell cultures
Cell cultures, grown on Thermanox plastic, were fixed with cold acetone (Chem‐Lab, Zedelgem, Belgium) at subconfluence and were stored in the freezer until use.
Immunostaining
The standard streptavidin‐biotin‐peroxidase complex method was performed on primary biopsies and tumour‐derived cell cultures, employing antibodies against hTERT (1 : 50 dilution, clone 44F12; Novocastra, Newcastle, UK) and Ki‐67 (1 : 50 dilution, clone MIB‐1; DAKO, Glostrup, Denmark). The percentage of positivity was defined as the ratio of cells showing appropriate immunoreactivity per 1000 tumour cells. Subsequently, samples were incubated with biotinylated rabbit antimouse antibody,1 : 200 dilution, for 30 min, followed by a third layer incubation with streptavidin‐biotin‐peroxidase complex, 1 : 200 dilution for 30 min. Visualization of the complex was performed by application of diaminobenzidine (DAB), followed by nuclear counterstaining with haematoxylin. A glioblastoma multiforme sample, already known to express hTERT, and with a highly proliferative nature, was used as a positive control, and an antigen‐free buffer was used as a negative control (Fig. 1).
Figure 1.
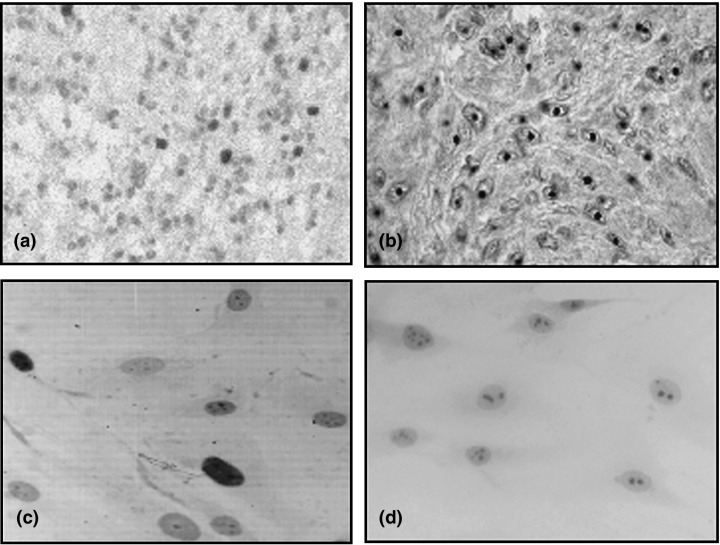
Immunohistochemical staining for Ki‐67 in vivo (a) versus in vitro (c) with total nuclear colouring and for hTERT in vivo (b) versus in vitro (d) with nucleolar colouring of human brain tumours.
Invasion model in vitro
By a multiple step method, putative invasiveness of tumour‐derived cells was evaluated in vitro as described by previously by de Ridder (de Ridder 2003). The succeeding paragraphs summarize.
Aggregates or spheroids of individual tumours were prepared by incubating flaps of tumour‐derived cells into small 5‐ml Erlenmeyer flasks with 3 ml MEM medium with Earl's salts, supplemented with glutamine and 10% FCS. These flasks were rotated for 24 h at 0.8 g under a steady gas flow of 95% air and 5% CO2 to form spheroids of these human tumour cells. Only spheroids with a mean diameter of 0.3–0.4 mm were selected. This was performed by selection under a light microscope. Samples of greater diameter are known to possibly have central necrosis.
At the same time, freshly cut fragments, obtained from 9‐day embryonic chick hearts, were cut into 1 × 1 × 1 mm blocks and incubated on a gyratory shaker for 3 days to obtain pre‐cultured heart spheroids. Their mean diameter was 0.4 mm, the core consisting of heart muscle cells surrounded by layers of fibroblastoid cells.
The next step consisted of confrontation of pre‐cultured heart cell spheroids with the spheroids of tumour‐derived cells. They attached firmly to each other after 2 h contact on a semisolid agar medium, covered by 3 ml MEM with Earl's salts and 10% FCS. Specimens were then transferred to 10 ml Erlenmeyer flasks filled with the same medium as described in the previous discussion. The confrontation specimens were incubated at 0.8 g on the gyratory shaker, for 1, 2, 4 and 7 days. Fixation in Zenker's solution and embedding in paraffin wax, to be cut into 5‐µm sections followed. All sections were stained with haematoxylin eosin and rat anti‐chicken antibody.
The confrontation specimen results were classified into two groups expressing different patterns, a non‐invasive pattern and an invasive pattern (Fig. 2). Into the ‘noninvasive’ category were placed specimens in which tumour‐derived cells survive in confrontation to, but not infiltrating into, heart fragments. Tumour cells were found separated from the heart tissue by a clear‐cut border. Into the ‘invasive’ category were placed specimens in which tumour‐derived cells infiltrate into and progressively replaced chick heart tissue (de Ridder & Calliauw 1992). In some specimens, tumour‐derived cells did not survive confrontation to chick heart and thus were not included.
Figure 2.
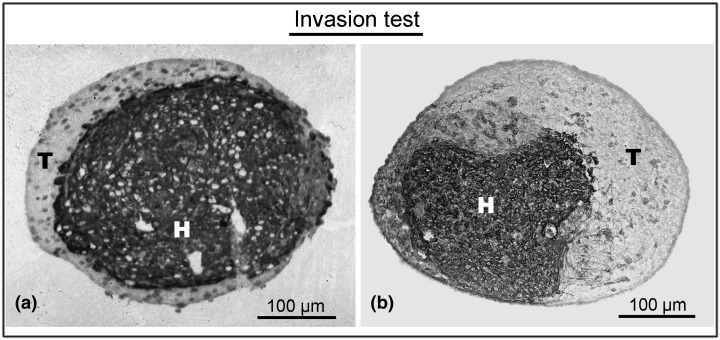
Histological sections: confrontation type. (a) Non‐invasive confrontation of meningioma‐derived cells (T) with host tissue (H) after 4‐day incubation. No invasion of tumour‐derived cells into the host tissue. (b) Invasive confrontation of tumour cells derived from a malignant brain tumour (T) with host tissue (H) after 4‐day incubation. Invasive pattern of tumour derived cells into the host tissue.
Statistical analysis
All statistical analyses were carried out using spss for Windows version 11.0 (SPSS Inc., Chicago, IL, USA). Values were expressed as mean ± SD and as percentages, when appropriate. A Wilcoxon signed rank test was used to analyse the difference between variables in vivo versus in vitro. A Mann–Whitney test was used to analyse associations for variables between the two groups of invasive and non‐invasive tumours. Categorized variables entered into the analysis were Ki‐67 labelling index (LI), hTERT LI and confrontation type. LI was determined as the percentage of positive cells per 1000 cells counted per cell culture specimen. The level of significance was set with the P value at less than 0.05. All reported P values were two‐tailed.
RESULTS
Primary brain tumour biopsies
Results can be found in 1, 2. As examples of cell proliferation capacity in vivo, Ki‐67 and hTERT immunostained cells were counted in sections of paraffin‐embedded tumour biopsies; the proliferation status of the brain tumours was obtained by staining with anti‐Ki‐67 antibodies. A total of 6 low‐grade astrocytomas, 8 glioblastomas and 13 meningiomas were collected and analysed immunohistochemically. Individual percentages are summarized in Table 1. Mean proliferation indices were 2.1 (SD = 1.9) for low‐grade astrocytomas and 22.3 (SD = 18.1) for glioblastomas. The group of benign meningiomas had a labelling index of 2.2 (SD = 2.7).
Table 1.
Histopathological and summarizing table with in vivo versus in vitro percentages for proliferation (Ki‐67 LI), labelling indexes for the subunit of telomerase (hTERT) and invasion in vitro (Y = invasion; N = no invasion)
| Nο | Tumour | WHO classification | Gender | Age | In vivo | In vitro | |||
|---|---|---|---|---|---|---|---|---|---|
| Proliferation | hTERT | Proliferation | hTERT | Invasion | |||||
| 1 | 1280 | Astrocytoma LG | M | 31 | 4.0 | 9.0 | 11.9 | 93.4 | N |
| 2 | 1285 | Astrocytoma LG | M | 9 | 5.0 | 26.6 | 4.9 | 96.6 | Y |
| 3 | 1316 | Astrocytoma LG | F | 8 | 1.3 | 7.5 | 0.5 | 91.3 | N |
| 4 | 1319 | Astrocytoma LG | M | 37 | 1.0 | 3.6 | 7.0 | 57.0 | Y |
| 5 | 1334 | Astrocytoma LG | F | 53 | 0.3 | 2.1 | 16.0 | 52.0 | N |
| 6 | 1356 | Astrocytoma LG | F | 34 | 1.0 | 12.4 | 4.6 | 96.6 | N |
| 7 | 149 | Glioblastoma multiforme | M | 59 | 9.0 | 38.0 | 25.8 | 84.9 | Y |
| 3 | 1292 | Glioblastoma multiforme | M | 42 | 25.0 | 60.0 | 12.2 | 89.8 | Y |
| 9 | 1306 | Glioblastoma multiforme | M | 65 | 10.0 | 49.0 | 22.4 | 94.9 | Y |
| 10 | 1309 | Glioblastoma multiforme | M | 52 | 14.8 | 88.6 | 11.5 | 95.2 | Y |
| 11 | 1343 | Glioblastoma multiforme | M | 56 | 63.0 | 23.0 | 33.3 | 97.4 | Y |
| 12 | 1345 | Glioblastoma multiforme | F | 12 | 23.1 | 5.0 | 17.8 | 95.2 | N |
| 13 | 1354 | Glioblastoma multiforme | F | 46 | 26.2 | 19.3 | 16.5 | 89.3 | Y |
| 14 | 1359 | Glioblastoma multiforme | M | 74 | 7.5 | 9.2 | 4.0 | 85.8 | Y |
| 15 | 1284 | Meningothelial meningioma | M | 60 | 1.8 | 5.6 | 5.4 | 8.4 | N |
| 16 | 1293 | Meningothelial meningioma | F | 50 | 0.8 | 11.9 | 10.4 | 85.0 | N |
| 17 | 1299 | Meningothelial meningioma | F | 42 | 5.0 | 0.0 | 17.4 | 0.6 | N |
| 18 | 1302 | Meningothelial meningioma | F | 79 | 0.7 | 0.0 | 5.0 | 2.6 | N |
| 19 | 1318 | Fibrous meningioma | F | 70 | 10.0 | 0.0 | 22.4 | 2.6 | N |
| 20 | 1342 | Fibrous meningioma | F | 50 | 0.3 | 0.0 | 2.4 | 0.6 | N |
| 21 | 1347 | Fibrous meningioma | F | 32 | 0.3 | 0.0 | 2.2 | 2.7 | N |
| 22 | 1362 | Meningothelial meningioma | M | 60 | 0.4 | 28.0 | 41.0 | 53.2 | Y |
| 23 | 1372 | Meningothelial meningioma | F | 38 | 1.5 | 1.1 | 7.0 | 1.2 | Y |
| 24 | 1390 | Tiansitional meningioma | F | 54 | 1.6 | 9.1 | 9.0 | 84.0 | N |
| 25 | 1396 | Secretory meningioma | F | 50 | 2.8 | 56.1 | 19.4 | 64.0 | N |
| 26 | 1400 | Transitional meningioma | F | 57 | 0.8 | 2.2 | 21.0 | 49.0 | N |
| 27 | 1402 | Meningothelial meningioma | F | 61 | 2.5 | 47.1 | 2.6 | 84.2 | N |
Table 2.
Mean proliferation (Ki‐67 LI) and hTERT (%) expression for our thee subgroups of low‐grade astrocytomas, glioblastomas multiforme and benign meningiomas
| WHO classification | Proliferation | hTERT | ||
|---|---|---|---|---|
| In vivo | In vitro | In vivo | In vitro | |
| Astrocytoma LG (n = 6) | 2.1 (SD = 1.9) | 7.5 (SD = 5.6) | 10.2 (SD = 8.9) | 81.2 (SD = 20.8) |
| Glioblastoma multiforme (n = 8) | 22.3 (SD = 18.1) | 17.9 (SD = 9.2) | 36.5 (SD = 28.4) | 91.6 (SD = 4.7) |
| Benign meningioma (n = 13) | 2.2 (SD = 2.7) | 12.7 (SD = 11.2) | 12.4 (SD = 19.2) | 33.7 (SD = 36.6) |
Detection of the hTERT subunit of telomerase was also visualized by immunohistochemistry. Mean percentages of stained cells varied from 10.2 (SD = 8.6) for low‐grade astrocytomas and 36.5 (SD = 28.4) for glioblastomas. The group of benign meningiomas had a labelling index of 12.4 (SD = 19.2).
Primary tumour‐derived cell cultures
Results can be found in 1, 2. As examples of cell proliferation capacity in vitro, Ki‐67 and hTERT primary tumour‐derived cell cultures were counted. To evaluate the putative progression of brain tumours by in vitro conditions, tumour‐derived cell cultures were analysed with the same histopathological marker of cell proliferation and presence of telomerase. Mean proliferation indices for the cultures were 7.5 (SD = 5.6) for low‐grade astrocytomas and 17.9 (SD = 9.2) for glioblastomas. Meningiomas had a mean proliferation level of 12.7 (SD = 11.2).
Mean percentages of cells stained for hTERT varied between 81.2 (SD = 20.8) for low‐grade astrocytomas, 91.6 (SD = 4.7) for glioblastomas and 33.7 (SD = 36.5) for benign meningiomas.
Cell invasiveness in vitro
Results can be found in 1, 3. To determine a putative statistical correlation between cell proliferation or hTERT expression with invasion, in vitro, tumour‐derived cell cultures were analysed for their invasive capacity by use of an invasion model (de Ridder 2003). In vitro, invasion was observed in one‐third of low‐grade astrocytomas (2/6), 87.5% of glioblastomas (7/8) and 15.4% of meningiomas (2/13).
Table 3.
Proliferation (Ki‐67 LI) and telomerase subunit expression (hTERT LI) in vivo versus in vitro regarding the invasive confrontation pattern between our subgroups of intracranial tumours
| WHO classification | Proliferation | hTERT | ||
|---|---|---|---|---|
| Invasion in vitro | In vivo | In vitro | In vivo | In vitro |
| Astrocytoma LG (n = 6) | ||||
| Non‐invasive tumours | 1.7 (SD = 1.6) | 8.2 (SD = 7.0) | 7.8 (SD = 4.3) | 83.3 (SD = 21.0) |
| Invasive tumours | 3.0 (SD = 2.8) | 6.0 (SD = 1.5) | 15.1 (SD = 16.3) | 76.8 (SD = 28.0) |
| Glioblastoma multiforme (n = 8) | ||||
| Non‐invasive tumours | 23.1 | 17.8 | 5.0 | 95.2 |
| Invasive tumours | 22.2 (SD = 19.5) | 18.0 (SD = 9.9) | 41.0 (SD = 27.4) | 91.0 (SD = 4.9) |
| Benign meningioma (n = 13) | ||||
| Non‐invasive tumours | 2.4 (SD = 2.9) | 10.7 (SD = 8.0) | 12.0 (SD = 20.1) | 34.9 (SD = 38.2) |
| Invasive tumours | 1.0 (SD = 0.8) | 24.0 (SD = 24.0) | 14.6 (SD = 19.0) | 27.2 (SD = 36.8) |
Statistical analysis
The Wilcoxon signed rank test was used to determine the difference between the histopatological markers in vivo versus in vitro. A significant difference was seen for the proliferation marker, Ki‐67, in vivo versus in vitro (P < 0.05) and for hTERT in vivo versus in vitro (P < 0.001).
Using Mann–Whitney statistical analysis, no difference was seen between the groups of invasive and non‐invasive tumour‐derived cell cultures for the histopathological markers Ki‐67 and hTERT (P > 0.05) in vitro.
DISCUSSION
This study investigated the putative progression of astrocytomas and meningiomas by measuring their cell proliferation and demonstration of telomerase in vitro. Tumour progression means that the tendency towards malignancy increases as genetic mutations accumulate, leading to increased cell proliferation and invasive capacity. Here, invasiveness in vitro was used as a parameter of malignant progression (de Ridder & Calliauw 1992).
Results show a significant difference between in vivo versus in vitro for both the level of cell proliferation and expression of the telomerase subunit. In the invasion test, there was no statistical correlation concerning cell proliferation or hTERT. The present study confirms that low cell proliferation rate and hTERT expression are found in both astrocytomas and benign meningiomas, compared to proliferation and hTERT expression of malignant gliomas (Cabuy & de Ridder 2001; Falchetti et al. 2002).
In carcinogenesis, cell immortalization and uncontrolled proliferation of the tumour cells are crucial phenomena. In cancer cells, telomere length is dependent on the balance between loss of telomeric repeats during DNA replication and elongation of telomeric repeats mediated by telomerase (Vogelstein & Kinzler 1993; Harada et al. 2000). Differences in levels of cell proliferation and telomerase expression between low‐grade astrocytomas and glioblastomas are intriguing, as secondary glioblastomas are thought to arise by progression from astrocytomas. It appears that this progression implies activation of telomeres and especially hTERT, prior to telomerase activity. Is hTERT expression a rate limiting factor of progression? It has been shown that hTERT mRNA is expressed in 100% of glioblastomas, irrespective of their positive or negative expression exhibited by the telomere repeat amplification protocol assay for telomerase activity (Cabuy & de Ridder 2001). On the other hand, there are benign meningiomas that are characterized by slow development; it is known that grading of meningiomas is controversial. In this respect, hTERT expression might be a marker of progression towards recurrence and malignancy of meningiomas.
As telomeres shorten with each cell division, if a tumour remains, the cell proliferation status is important. The low incidence of hTERT expression in vivo in meningiomas and some astrocytomas suggests that telomerase activity is not an early event in the molecular genesis of carcinogenesis, but is rather a late event in malignancy. However, several researchers have suggested that hTERT gene expression represents an early event in the multistep process of carcinogenesis, already detectable at the stage of pre‐cancerous changes in cervical and oral tumours (Zheng et al. 1997; Luzar et al. 2004). In addition, hTERT expresses antiapoptotic activity (Yan et al. 2004; Rahman et al. 2005). Not all normal primary cells and telomerase‐negative cancer cell lines lack hTERT protein expression, suggesting that the activity of telomerase is determined by complex mechanisms, not simply by the presence of the hTERT protein (Aisner et al. 2002; Yang et al. 2002; Kyo et al. 2003). For this, Shay and colleagues (Shay et al. 1994; Shay 1997; Holt & Shay 1999) proposed the telomere‐telomerase hypothesis of ageing and cancer, and commented that there are at least two different cellular mechanisms that must be overcome before cell immortalization occurs. The first generally requires inactivation of the pathways involving two tumour suppressor genes, p53 and pRb, and the second step almost always involves the activation of telomerase (Vogelstein & Kinzler 1993).
Cultures of tumour‐derived cells expressed a clear cut difference in proliferation and hTERT positivity compared to those of the tumour biopsy. The proliferation index for glioblastomas was at a steady state in vivo versus in vitro probably because the tumour cells had divided already to the maximum level, explaining the uncontrolled proliferation of these cells. Astrocytomas and meningiomas expressed a higher proliferation index in vitro than in the biopsy, resulting probably from the ‘progression’ potential in culture circumstances. The culture environment in vitro for those tumour cells without tumour inhibitors and full access to medium might stimulate astrocytomas and meningiomas to proliferate at a higher level. Also, hTERT expression was higher in vitro than in vivo. In vitro, the astrocytomas had the same proliferation indices as glioblastomas, and meningioma cells also proliferated more and expressed higher hTERT levels, but at a considerably lower level than gliomas and astrocytomas. This suggests the less progressive status towards malignancy of meningiomas.
Concerning hTERT in vitro, the higher number of hTERT‐positive cells points to the selection of hTERT‐positive tumour cells in our cell cultures. hTERT‐positive cells may have a higher viability and lifespan in comparison to hTERT‐negative cells. Our hypothesis for this phenomenon is that hTERT probably protects the chromosomal telomeres from apoptotic promoters, such as p53. By this connection, the tumour cells can pass the mortality stage 1 (M1) phase of the Hayflick limit and can divide for several more cell cycles (Klapper et al. 2001). Finally, the telomeres are so short that the protecting telomeric loop can no longer be formed and also interaction between hTERT and telomeres is no longer granted. The tumour cells will reach mortality stage 2 (M2) of the Hayflick limit. Double‐stand breaks (DSB) are a cause of the loss of the protective capacity of telomeres; and apoptotic promoters recognize the shortened telomere, which leads to apoptosis and the cells consequently disappear. However, a certain number of cells probably begin to express active telomerase and become immortal cells. This can be supported by the higher appearance of telomerase activity in secondary glioblastomas than primary ones. Secondary glioblastomas, derived from astrocytomas, have gone through more passages of malignancy and therefore already express telomerase (Harada et al. 2000). In agreement with our results, no significant association has been found between telomerase activity levels and those of cell proliferation (Bednarek et al. 1997; Dome et al. 1999; Maes et al. 2005).
Thus, it seems that the change of environment with the lack of host factors and tumour inhibitors is of crucial importance to progression of tumour cells towards invasive malignancy. On the other hand, we have evaluated invasion in vitro. Tumour cell invasion in vitro is a time‐dependent and progressive process. In malignant tumours such as glioblastomas, it is possible that all confrontation cultures might not yet demonstrate invasion in vitro; this is because at present the confrontation period in vitro is limited to 7 days (see Table 1). It was clear in our experiments that hTERT expression and cell proliferation level did not correlate with invasion in vitro. This suggests that invasion is a later event in the progression towards malignancy, and is not an initial step in the development of a malignant tumour. Yet, the invasion test can help to predict tumour behaviour in vivo, a useful tool for neurosurgeons prior to patient surgery (Corcoran et al. 2003). As previously described (Kalala et al. 2004), there was no direct link shown between cell proliferation levels and confrontation status. The same seems true for hTERT, which is also seems to not be linked to invasion.
In conclusion, tumorigenesis is a multistep process that may be called ‘progression’. During this process, the activation of hTERT is a crucial event in cell immortalization, and elevated expression of hTERT and Ki‐67 in vitro provides a potential prognostic tool to demonstrate progression status of cancer cells. As tumour cells require telomerase for continued proliferation, expression of hTERT may mark incipient immortality leading to an indefinite lifespan. On the other hand, hTERT and cell proliferation are not directly linked to tumour cell invasion.
ACKNOWLEDGEMENTS
We would like to thank the neurosurgeons from the University Hospital, St Lucas Hospital and Maria‐Middelares in Ghent, who kindly provided us with tumour samples; N. François, R. De Vos, L. Pieters and J. Aernoudt from our laboratory for technical assistance. Thanks for linguistic corrections to S. Sutch.
REFERENCES
- Aisner DL, Wright WE, Shay JW (2002) Telomerase regulation: not just flipping the switch. Curr. Opin. Genet. Dev. 12, 80–85. [DOI] [PubMed] [Google Scholar]
- Bednarek AK, Sahin A, Brenner AJ, Johnston DA, Aldaz CM (1997) Analysis of telomerase activity levels in breast cancer: positive detection at the in situ breast carcinoma stage. Clin. Cancer Res. 3, 11–16. [PubMed] [Google Scholar]
- Cabuy E, De Ridder L (2001) Telomerase activity and expression of telomerase reverse transcriptase correlated with cell proliferation in meningiomas and malignant brain tumors in vivo . Virchows Arch. 439, 176–184. [DOI] [PubMed] [Google Scholar]
- Carroll T, Maltby E, Brock I, Royds J, Timperley W, Jellinek D (1999) Meningiomas, dicentric chromosomes, gliomas, and telomerase activity. J. Pathol. 188, 395–399. [DOI] [PubMed] [Google Scholar]
- Cavalla P, Schiffer D (1997) Cell cycle and proliferation markers in neuroepithelial tumors. Anticancer Res. 17, 4135–4144. [PubMed] [Google Scholar]
- Corcoran A, De Ridder LI, Del Duca D, Kalala OJ, Lah T, Pilkington GJ, Del Maestro RF (2003) Evolution of the brain tumour spheroid model: transcending current model limitations. Acta Neurochir. (Wien) 145, 819–824. [DOI] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, Le Feuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dome JS, Chung S, Bergemann T, Umbricht CB, Saji M, Carey LA, Grundy PE, Perlman EJ, Breslow NE, Sukumar S (1999) High telomerase reverse transcriptase (hTERT) messenger RNA level correlates with tumor recurrence in patients with favourable histology Wilms’ tumor. Cancer Res. 59, 4301–4307. [PubMed] [Google Scholar]
- Falchetti ML, Larocca LM, Pallini R (2002) Telomerase in brain tumours. Childs Nerv. Syst. 18, 112–117. [DOI] [PubMed] [Google Scholar]
- Harada K, Kurisu K, Tahara H, Tahara E, Ide T, Tahara E (2000) Telomerase activity in primary and secondary glioblastomas multiforme as a novel molecular tumor marker. J. Neurosurg. 93, 618–625. [DOI] [PubMed] [Google Scholar]
- Holt SE, Shay JW (1999) Role of telomerase in cellular proliferation and cancer. J. Cell. Physiol. 180, 10–18. [DOI] [PubMed] [Google Scholar]
- Jaaskelainen J, Haltia M, Servo M (1986) Atypical and anaplastic meningiomas: radiology, surgery, radiotherapy, and outcome. Surg. Neurol. 25, 233–242. [DOI] [PubMed] [Google Scholar]
- Kalala JP, Benoit D, De Ridder L (2004) Can recurrence of meningiomas be predicted? Anticancer Res. 24, 2319–2324. [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994) Specific associations of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Klapper W, Parwaresch R, Krupp G (2001) Telomere biology in human aging and aging syndromes. Mech. Ageing Dev. 122, 695–712. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Cavenee WK (2000). Pathology and genetics of tumours of the nervour system. World Health Organisation Classification of Tumours, pp. 176–184. Lyon: IARC Press. [Google Scholar]
- Kyo S, Masutomi K, Maida Y, Kanaya T, Yatabe N, Nakamura M, Tanaka M, Takarada M, Sugawara I, Murakami S, Taira T, Inoue M (2003) Significance of immunological detection of human telomerase reverse transcriptase: re‐evaluation of expression and localization of human telomerase reverse transcriptase. Am. J. Pathol. 163, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig AJ (1999) Crisis intervention: the role of telomerase. Proc. Natl. Acad. Sci. USA 96, 3339–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzar B, Poljak M, Marin IJ, Eberlink A, Klopcic U, Gale N (2004) Human telomerase catalytic subunit re‐expression is an early event in oral carcinogenesis. Histopathology 45, 13–19. [DOI] [PubMed] [Google Scholar]
- Maes L, Lippens E, Kalala JPO, De Ridder L (2005) The hTERT‐protein and Ki‐67 labelling index in recurrent and non‐recurrent meningiomas. Cell Prolif. 38, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman R, Latonen L, Wiman KG (2005) hTERT antagonizes p53‐induced apoptosis independently of telomerase activity. Oncogene 24, 1320–1327. [DOI] [PubMed] [Google Scholar]
- De Ridder L (2003) Sphéroids et cultures en agrégats In: Barlovatz‐Meimon G, Adolphe M, eds. Culture de Cellules Animales: Méthodologies‐Applications, pp. 95–110. Editions Inserm, Paris, France. [Google Scholar]
- De Ridder L, Calliauw L (1992) Invasiveness of primary and secondary brain tumours in vitro correlated with clinical results. Neurosurgery 31, 1043–1047. [DOI] [PubMed] [Google Scholar]
- Shay JW (1997) Telomerase in human development and cancer. J. Cell Physiol. 173, 266–270. [DOI] [PubMed] [Google Scholar]
- Shay JW, Werbin H, Wright WE (1994) Telomere shortening may contribute to aging and cancer: a perspective. Mol. Cell. Differ. 2, 1–21. [Google Scholar]
- Vogelstein B, Kinzler K (1993) The multistep nature of cancer. Trends Genet. 9, 138–141. [DOI] [PubMed] [Google Scholar]
- Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lchtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, Morin GB (1997) Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 17, 498–502. [DOI] [PubMed] [Google Scholar]
- Yan P, Benhattar J, Seelentag W, Stehle JC, Bosman FT (2004) Immunohistochemical localization of hTERT protein in human tissues. Histochem. Cell Biol. 121, 391–397. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chen Y, Zhang C, Huang H, Weissman SM (2002) Nucleolar localization of hTERT protein is associated with telomerase function. Exp. Cell Res. 277, 201–209. [DOI] [PubMed] [Google Scholar]
- Zheng PS, Iwasaka T, Yokoyama M, Nakao Y, Pater A, Sugimori H (1997) Telomerase activation in in vitro and in vivo cervical carcinogenesis. J . Gynaecol. Oncol. 66, 222–226. [DOI] [PubMed] [Google Scholar]