

Abstract
Accurate chromosome segregation relies on activity of the spindle assembly checkpoint, a surveillance mechanism that prevents premature anaphase onset until all chromosomes are properly attached to the mitotic spindle apparatus and aligned at the metaphase plate. Defects in this mechanism contribute to chromosome instability and aneuploidy, a hallmark of malignant cells. Here, we review the molecular mechanisms of activation and silencing of the spindle assembly checkpoint and its relationship to tumourigenesis.
Introduction
Mitosis is a complex and highly regulated event during which eukaryotic somatic cells face the task of accurately segregating sister‐chromatids (replicated in S phase of the cell cycle), to the two daughter cells. Failure in chromosome segregation may lead to loss or gain of one or more chromosomes, a condition known as aneuploidy, a hallmark of malignant cells (1, 2). Correct chromosome segregation requires that each chromosome establishes bipolar attachments, through its sister‐kinetochores, to microtubules emanating from opposite poles of the mitotic spindle, and becomes aligned at the metaphase plate (3). Given the stochastic and asynchronous nature of chromosome attachments to the spindle, chromosomes already aligned at the metaphase plate must wait for still unaligned chromosomes before anaphase can be initiated. Eukaryotic cells have evolved a ‘wait anaphase’ mechanism, named spindle assembly checkpoint (SAC), that inhibits metaphase to anaphase transition until the last chromosome reaches the metaphase plate (4). This sophisticated surveillance mechanism detects inappropriate kinetochore‐microtubule attachments during chromosome congression from prometaphase to metaphase and delays mitotic exit, allowing sufficient time for error correction and chromosome bi‐orientation. Inhibition exerted by the SAC involves Mad (mitotic arrest deficient, Mad1 and Mad2) and Bub (budding uninhibited by benzimidazole, Bub1, Bub3 and BubR1/Mad3) proteins that prevent Cdc20 protein from activating anaphase promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase (5). Activation of APC/C is needed to target Securin and Cyclin B for degradation by the 26S proteasome to promote anaphase onset and mitotic exit.
In this article, we review the spindle assembly checkpoint mechanism, focusing on its role in kinetochore–microtubule interactions, in higher eukaryotes. By addressing this topic, we will emphasize aspects related to kinetochore attachment, SAC activation, error correction and SAC silencing. Furthermore, as defects in SAC mechanism are thought to contribute to chromosome instability, current understanding of the relationship between SAC and tumourigenesis is presented.
The ‘search and capture’ mechanism of kinetochore attachment to spindle microtubules
After nuclear envelope breakdown, which marks transition from prophase to prometaphase, chromosomes are released into the cytosol, and become accessible to microtubules of the mitotic spindle. At this stage, microtubules probe the cytoplasm, through episodes of lengthening and shortening of their plus ends, to search and capture chromosomes (6). Each chromosome has two sister‐kinetochores, proteinaceous complexes assembled on centromeric DNA on each sister‐chromatid that serve as attachment sites of chromosomes to spindle microtubules. Over the past few years, functional and proteomic‐based analysis of the kinetochore‐microtubule interface has increased our understanding of molecular mechanisms of chromosome attachment to spindle and has shed light on kinetochore bi‐orientation (Fig. 1) (7).
Figure 1.
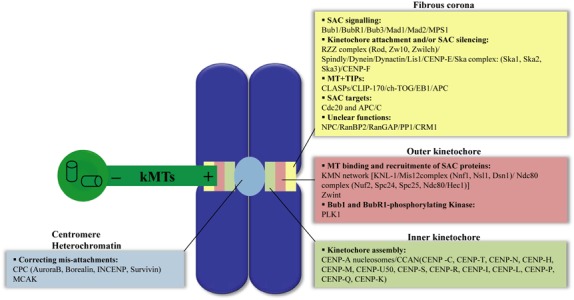
Overview of protein complexes that build the kinetochore in animal cells. The kinetochore is built on the centromere as a trilaminar protein‐rich structure: the inner kinetochore, the outer kinetochore and the fibrous corona. Proteins that compose each kinetochore layer are grouped by function (APC/C, anaphase promoting complex/cyclosome; Bub1BubR1‐Bub3, budding uninhibited by benzimidazole; Cdc20, cell division cycle 20; CENP, centromere protein; CLASP, CLIP‐associating protein; CLIP170, cytoplasmic linker protein‐170; CPC, chromosome passenger complex; EB1, end‐binding protein‐1; INCENP, inner centromere protein; kMTs, kinetochore microtubules; LIS1, lissencephaly‐1; Mad1‐Mad2, mitotic‐arrest deficient; MCAK, mitotic centromere‐associated kinesin; MPS1, multipolar spindle‐1; MT, microtubules; NPC, nuclear pore complex; PLK1, polo‐like kinase‐1; RanBP2, Ran‐binding protein 2; RanGAP, Ran‐GTPase‐activating protein; RZZ, Rod (rough deal); SAC, spindle assembly checkpoint; Ska1–3, spindle and kinetochore‐associated proteins; Zw10, zeste white 10‐Zwilch complex; Zwint, Zw10 interactor. For details of dynamic localization of kinetochore proteins, see references (4, 92).
Different studies have demonstrated that plus ends of microtubules bind to kinetochores through the KMN protein network, a crucial constituent of the outer kinetochore. This structural core of the kinetochore is composed of KNL‐1 protein (Blinkin/Spc105 in human and budding yeast, respectively) and the two subcomplexes Mis12, composed of four proteins Mis12/Nnf1/Nsl1/Dsn1, and Ndc80, containing four proteins Ndc80 (Hec1 in mammals)/Nuf2/Spc24/Spc25 (Fig. 1) (7). Removal of any of the components of the KMN network leads to disruption of binding scaffolds for microtubules at outer kinetochore plates (8, 9).
Initial capture results in binding of one kinetochore to the lateral surface of a microtubule, followed by rapid poleward movements of attached chromosomes along microtubules (Fig. 2).These movements are probably powered by the motor activity of cytoplasmic dynein (10, 11, 12, 13, 14), recruited to kinetochores by RZZ complex [composed of Rough‐deal (ROD), Zeste‐white 10 (ZW10), and Zwilch] via Spindly (SPDL‐1 in Caenorhabditis elegans) (15, 16, 17, 18, 19, 20). High density of microtubules near spindle poles contribute to conversion of the lateral attachments to ‘end‐on’ attachments (Fig. 2). In C. elegans, RZZ complex and Spindly/SPDL‐1 have been reported to be required for this conversion (18). Furthermore, a complex of three proteins, Ska1, Ska2 and Ska3, have also been shown to be involved in stable end‐on kinetochore‐microtubule attachments, in vertebrate cells (21, 22, 23). Due to polar ejection forces, the now mono‐attached chromosome is forced to move towards the spindle equator (a process known as chromosome congression), with the unattached sister‐kinetochore facing microtubules from the opposing pole, resulting in its end‐on attachment (Fig. 2) (24). Besides this mechanism, in metazoan cells, mono‐oriented chromosomes can be transported towards the spindle equator by gliding alongside microtubules attached to other already bi‐oriented chromosomes, driven by kinetochore‐bound CENP‐E, a plus end‐directed microtubule motor of the kinesin‐7 family (25). Chromosomes are aligned at the metaphase plate once they become bi‐oriented, a condition known as amphitelic attachment, with full microtubule occupancy (Fig. 2).
Figure 2.
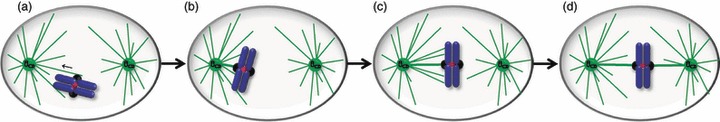
Chromosome bi‐orientation during prometaphase. When the nuclear envelope breaks down, the kinetochore is captured by lateral surfaces of microtubules emanating from a spindle pole (a), resulting in its transport towards that pole (arrow). High density of microtubules near the pole contributes to maturation of the lateral attachment to end‐on attachment, with the kinetochore tethered at the plus end of the microtubules (b). Polar ejection forces and/or gliding alongside microtubules attached to other already bi‐orientated chromosomes (not depicted in the figure) drive the mono‐orientated chromosome towards the metaphase plate (c), leading to its bi‐orientation (d). Attachment errors depicted in Fig. 3 can happen and are detected and corrected to bi‐oriented attachments.
Correcting kinetochore‐microtubule mis‐attachments
It is known that the amphitelic attachment, achieved when sister kinetochores are attached to opposite poles of the spindle, is the only geometry that ensures accurate segregation of sister‐chromatids to daughter cells, at anaphase. However, due to the stochastic nature of the widely accepted ‘search and capture’ mechanism, to chromosome position within the cell and geometry of their sister‐kinetochores relative to microtubules at the onset of prometaphase, other connections can occur and compromise correct segregation of chromosomes. There are three possibilities for kinetochore‐microtubule mis‐attachment: monotelic, syntelic and merotelic abberations (Fig. 3) (3, 26).
Figure 3.

Bi‐orientation and kinetochore attachment errors. (a) In amphitelic attachment, sister kinetochores are correctly attached to microtubules emanating from opposite poles of the spindle, leading to chromosome bi‐orientation. (b) In monotelic attachment, the chromosome is mono‐oriented as one kinetochore is attached to the microtubules from one spindle pole, while its sister is unattached. (c) In syntelic attachment, the chromosome is mono‐oriented but, in this case, both sister kinetochores are attached to microtubules from the same spindle pole. (d) In merotelic attachment, one sister kinetochore is attached to microtubules from both spindle poles, the chromosome is improperly bi‐oriented and, if left uncorrected, can produce an anaphase lagging chromosome.
Monotelic kinetochore attachment occurs when one sister‐kinetochore is unattached, while the other is attached to microtubules from just one pole. This is common in early mitosis and it is a normal condition at the very beginning of prometaphase (Fig. 3). Syntelic attachment is observed, although rarely, when the two sister kinetochores are bound to microtubules from the same spindle pole. Both monotelic and syntelic attachments activate the SAC, due to reduced tension at sister‐kinetochores, and are generally corrected and converted into amphitelic configurations. Merotelic attachments occur when one sister kinetochore binds to microtubules from both poles, frequently in early prometaphase. These attachments do not interfere with chromosome alignment during prometaphase and are not always detected by the SAC. Nevertheless they cause chromosome mis‐segregation rarely, as they are usually corrected by an Aurora B‐dependent mechanism, before anaphase onset (3, 26, 27).
How are mis‐attachments distinguished from amphitelic attachments and corrected? Appropriate tension across sister kinetochores contributes to detection and correction of merotelic and syntelic attachment errors. Sister kinetochores in amphitelic attachments are under tension, which results from pulling forces of spindle microtubules in opposite directions. Pioneering micromanipulation experiments from Nicklas and co‐workers has suggested that mechanical tension at kinetochores increases occupancy of microtubule attachment sites, which contributes to stabilizing kinetochore to microtubule attachments (28, 29). It is widely accepted that tension is the signal that distinguishes different attachment states of sister kinetochores and that Aurora B kinase (mammalian homologue of budding yeast Ipl1 kinase), acts as tension sensor to correct mis‐attachments by destabilizing them (27, 30). Aurora B localizes to inner centromeres and regulates interactions between kinetochores and microtubules through phosphorylation of Ndc80 complex and MCAK (mitotic centromere‐associated kinesin) a member of the kinesin‐13 family of microtubule depolymerases (31). A spatial separation model has been proposed that might explain how tension mediates correction of mis‐attachments by Aurora B kinase (32). Low tension at syntelically attached kinetochores and unbalanced tension resulting from merotelic orientation would locate kinetochores close to a peak of Aurora B kinase activity in the inner centromere, which releases microtubules as a consequence of Ndc80 and MCAK phosphorylation. Phosphorylation of Ndc80 complex weakens its affinity to microtubules, while phosphorylated MCAK catalyses depolymerization at ends of microtubules (33, 34). Selective destabilization of incorrect chromosome attachments provides a further opportunity for the chromosome to bi‐orientate. Bi‐orientation increases distance between kinetochores and the inner centromere, due to forces exerted by spindle microtubules in opposite directions. As a consequence, Aurora B becomes spatially separated from its substrates and attachments are stabilized. In this spatial separation model, a constitutively active phosphatase, such as PP1 (protein phosphatase 1) in budding yeast and PP1γ and PP2A in vertebrates, dephosphorylates Aurora B substrates allowing for rapid re‐attachment (30). Other models are possible for mechanisms by which Aurora B regulates kinetochore‐microtubule attachments, stressing the need to clarify the molecular nature of processes through which inappropriate attachments can be detected and corrected (27, 30).
The spindle assembly checkpoint
The spindle assembly checkpoint is a constitutive surveillance mechanism in eukaryotic dividing cells that is extremely sensitive to defects in kinetochore attachment. It prevents chromosome mis‐segregation by delaying metaphase to anaphase transition until all chromosomes are correctly connected to spindle microtubules, bi‐orientated and aligned at the metaphase plate (4). The SAC consists of a signalling cascade that represents primary cell‐cycle control mechanisms in mitosis and is activated immediately after entrance into mitosis (or meiosis), every cell cycle. Accurate activity of this checkpoint mechanism is crucial for equal segregation of the genetic material into the two daughter cells and thus, for effective reduction of error rate during cell division. Failure in SAC function has been suggested as a possible cause of aneuploidy in several tumour types (35, 36). Moreover, SAC contributes to temporal organization of the cell cycle, since the cell only progresses to the next phase when SAC’s requirements are satisfied. SAC molecular pathway involves detection of attachment errors and generation of the signal that inhibits mitotic progression, error correction and SAC silencing (see below).
Controversy around the signal detected by SAC
Chromosome mis‐attachment defects that trigger SAC response are still a matter of debate (37, 38). Two models are currently proposed: (i) attachment model, which proposes that SAC senses level of kinetochore occupancy by microtubules; and (ii) tension model, which suggests that SAC senses lack of tension across sister kinetochores. Experimental data seem to support proponents of both models. Tension artificially applied using a microneedle on the last tensionless chromosome revealed anaphase delay and induced completion of division, in praying mantid spermatocytes (39), thus arguing in favour of the tension hypothesis. Moreover, engineered budding yeast cells with attached but tensionless kinetochores of unreplicated mitotic chromatids were unable to satisfy SAC, demonstrating that tension is required to turn off the checkpoint (40). HeLa cells treated with low doses of vinblastine (a microtubule‐depolymerizing drug, that reduces tension across kinetochores without affecting microtubule attachments), arrested in mitosis, indicating that microtubule attachments were not sufficient to override the checkpoint (41). On the other hand, laser ablation of the last unattached kinetochore of a tensionless mono‐oriented chromosome caused PtK cells to enter anaphase, arguing in favour of the attachment model (42). Furthermore, Mad2 removed from kinetochores on attachment, a sign of SAC inactivation, indicates that the checkpoint is turned off by microtubule attachment and not by tension.
Individual analysis of each model is made difficult by interdependency between attachment and tension. Indeed, tension is needed to promote stable kinetochore‐microtubule attachments (29), while kinetochore occupancy by microtubules provides necessary forces to generate tension across sister kinetochores (43). In this respect, a ‘partitioned checkpoint’ hypothesis has also been proposed. In this model, the wait anaphase signal can be generated by specific signalling molecules that differentially signal absence of attachment or tension (38, 44, 45). Mad2 is enriched at unattached kinetochores and could be, in association with its kinetochore partner Mad1, one of the signalling molecules of the kinetochore attachment state (45) and state of interkinetochore tension can be monitored by kinetochore localization of BubR1 and Bub1 together with yet‐unidentified kinetochore phosphoepitopes recognized by 3F3/2 antibody (38, 44).
Recent studies have reported that intrakinetochore stretchs, rather than interkinetochore stretchs, are sufficient to satisfy SAC, ‘introducing a new kind of tension to the debate’ (46). According to the intrakinetochore stretch model, SAC satisfaction depends on molecular rearrangements within the kinetochore structure, induced in part by microtubule attachments and dynamics, and not on tension across kinetochores (47).
One is tempted to suggest that the attachment versus tension controversy is a false debate. Our group shares the opinion of Khodjakov and Rieder that presence of free kinetochores is the only signal that triggers SAC response (48). Free kinetochores appear in early prometaphase or can be created during correction of erroneous attachments. For example, during syntelic attachment correction, one kinetochore is disconnected from its associated microtubules by Aurora B kinase activity, and is converted into a free kinetochore, which prevents SAC release. Taken together, there is no doubt that the controversial models (described above) require presence of free kinetochores to trigger SAC response and anaphase delay. Interestingly, free kinetochores are rarely generated in merotelic attachments during prometaphase, which is why these erroneous attachments are not detected by SAC. In this context, attachment and interkinetochore/intrakinetochore stretches by themselves would not represent SAC triggers but instead, would be part of the correction mechanism and act to regulate physical contact between Aurora B and its substrates, by modulating distance between inner and outer kinetochore regions.
Molecular pathway of SAC and mechanism of anaphase delay
Although not consensual, proteins involved in SAC molecular pathways are often divided into two groups: (i) proteins that form ‘bona fide SAC components’ these include Bub1, BubR1, Bub3, Mad1, Mad2 and Mps1; and (ii) proteins of the attachment, APC/C regulatory, correction and SAC silencing machinery, with which true SAC proteins must interact to monitor attachments and cell cycle progression (7). The distinction between the two groups has been elegantly expressed in a recent article (48). In this section, we will focus on bona fide SAC proteins, as proteins involved in attachment and cell cycle progression are discussed throughout the text.
Bona fide SAC proteins, true SAC components, are comprised of Mad1, Mad2, Mad3 (BubR1 in higher eukaryotes), Bub1, Bub3 and Mps1, which have been initially identified in budding yeast (4, 7). Homologues for these proteins have also been identified in higher organisms, including mammals. These proteins have been shown to share a high degree of homology at both the sequence and functional levels with their yeast counterparts, as functional disruption studies through dominant‐negative mutants, antibody injection or RNA interference, completely compromised spindle checkpoint activity, causing chromosome mis‐segregation, aneuploidy and escape from mitotic arrest in presence of microtubule poisons such as nocodazole and taxol (49).
Whenever unattached kinetochores are present, Mad2, Mad3/BubR1 and Bub3 proteins localize there to generate the mitotic checkpoint complex (MCC) (50), a ‘wait anaphase’ signal that diffuses through the cytosol to inhibit Cdc20, an activator of APC/C (Fig. 4a) (5, 51). This keeps APC/C (ubiquitin ligase that regulates many cell‐cycle processes) inhibited, preventing it from ubiquitinating Securin (Pds1 in budding yeast) and Cyclin B and thus, from targeting them for destruction by the 26S proteasome. By preventing Securin and Cyclin B degradation, sister‐chromatid cohesion and the mitotic state are maintained respectively. A ‘Mad2‐template’ model has been proposed as the mechanism by which cytosolic inhibitory signals are propagated away from the kinetochore (Fig. 4b) (52). According to this model, Mad2 can adopt either an open conformation (O‐Mad2) or a closed form (C‐Mad2) (52, 53, 54). Constitutively C‐Mad2 bound to Mad1 serves as template or receptor at unattached kinetochores for cytosolic O‐Mad2 to switch this latter to C‐Mad2 bound to Cdc20 form. C‐Mad2/Cdc20 complex leaves kinetochores and acts as structural equivalent of Mad1/Mad2 to convert more O‐Mad2 into Cdc20 bound C‐Mad2 in the cytosol, resulting in signal amplification (Fig. 4b) (52, 55). This model starts to be initiated early as nuclear envelope breakdown when level of MCC complex is not yet sufficient to prevent anaphase. Once the last chromosome becomes bi‐oriented, ‘wait anaphase’ is no longer produced and Cdc20 is released to trigger APC/C activation, which in turn ubiquitinates Securin and Cyclin B, targeting them to degradation. Degradation of Securin, an inhibitor of the protease Separase, leads to cohesin proteolysis and sister‐chromatid separation, whereas Cyclin B degradation leads to inactivation of cyclin‐dependent kinase 1, which drives mitotic exit.
Figure 4.
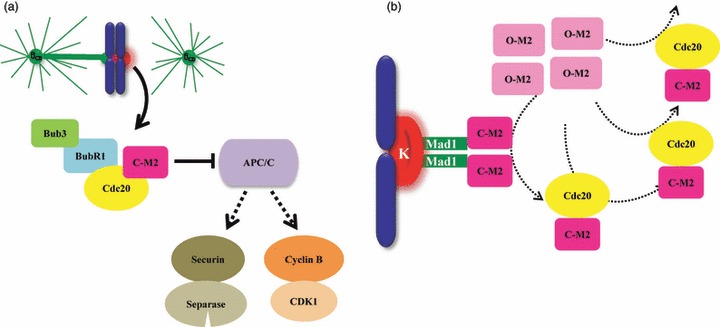
Model of spindle assembly checkpoint signalling. (a) Unattached kinetochore (K) serves as a platform for SAC proteins BubR1, Bub3, and Mad2 to generate the mitotic checkpoint complex (50) that binds to Cdc20 preventing it from activating APC/C, the E3 ubiquitin ligase that targets Securin and Cyclin B for degradation by the 26S proteasome, thereby inhibiting anaphase onset. (b) According to the Mad2 template model, a constitutively closed conformation of Mad2 (C‐M2) bound to Mad1 serves as receptor at unattached kinetochore for cytosolic open form of Mad2 (O‐M2) to switch this latter to C‐Mad2 bound to Cdc20. C‐Mad2/Cdc20 complex acts as a structural equivalent of Mad1/Mad2 to convert more O‐Mad2 into Cdc20 bound C‐Mad2 in the cytosol, leading to signal amplification (93, 94).
Protein phosphorylation and dephosphorylation probably have major roles in transduction and amplification of SAC signals. In this respect, however, the exact role of kinase activity of checkpoint proteins Bub1, BubR1 and (to a lesser extent) Mps1, in SAC signalling, has long been controversial. Contradictory results have been reported concerning requirement of these checkpoint kinases in SAC, probably due to variability between different assays used to assess SAC response or inefficient depletion of endogenous proteins (56, 57). Bub1 has been reported to phosphorylate Cdc20, inhibiting its ability to activate APC/C (58), suggesting a model in which Bub1 kinase contributes to amplify or strengthen SAC signals in presence of few unattached kinetochores (59). Other studies have shown that Bub1 kinase activity is not sufficient for complete SAC function (60). Less certain is the contribution of BubR1 kinase activity to SAC. It has been reported that BubR1 kinase activity is activated by binding to CENP‐E tail and inactivated upon CENP‐E binding to microtubules (61). Conflicting studies have reported mixed results concerning whether BubR1 kinase activity is required for efficient chromosome capture and congression (62, 63, 64). Requirement of Mps1 kinase activity is essential for SAC activity, as its inhibition overrides SAC (65, 66). Mps1 kinase activity has also been shown to be involved in error correction during chromosome bi‐orientation (67); moreover, Mps1 phosphorylates Borealin that in turn directs activity of Aurora B (68), in agreement with its role in regulating chromosome attachment and alignment. Phosphorylation of Mad1 has been reported to be Mps1‐dependent (69), but the role of Mad1 phosphorylation in SAC remains to be determined.
SAC silencing
SAC silencing implies preventing generation of the ‘wait anaphase’ signal once correct chromosome attachment is achieved. This presumes existence of a regulatory link between chromosome bi‐orientation and silencing mechanisms. Several models of SAC silencing mechanism have been proposed (70, 71). The first model suggests that production of MCC is halted by Dynein‐dependent stripping of SAC components from the attached kinetochore (72). Upon kinetochore‐microtubule attachment, the minus‐end directed motor Dynein actively transports SAC proteins such as Mad2 and BubR1, along spindle microtubules, away from attached kinetochores, towards spindle poles. Consistent with this mechanism, cells arrest in mitosis with high kinetochore‐associated Mad2 levels following depletion of Dynein‐light intermediate chain 1 or after microinjection of 70.1 anti‐Dynein antibodies (72, 73). Another silencing mechanism is inhibition exerted by p31comet protein on Mad2, preventing it from inhibiting APC/CCdc20 in mammalian cells (74, 75). By binding dimerization interface of Mad2, p31comet protein prevents Mad2 activation and promotes dissociation of Mad2/Cdc20 complex (75). Indeed, HeLa cells that recover from SAC‐dependent nocodazole‐induced block are delayed in mitosis under conditions of low p31comet expression. Accordingly, over‐expression of p31comet abrogates SAC‐dependent mitotic arrest in HeLa cells treated with microtubule poisons (75). In addition, phosphorylation of Mad2 has been reported to inhibit its interaction with APC/CCdc20 or Mad1, suggesting its implication in SAC silencing (76). Although the regulatory mechanism whereby Mad2 becomes phosphorylated and silences SAC upon kinetochore attachment, is still unknown, it is possible that phosphorylated Mad2 facilitates its binding by p31comet and/or makes it competent to be transported by Dynein during kinetochore stripping. Recently, an alternative silencing mechanism mediated by kinetochore‐associated protein phosphatase 1 (PP1) was proposed in fission yeast (77). Independent of its direct role in kinetochore‐microtubule error correction, PP1 promotes SAC silencing by reversing phosphorylation of Aurora kinase substrates at kinetochores. Identity of these substrates is unknown and it remains to be proven whether mammalian PP1γ isoform also operates in a similar silencing mechanism.
Elusive relationship between SAC and tumourigenesis
The discovery of SAC and its relevance to genetic stability, together with that many cancer cells exhibit weakened SAC activity, had initially prompted many scientists to search for mutations in SAC genes (in several tumours), to establish a relationship between SAC and tumourigenesis and, eventually, to anticipate prevention, diagnosis and cancer treatment (36). Although the first identification of mutations in SAC genes BUB1 and BUB1B in human colorectal cancer cell lines was encouraging (78), genetic lesions on SAC components were revealed to be quite rare in a large number of aneuploid cancers with weakened SAC activity, suggesting that epigenetic alterations might be responsible for SAC impairment (79). Many studies have reported altered expression of SAC components in various tumours. Moreover, mice with heterozygous SAC genes, hence with low levels of SAC proteins, have weakened SAC activity, exhibit high frequency of aneuploid cells and develop tumours (80, 81, 82). Although mutations or altered expression levels of SAC genes have been reported in many aneuploid cancers, it remains to be elucidated whether these alterations are directly responsible for SAC weakening. It is likely that decreased levels of some SAC components, known to have roles in chromosome congression, may contribute to aneuploidy in cancer cells. For instance, Bub1‐, BubR1‐ or Bub3‐depleted cells have been reported to exhibit chromosome congression defects (83, 84, 85, 86).
While presence of compromised SAC and its contribution to aneuploidy in many tumours had gained widespread acceptance, a number of studies have reported that SAC is fully functional in most aneuploid cancer cells (87, 88). Aneuploid cell lines were shown to arrest in response to microtubule damage for longer than non‐transformed cells and, interestingly, they only rarely entered anaphase in presence of non‐aligned chromosomes (88). One possible explanation to this controversy is that SAC status varies between cancer types depending on putative underlying molecular alterations. For instance, different expression profiles of SAC genes have been reported in different cancer lines, with the same genes showing increased expression in some cancers and decreased expression in others. Moreover, efficient SAC activity is based on equilibrium between its components and their expression levels; thus, SAC status in a given tumour would be influenced by extent to which this equilibrium is affected by overall alterations in expression profiles of all SAC genes in that tumour. Taken together, it appears that SAC activity in aneuploid cancer cells is sufficient to prevent premature anaphase under normal proliferative conditions. However, its ability to sustain artificially prolonged arrest, such as the one imposed by microtubule poisons, would depend on the nature of molecular alterations in SAC components or in components of other mechanisms that allow premature satisfaction of SAC, such as those responsible for microtubule dynamics or for correcting chromosome attachment errors.
Independent of the controversy around SAC status in cancer cells and its role in occurrence of chromosome instability and tumourigenesis, there is no doubt that complete SAC inactivation is lethal to cells, due to massive chromosome mis‐segregation (89, 90). As SAC is only required during mitosis, its targeting obviously represents a promising therapeutic strategy to selectively kill dividing cells, which could circumvent resistance to or side effects of anti‐cancer agents currently in use, such as those that target microtubules. In this respect, SAC components with no functional roles outside mitosis constitute suitable targets (91).
Conclusion
In the present review, we have summarized our current knowledge on chromosome attachment to spindle microtubules, attachment error detection and correction, and SAC activation and silencing. Significant progress has been made concerning relationships between SAC and kinetochore‐microtubule attachment interface, contributing to our understanding of how kinetochore attachment to spindle microtubules is linked to SAC activation and silencing, both at dynamic and at molecular levels. However, many gaps between attachment state, activation of SAC and its silencing, still need to be filled. For instance, how is presence of unattached kinetochores signalled to SAC to generate the MCC inhibitory complex? Some bona fide SAC proteins were themselves implicated in kinetochore‐to‐microtuble attachment (84); how is this function integrated in our current understanding of SAC activation and silencing? How is the state of chromosome bi‐orientation signalled to Dynein to proceed to SAC protein stripping? What are the substrates of checkpoint protein kinases and phosphatases, and how does the phosphorylated state of theses substrates modulate SAC activity? Are unattached kinetochores required to regulate p31comet activity? Different silencing mechanisms were proposed; do they constitute parallel networks or are they branches of a common pathway? Answers to these questions will significantly advance our understanding of SAC signalling. Finally, understanding how abnormalities in SAC function are linked to the process of tumourigenesis will provide important clues to promising therapeutic strategies that target SAC to kill cancer cells.
Acknowledgements
This work was supported by grant 04‐GBMC‐CICS‐09, from Cooperativa de Ensino Superior Politécnico e Universitário (CESPU), and by grant PTDS/SAU‐FCF/100930/2008 from Fundação para a Ciência e Tecnologia (FCT).
References
- 1. Sen S (2000) Aneuploidy and cancer. Curr. Opin. Oncol. 12, 82–88. [DOI] [PubMed] [Google Scholar]
- 2. Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanaka TU (2008) Bi‐orienting chromosomes: acrobatics on the mitotic spindle. Chromosoma 117, 521–533. [DOI] [PubMed] [Google Scholar]
- 4. Musacchio A, Salmon ED (2007) The spindle‐assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393. [DOI] [PubMed] [Google Scholar]
- 5. Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holy TE, Leibler S (1994) Dynamic instability of microtubules as an efficient way to search in space. Proc. Natl. Acad. Sci. USA 91, 5682–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore‐microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46. [DOI] [PubMed] [Google Scholar]
- 8. DeLuca JG, Dong Y, Hergert P, Strauss J, Hickey JM, Salmon ED et al. (2005) Hec1 and nuf2 are core components of the kinetochore outer plate essential for organizing microtubule attachment sites. Mol. Biol. Cell 16, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kline SL, Cheeseman IM, Hori T, Fukagawa T, Desai A (2006) The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J. Cell Biol. 173, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharp DJ, Rogers GC, Scholey JM (2000) Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat. Cell Biol. 2, 922–930. [DOI] [PubMed] [Google Scholar]
- 11. Vaisberg EA, Koonce MP, McIntosh JR (1993) Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J. Cell Biol. 123, 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaughan KT, Mikami A, Paschal BM, Holzbaur EL, Hughes SM, Echeverri CJ et al. (1996) Multiple mouse chromosomal loci for dynein‐based motility. Genomics 36, 29–38. [DOI] [PubMed] [Google Scholar]
- 13. Vorozhko VV, Emanuele MJ, Kallio MJ, Stukenberg PT, Gorbsky GJ (2008) Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma 117, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Z, Tulu US, Wadsworth P, Rieder CL (2007) Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr. Biol. 17, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karess R (2005) Rod‐Zw10‐Zwilch: a key player in the spindle checkpoint. Trends Cell Biol. 15, 386–392. [DOI] [PubMed] [Google Scholar]
- 16. Starr DA, Williams BC, Hays TS, Goldberg ML (1998) ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griffis ER, Stuurman N, Vale RD (2007) Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J. Cell Biol. 177, 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gassmann R, Essex A, Hu JS, Maddox PS, Motegi F, Sugimoto A et al. (2008) A new mechanism controlling kinetochore‐microtubule interactions revealed by comparison of two dynein‐targeting components: SPDL‐1 and the Rod/Zwilch/Zw10 complex. Genes Dev. 22, 2385–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan YW, Fava LL, Uldschmid A, Schmitz MH, Gerlich DW, Nigg EA et al. (2009) Mitotic control of kinetochore‐associated dynein and spindle orientation by human Spindly. J. Cell Biol. 185, 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barisic M, Sohm B, Mikolcevic P, Wandke C, Rauch V, Ringer T et al. (2010) Spindly/CCDC99 is required for efficient chromosome congression and mitotic checkpoint regulation. Mol. Biol. Cell 21, 1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hanisch A, Sillje HH, Nigg EA (2006) Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J. 25, 5504–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaitanos TN, Santamaria A, Jeyaprakash AA, Wang B, Conti E, Nigg EA (2009) Stable kinetochore‐microtubule interactions depend on the Ska complex and its new component Ska3/C13Orf3. EMBO J. 28, 1442–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guimaraes GJ, Deluca JG (2009) Connecting with Ska, a key complex at the kinetochore‐microtubule interface. EMBO J. 28, 1375–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kops GJ, Saurin AT, Meraldi P (2010) Finding the middle ground: how kinetochores power chromosome congression. Cell. Mol. Life Sci. 67, 2145–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED et al. (2006) Chromosomes can congress to the metaphase plate before biorientation. Science 311, 388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanaka TU (2005) Chromosome bi‐orientation on the mitotic spindle. Philos. Trans. R Soc. Lond. B Biol. Sci. 360, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly AE, Funabiki H (2009) Correcting aberrant kinetochore microtubule attachments: an Aurora B‐centric view. Curr. Opin. Cell Biol. 21, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nicklas RB, Ward SC, Gorbsky GJ (1995) Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J. Cell Biol. 130, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. King JM, Nicklas RB (2000) Tension on chromosomes increases the number of kinetochore microtubules but only within limits. J. Cell Sci. 113(Pt 21), 3815–3823. [DOI] [PubMed] [Google Scholar]
- 30. Liu D, Lampson MA (2009) Regulation of kinetochore‐microtubule attachments by Aurora B kinase. Biochem. Soc. Trans. 37, 976–980. [DOI] [PubMed] [Google Scholar]
- 31. Ruchaud S, Carmena M, Earnshaw WC (2007) The chromosomal passenger complex: one for all and all for one. Cell 131, 230–231. [DOI] [PubMed] [Google Scholar]
- 32. Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E et al. (2002) Evidence that the Ipl1‐Sli15 (Aurora kinase‐INCENP) complex promotes chromosome bi‐orientation by altering kinetochore‐spindle pole connections. Cell 108, 317–329. [DOI] [PubMed] [Google Scholar]
- 33. Cheeseman IM, Chappie JS, Wilson‐Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule‐binding site of the kinetochore. Cell 127, 983–997. [DOI] [PubMed] [Google Scholar]
- 34. Lan W, Zhang X, Kline‐Smith SL, Rosasco SE, Barrett‐Wilt GA, Shabanowitz J et al. (2004) Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr. Biol. 14, 273–286. [DOI] [PubMed] [Google Scholar]
- 35. Weaver BA, Cleveland DW (2005) Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell 8, 7–12. [DOI] [PubMed] [Google Scholar]
- 36. Thompson SL, Bakhoum SF, Compton DA (2010) Mechanisms of chromosomal instability. Curr. Biol. 20, R285–R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pinsky BA, Biggins S (2005) The spindle checkpoint: tension versus attachment. Trends Cell Biol. 15, 486–493. [DOI] [PubMed] [Google Scholar]
- 38. Logarinho E, Bousbaa H, Dias JM, Lopes C, Amorim I, Antunes‐Martins A et al. (2004) Different spindle checkpoint proteins monitor microtubule attachment and tension at kinetochores in Drosophila cells. J. Cell Sci. 117, 1757–1771. [DOI] [PubMed] [Google Scholar]
- 39. Li X, Nicklas RB (1995) Mitotic forces control a cell‐cycle checkpoint. Nature 373, 630–632. [DOI] [PubMed] [Google Scholar]
- 40. Stern BM, Murray AW (2001) Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr. Biol. 11, 1462–1467. [DOI] [PubMed] [Google Scholar]
- 41. Skoufias DA, Andreassen PR, Lacroix FB, Wilson L, Margolis RL (2001) Mammalian mad2 and bub1/bubR1 recognize distinct spindle‐attachment and kinetochore‐tension checkpoints. Proc. Natl. Acad. Sci. USA 98, 4492–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130, 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou J, Yao J, Joshi HC (2002) Attachment and tension in the spindle assembly checkpoint. J. Cell Sci. 115, 3547–3555. [DOI] [PubMed] [Google Scholar]
- 44. Gorbsky GJ, Ricketts WA (1993) Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J. Cell Biol. 122, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Waters JC, Chen RH, Murray AW, Salmon ED (1998) Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141, 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maresca TJ, Salmon ED (2010) Welcome to a new kind of tension: translating kinetochore mechanics into a wait‐anaphase signal. J. Cell Sci. 123, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McEwen BF, Dong Y (2009) Releasing the spindle assembly checkpoint without tension. J. Cell Biol. 184, 355–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khodjakov A, Rieder CL (2009) The nature of cell‐cycle checkpoints: facts and fallacies. J. Biol. 8, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bharadwaj R, Yu H (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23, 2016–2027. [DOI] [PubMed] [Google Scholar]
- 50. McCurdy JD, McElroy JS, Kopsell DA, Sams CE, Sorochan JC (2008) Effects of mesotrione on perennial ryegrass (Lolium perenne L.) carotenoid concentrations under varying environmental conditions. J. Agric. Food Chem. 56, 9133–9139. [DOI] [PubMed] [Google Scholar]
- 51. Kramer ER, Gieffers C, Holzl G, Hengstschlager M, Peters JM (1998) Activation of the human anaphase‐promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol. 8, 1207–1210. [DOI] [PubMed] [Google Scholar]
- 52. De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L et al. (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214–225. [DOI] [PubMed] [Google Scholar]
- 53. Luo X, Yu H (2008) Protein metamorphosis: the two‐state behavior of Mad2. Structure 16, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Skinner JJ, Wood S, Shorter J, Englander SW, Black BE (2008) The Mad2 partial unfolding model: regulating mitosis through Mad2 conformational switching. J. Cell Biol. 183, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu H (2006) Structural activation of Mad2 in the mitotic spindle checkpoint: the two‐state Mad2 model versus the Mad2 template model. J. Cell Biol. 173, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolanos‐Garcia VM, Blundell TL (2010) BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem. Sci. 36, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zich J, Hardwick KG (2010) Getting down to the phosphorylated ‘nuts and bolts’ of spindle checkpoint signalling. Trends Biochem. Sci. 35, 18–27. [DOI] [PubMed] [Google Scholar]
- 58. Tang Z, Shu H, Oncel D, Chen S, Yu H (2004) Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell 16, 387–397. [DOI] [PubMed] [Google Scholar]
- 59. Chen RH (2004) Phosphorylation and activation of Bub1 on unattached chromosomes facilitate the spindle checkpoint. EMBO J. 23, 3113–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamaguchi S, Decottignies A, Nurse P (2003) Function of Cdc2p‐dependent Bub1p phosphorylation and Bub1p kinase activity in the mitotic and meiotic spindle checkpoint. EMBO J. 22, 1075–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mao Y, Abrieu A, Cleveland DW (2003) Activating and silencing the mitotic checkpoint through CENP‐E‐dependent activation/inactivation of BubR1. Cell 114, 87–98. [DOI] [PubMed] [Google Scholar]
- 62. Huang H, Yen TJ (2009) BubR1 is an effector of multiple mitotic kinases that specifies kinetochore: microtubule attachments and checkpoint. Cell Cycle 8, 1164–1167. [DOI] [PubMed] [Google Scholar]
- 63. Malureanu LA, Jeganathan KB, Hamada M, Wasilewski L, Davenport J, van Deursen JM (2009) BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev. Cell 16, 118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang J, Ahmad S, Mao Y (2007) BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J. Cell Biol. 178, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dorer RK, Zhong S, Tallarico JA, Wong WH, Mitchison TJ, Murray AW (2005) A small‐molecule inhibitor of Mps1 blocks the spindle‐checkpoint response to a lack of tension on mitotic chromosomes. Curr. Biol. 15, 1070–1076. [DOI] [PubMed] [Google Scholar]
- 66. Schmidt M, Budirahardja Y, Klompmaker R, Medema RH (2005) Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 6, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM et al. (2008) Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132, 233–246. [DOI] [PubMed] [Google Scholar]
- 69. Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW (1996) Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 273, 953–956. [DOI] [PubMed] [Google Scholar]
- 70. Fuller BG, Stukenberg PT (2009) Cell division: righting the check. Curr. Biol. 19, R550–R553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vanoosthuyse V, Hardwick KG (2009) Overcoming inhibition in the spindle checkpoint. Genes Dev. 23, 2799–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL et al. (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155, 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sivaram MV, Wadzinski TL, Redick SD, Manna T, Doxsey SJ (2009) Dynein light intermediate chain 1 is required for progress through the spindle assembly checkpoint. EMBO J. 28, 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H (2004) Conformation‐specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 23, 3133–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H et al. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131, 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wassmann K, Liberal V, Benezra R (2003) Mad2 phosphorylation regulates its association with Mad1 and the APC/C. EMBO J. 22, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vanoosthuyse V, Hardwick KG (2009) A novel protein phosphatase 1‐dependent spindle checkpoint silencing mechanism. Curr. Biol. 19, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD et al. (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303. [DOI] [PubMed] [Google Scholar]
- 79. Kops GJ, Weaver BA, Cleveland DW (2005) On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 5, 773–785. [DOI] [PubMed] [Google Scholar]
- 80. Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W et al. (2001) MAD2 haplo‐insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409, 355–359. [DOI] [PubMed] [Google Scholar]
- 81. Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang‐Decker N, van Deursen JM (2003) Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S et al. (2004) Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 64, 440–445. [DOI] [PubMed] [Google Scholar]
- 83. Logarinho E, Bousbaa H (2008) Kinetochore‐microtubule interactions “in check” by Bub1, Bub3 and BubR1: the dual task of attaching and signalling. Cell Cycle 7, 1763–1768. [DOI] [PubMed] [Google Scholar]
- 84. Logarinho E, Resende T, Torres C, Bousbaa H (2008) The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore‐microtubule attachments. Mol. Biol. Cell 19, 1798–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Meraldi P, Sorger PK (2005) A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J. 24, 1621–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lampson MA, Kapoor TM (2005) The human mitotic checkpoint protein BubR1 regulates chromosome‐spindle attachments. Nat. Cell Biol. 7, 93–98. [DOI] [PubMed] [Google Scholar]
- 87. Tighe A, Johnson VL, Albertella M, Taylor SS (2001) Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gascoigne KE, Taylor SS (2008) Cancer cells display profound intra‐ and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122. [DOI] [PubMed] [Google Scholar]
- 89. Basu J, Bousbaa H, Logarinho E, Li Z, Williams BC, Lopes C et al. (1999) Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J. Cell Biol. 146, 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kops GJ, Foltz DR, Cleveland DW (2004) Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA 101, 8699–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bolanos‐Garcia VM (2009) Assessment of the mitotic spindle assembly checkpoint (SAC) as the target of anticancer therapies. Curr. Cancer Drug Targets 9, 131–141. [DOI] [PubMed] [Google Scholar]
- 92. Przewloka MR, Glover DM (2009) The kinetochore and the centromere: a working long distance relationship. Annu. Rev. Genet. 43, 439–465. [DOI] [PubMed] [Google Scholar]
- 93. Kops GJ (2008) The kinetochore and spindle checkpoint in mammals. Front Biosci. 13, 3606–3620. [DOI] [PubMed] [Google Scholar]
- 94. Santaguida S, Musacchio A (2009) The life and miracles of kinetochores. EMBO J. 28, 2511–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]