

Abstract
The relationship of cyclin‐dependent kinase inhibitor 3 (CDKN3) with tumours has previously been presented in a number of publications. However, the molecular network and interpretation of CDKN3 through the cell cycle between non‐malignancy associated hepatitis/cirrhosis and hepatocellular carcinoma (HCC) have remained to be elucidated. Here, we have constructed and analysed significant high expression gene CDKN3 activated and inhibited cell cycle networks from 25 HCC versus 25 non‐malignancy associated hepatitis/cirrhosis patients (viral infection HCV or HBV) in GEO Dataset GSE10140‐10141, by combination of a gene regulatory network inference method based on linear programming, and decomposition procedure using CapitalBio MAS 3.0 software, based on integration of public databases including Gene Ontology, KEGG, BioCarta, GenMapp, Intact, UniGene, OMIM, and others. Comparing the same and differently activated and inhibited CDKN3 networks with GO analysis, between non‐malignancy associated hepatitis/cirrhosis and HCC, our results suggest a CDKN3 cell cycle network (i) with stronger DNA replication and with weaker ubiquitin‐dependent protein catabolism as common characteristics in both non‐malignancy associated hepatitis/cirrhosis and HCC; (ii) with more cell division and weaker mitotic G2 checkpoint in non‐malignancy associated hepatitis/cirrhosis; (iii) with stronger cell cycle and weaker cytokinesis, as a result forming multinucleate cells in HCC. Thus, it is useful to identify CDKN3 cell cycle networks for comprehension of molecular mechanism between non‐malignancy associated hepatitis/cirrhosis and HCC transformation.
Introduction
Cyclin‐dependent kinase inhibitor 3 (CDKN3) is one of the genes our group has identified as highly expressed in 25 HCC versus 25 non‐malignancy associated hepatitis/cirrhosis cases. We data‐mined CDKN3 roles in cell components, molecular functions, biological processes, KEGG, GenMAPP, BioCarta and disease, from CapitalBio MAS 3.0 software (CapitalBio Corporation, Beijing, China). CDKN3 cell component is localized in perinuclear regions, molecular function is comprised of protein serine/threonine phosphatase activity, protein tyrosine phosphatase activity, protein binding, protein tyrosine • serine • threonine phosphatase activity and hydrolase activity, biological process includes regulation of cyclin‐dependent protein kinase activity, G1/S transition after mitosis in the cell cycle, cell cycle in general, cell cycle arrest, negative regulation of cell proliferation, dephosphorylation (GO (http://www.geneontology.org)). CDKN3 is relative to hs_1‐tissue‐endocrine_and_cns, regulation of protein kinase activity, regulation of transferase activity, protein amino acid dephosphorylation, negative regulation of cell proliferation and regulation of cell proliferation (GenMAPP (http://www.genmapp.org/)). Studies into CDKN3 relationships in tumours has already been presented in a number of papers (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13). However, distinct molecular networks and interpretation concerning CDKN3 in the cell cycle during non‐malignancy associated hepatitis/cirrhosis and hepatocellular carcinoma (HCC) transformation have remained to be elucidated.
HCC is one of the most common causes of cancer‐related death the world over. Thus, to develop novel drugs for treatment of HCC has become a challenge for biologists. Here, we have constructed and analysed significantly higher expression of the gene CDKN3 activated and inhibited in cell cycle networks from 25 HCC versus 25 non‐malignancy associated hepatitis/cirrhosis patients (viral infection HCV or HBV) in GEO Dataset GSE10140‐10141, by combination of gene regulatory network inference methods based on linear programming and decomposition procedures, with the CapitalBio MAS 3.0 software, based on integration of public databases including Gene Ontology, KEGG, BioCarta, GenMapp, Intact, UniGene, OMIM, and more.
Mechanisms that inhibit a signal are as important as mechanisms that initiate it. The GRNInfer tool (14) identifies molecular activation and inhibition relationships based on a novel mathematic method called Gene Network Reconstruction tool (GNR), using linear programming and a decomposition procedure for inferring gene networks. The method theoretically ensures derivation of the most consistent network structures with respect to all the datasets, thereby not only significantly alleviating the problem of data scarcity but also remarkably improving reconstruction reliability.
Here, we have compared the same and different, activated and inhibited, CDKN3 networks with GO analysis, between non‐malignancy associated hepatitis/cirrhosis and HCC. Our results suggest CDKN3 cell cycle network (i) with stronger DNA replication and weaker ubiquitin‐dependent protein catabolism, as common characteristics in both non‐malignancy associated hepatitis/cirrhosis and HCC; (ii) with the stronger cell division and the weaker G2 phase checkpoint in non‐malignancy associated hepatitis/cirrhosis; (iii) with stronger ell cycle and the weaker cytokinesis, as a result forming multinucleate cells in HCC. Thus, it is useful to be able to identify the CDKN3 cell cycle network for understanding molecular mechanisms between non‐malignancy associated hepatitis/cirrhosis and HCC transformation.
Materials and methods
Microarray data
We used microarrays containing 6144 genes from 25 non‐malignancy associated hepatitis/cirrhosis and 25 HCC patients, in the same GEO Dataset GSE10140‐10141 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10140, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10141) (15) and we pre‐processed raw microarray data as log 2.
Gene selection algorithms
Potential HCC molecular markers were identified using significant analysis of microarrays (SAM), which is a statistical technique for finding significant genes in a set of microarray experiments. Input to SAM comprised gene expression measurements from a set of microarray experiments, as well as a response variable from each experiment. The response variable might be a grouping such as untreated, treated, groups and so on. SAM computes a statistic d i for each gene i, and measures strength of relationship between gene expression and response variable. It uses repeated permutations of data to determine whether expression of any genes is significantly related to the response. Cut‐off for significance is determined by a tuning parameter delta (chosen by the user) based on the false positive rate. We normalized data by log 2, selected two classes, paired and minimum fold change ≥2, and chose significant (high expression genes of HCC compared to non‐malignancy associated hepatitis/cirrhosis) genes under the false‐discovery rate and q‐value being 0%. q‐value [invented by John Storey (16)] is similar to the well‐known P‐value, but adapted to multiple‐testing situations.
Molecule annotation system
Molecule Annotation System (MAS) is a web‐based software toolkit for whole data mining and function annotation solution, to extract & analyse biological molecule relationships from public database. MAS uses relational databases of biological networks created from millions of individually modelled relationships between genes, proteins, complexes, cells, disease and tissues. MAS allows view on our data, integrated in biological networks according to different kinds of biological context. This unique feature results from multiple lines of evidence, integrated in MASCORE. MAS helps comprehension of relationships of gene expression data through the given molecular symbols list, and provides thorough, unbiased and visible results.
Primary databases of MAS integrated various well‐known biological resources including Genbank, EMBL, SwissProt, Gene Ontology, KEGG, BioCarta, GenMapp, mirBase, EPD, HPRD, MIND, BIND, Intact, TRANSFAC, UniGene, dbSNP, OMIM, InterPro, HUGO, MGI and RGD. MAS offers various query entries and graphical results. The system represents an alternative approach to mining biological signification for high‐throughput array data (17).
Network establishment of candidate genes
The entire network was constructed using GRNInfer (42) and GVedit tools (http://www.graphviz.org/About.php). GRNInfer is a novel mathematical method called GNR based on linear programming and a decomposition procedure for inferring gene networks. The method theoretically ensures derivation of the most consistent network structure with respect to all of the datasets, thereby not only significantly alleviating the problem of data scarcity but also remarkably improving reconstruction reliability. Equation (1) represents all of the possible networks for the same dataset.
| (1) |
where J = (J ij)n×n = ∂f (x)/ ∂x is an n × n Jacobian matrix or connectivity matrix, X = (x(t 1), …, x(t m)), A = (a(t 1), …, a(t m) and X′ = (x′(t 1), …, x′(t m)) are all n × m matrices with x′i(t j) = [x i(t j + 1) − x i(t j)]/[t j +1 − t j] for i = 1, …, n; j = 1, …, m. X(t) = (x 1(t), …, x n(t))T∈R n, a = (a 1, …, a n)T∈R n, x i(t) is the expression level (mRNA concentrations) of gene i at time instance t. y = (y ij) is an n × n matrix, where y ij is zero if e j ≠ 0 and is otherwise an arbitrary scalar coefficient. ∧−1 = diag (1/e i) and 1/e is set to be zero if e i = 0. U is a unitary m × n matrix of left eigenvectors, ∧ = diag (e 1, …, e n) is a diagonal n × n matrix containing the n eigenvalues and V T is the transpose of a unitary n × n matrix of right eigenvectors. We established network based on the fold change ≥2 distinguished genes and selected parameters as lambda 0.0 because we used one dataset and tried several thresholds 1, 0.5, 0.1, 0.001, 0.0001, 0.000001, 0.0000001, 0.00000001, 0.000000001. Lambda was a positive parameter, which balanced the matching and sparsity terms in the objective function. Using different thresholds, we could predict various networks with different edge density.
Results
Identification of HCC novel molecular markers
We normalized data by log 2, selected two classes paired and minimum fold change ≥2, and chose the significant (high expression genes of HCC compared non‐malignancy associated hepatitis/cirrhosis) genes under false‐discovery rate and q‐value being 0%. We obtained 225 significant high expression molecules (fold change ≥2) from 6144 genes of 25 HCC versus 25 non‐malignancy associated hepatitis/cirrhosis in the same GEO Dataset GSE10140‐10141 containing NEK2, NUSAP1, CAD, DLGAP5, LCN2, SFRP4, RRM2, HIST1H3H, TAGLN2, MYBL2, TK1, PRCC, E2F1, ACTN2, etc. as shown in Table 1.
Table 1.
Abbreviations of 225 significant high expression genes (fold≥2) of HCC compared with human non‐malignancy associated hepatitis/cirrhosis
| Gene | Gene | Gene | Gene | Gene | Gene | Gene | Gene | Gene | Gene |
|---|---|---|---|---|---|---|---|---|---|
| ACTG2 | CAMK1 | CHST1 | DKK1 | GJA5 | KCNQ3 | MAN2A1 | MYBL2 | PAGE4 | RBCK1 |
| ACTN2 | CBX5 | CIAO1 | DLGAP5 | GML | KCTD2 | MAOA | MYCN | PCOLCE2 | RBM34 |
| ADAMDEC1 | CCL20 | CKS1B | E2F1 | GNAZ | KIAA0101 | MAP2 | MYH6 | PHLDA2 | REG1A |
| AFP | CCNA2 | CLIC1 | ECT2 | GNG10 | KIAA0513 | MAP2K6 | MYOM1 | PIGC | REG3A |
| AKR1B10 | CCNB1 | CNTNAP2 | EFNA1 | GPC3 | KIAA0859 | MAP4K4 | NAT9 | PLA2G1B | RFC4 |
| ALDH3A1 | CCNB2 | CORO2A | EIF1AX | GPSM2 | KLHL35 | MAPK3 | NCAPH | PLK4 | RIMS3 |
| ALK | CCNE2 | CPD | ELAVL3 | GRM1 | KLRC3 | MAPT | NEK2 | POLD1 | RNF185 |
| AMELY | CD34 | CRYGA | ENAH | HIST1H2AD | KPNA2 | MCM2 | NFKBIB | PPP1R12B | RNF2 |
| ARHGDIG | CDC2 | CSPG4 | EPHA4 | HIST1H2AG | LAPTM4B | MCM4 | NINJ2 | PRCC | ROBO1 |
| B4GALNT2 | CDC20 | CST6 | ESM1 | HIST1H2BJ | LCN2 | MCM7 | NKX2‐5 | PRKCG | RRM2 |
| BAP1 | CDC6 | CSTB | ESPL1 | HIST1H3H | LEF1 | MDK | NOTCH3 | PRKG2 | RRP1B |
| BCAT1 | CDH13 | CSTF2 | EYA1 | HMGB2 | LGALS3 | MELK | NOTCH4 | PROK1 | S100P |
| BIRC5 | CDKN2C | CTHRC1 | F13A1 | HOMER2 | LLGL2 | METAP2 | NQO1 | PRSS1 | SBF1 |
| BLVRA | CDKN3 | CYP17A1 | FGF9 | HOXA5 | LOX | MKRN3 | NR5A1 | PSMC3IP | SCGB1D2 |
| BRCA1 | CEBPA | CYP21A2 | FKBP1B | HOXD4 | LTBP2 | MMP11 | NRXN3 | PTH2R | SCML2 |
| BUB1B | CELSR2 | CYP27B1 | FOLR1 | IGF2BP3 | LTBP3 | MMP9 | NTN1 | PTHLH | SEMA3B |
| C4orf8 | CENPF | CYP51A1 | FOXM1 | IRF5 | LYPD3 | MRPL49 | NUP62 | PTTG1 | SERPINB2 |
| C9orf127 | CHAF1A | DDX10 | GALK1 | ISG20 | MAGI1 | MS4A1 | NUSAP1 | PVRL2 | SFRP4 |
| CAD | CHL1 | DDX11 | GAS7 | ITGA2 | ZNF43 | MS4A2 | OCRL | RAB3B | SFTPA2B |
| ZIC2 | CHRNA4 | DFFB | GDPD5 | KATNB1 | ZWINT | MUTYH | ORC1L | RABGGTA | SLC16A3 |
| ORC6L | TPSD1 | TRAF2 | TRIP13 | TSHB | TSTA3 | TUBG1 | UNG | VDR | XRCC2 |
| TOP2A | TPST2 | TRIM26 | TROAP | TSR1 | TTK | UBE2C | VCAN | WDR1 | YWHAE |
| SLC4A3 | SORT1 | SPINK1 | SQLE | ST6GALNAC2 | STX1A | SULT1C4 | TAGLN2 | TBL3 | TK1 |
| SLC6A12 | SOX2 | SPON2 | SSTR5 | STMN1 | SULT1C2 | SYN2 | TANK | TCAP | TNFRSF9 |
| TP53I11 |
Candidate novel activated and inhibited genes of CDKN3 upstream and downstream network in human non‐malignancy associated hepatitis/cirrhosis and HCC by GRNInfer
We assayed GRNInfer at several thresholds 1, 0.5, 0.1, 0.001, 0.0001, 0.000001, 0.0000001, 0.00000001, 0.000000001 and finally, selected threshold 0.000000001 as its results covered CDKN3 pathway by CapitalBio MAS 3.0 software, from the published data. We identified candidate genes of CDKN3 upstream and downstream networks from our constructed total network between non‐malignancy associated hepatitis/cirrhosis and HCC by GRNInfer separately, as shown in Table 2.
Table 2.
Candidate upstream and downstream genes of CDKN3 networks in human non‐malignancy associated hepatitis/cirrhosis and HCC transformation by GRNInfer respectively. control: human non‐malignancy associated hepatitis/cirrhosis, experiment: HCC patients
| Control | Experiment |
|---|---|
| Upstream | |
| NUSAP1, DLG7, LCN2, SFRP4, RRM2, TROAP, HIST1H3H, TAGLN2, MYBL2, TK1, PRCC, E2F1, BRCA1, CENPF, SCML2, BIRC5, TOP2A, SPINK1, MDK, GNG10, FOXM1, TRAF2, SPON2, RAB3B, ECT2, KIAA0859, PIGC, TTK, CDC20, GPSM2, PLK4, TSTA3, VDR, MCM2, CIAO1, XRCC2, MELK, MAP4K4, CCNE2, AMELY, CDC6, SEMA3B, TUBG1, KIAA0513, CSTF2, TNFRSF9, IGF2BP3, NCAPH, YWHAE, SBF1, PVRL2, ORC1L, EIF1AX, ZWINT, GDPD5, GRM1, ARHGDIG, MCM4, BUB1B, ACTN2, RBCK1, DDX10, C9orf127, PTTG1, ACTG2, CLIC1, RNF185, CCNA2, RBM34, LTBP2, SORT1, ZIC2, CDH13, GPC3, ST6GALNAC, CYP21A2, CEBPA, PRKCG, HIST1H2AG, UBE2C, CCNB2, CST6, NAT9, MYH6, HOXD4, CAMK1, CNTNAP2, MUTYH, PSMC3IP, SFTPA2, ALK, CDC2, CD34, TP53I11, CHST1, NOTCH3, HMGB2, BLVRA, LGALS3, MKRN3, CHAF1A, KPNA2, ITGA2, CELSR2, SLC16A3, GNAZ, KCTD2, MAPK3, GJA5, RRP1B, BAP1, CAD, HIST1H2BJ, REG3A, ESM1, WDR1, TRIP13, GML, CKS1B, ROBO1, ORC6L, FGF9, SYN2, MYCN, AFP, CCL20, LEF1, SERPINB2, IRF5, BCAT1, PRKG2, REG1A, TPST2, S100P, NR5A1, HIST1H2AD, MMP9, PTHLH, SQLE, PHLDA2, CSPG4, CCNB1, CRYGA, STX1A, UNG, SULT1C2, MYOM1, MAOA, ZNF43, FKBP1B, HOXA5, GAS7, CYP51A1, EYA1, PRSS1, CHRNA4, KATNB1, MAN2A1, OCRL, ALDH3A1, F13A1, VCAN, STMN1, FLJ33790, EFNA1, PLA2G1B, KCNQ3, PAGE4, TCAP, CDKN2C, NQO1, CYP17A1, KIAA0101, TSHB, KLRC3, ESPL1, MRPL49, RIMS3, NKX2‐5, PTHR2, NTN1, CTHRC1, TBL3, MMP11, PROK1, TSR1, ADAMDEC1, MS4A2, SCGB1D2, PCOLCE2, NUP62 NINJ2 LAPTM4B DMN RABGGTA SOX2 SSTR5 NRXN3 CDKN3 | NEK2, NUSAP1, DLG7, LCN2, SFRP4, RRM2, TROAP, HIST1H3H, TAGLN2, MYBL2, TK1, PRCC, E2F1, BRCA1, CENPF, SCML2, TOP2A, SPINK1, MDK, GNG10, FOXM1, TRAF2, SPON2, RAB3B, ECT2, ENAH, SLC4A3, TTK, CDC20, GPSM2, PLK4, TSTA3, MAPT, VDR, MCM2, MELK, MAP4K4, CCNE2, MAP2, AMELY, SEMA3B, TUBG1, CSTF2, CPD, TNFRSF9, IGF2BP3, NCAPH, YWHAE, SBF1, PVRL2, EIF1AX, ZWINT, GDPD5, GRM1, ARHGDIG, BUB1B, GALK1, ACTN2, RBCK1, DDX10, C9orf127, PTTG1, ACTG2, CLIC1, RNF185, CCNA2, RBM34, LTBP2, SORT1, ZIC2, CDH13, GPC3, CYP21A2, CEBPA, PRKCG, HIST1H2AG, UBE2C, CCNB2, CST6, NAT9, MYH6, SFTPA2, ALK, CDC2, CD34, NFKBIB, TP53I11, NOTCH3, HMGB2, FOLR1, BLVRA, LGALS3, RFC4, MKRN3, CHAF1A, ITGA2, EPHA4, CELSR2, SLC16A3, GNAZ, MAPK3, TPSD1, GJA5, BAP1, CAD, HIST1H2BJ, PPP1R12B, REG3A, ESM1, WDR1, LYPD3, GML, CKS1B, ROBO1, FGF9, MYCN, ELAVL3, AFP, CCL20, CBX5, LEF1, IRF5, PRKG2, REG1A, C4orf8, TPST2, S100P, NR5A1, MAP2K6, CHL1, HIST1H2AD, MMP9, PTHLH, SQLE, PHLDA2, CSPG4, CCNB1, CRYGA, STX1A, SULT1C2, MYOM1, MAOA, ZNF43, GAS7, EYA1, PRSS1, CHRNA4, KATNB1, MAN2A1, OCRL, ALDH3A1, F13A1, STMN1, EFNA1, PLA2G1B, KCNQ3, PAGE4, TCAP, CDKN2C, NQO1, CYP17A1, KIAA0101, TSHB, KLRC3, DKK1, RIMS3, NKX2‐5, PTHR2, NTN1, TBL3, MMP11, PROK1, TSR1, ADAMDEC1, PCOLCE2, NUP62, NINJ2, LAPTM4B, RABGGTA, SOX2, SSTR5, NRXN3, CDKN3 |
| Downstream | |
| NEK2, NUSAP1, DLG7, LCN2, SFRP4, RRM2, TROAP, TAGLN2, MYBL2, TK1, PRCC, E2F1, BRCA1, CENPF, SCML2, BIRC5, TOP2A, SPINK1, MDK, GNG10, FOXM1, TRAF2, SPON2, RAB3B, ECT2, KIAA0859, ENAH, SLC4A3, TTK, CDC20, GPSM2, PLK4, TSTA3, MAPT, VDR, MCM2, CIAO1, XRCC2, MELK, MAP4K4, CCNE2, MAP2, AMELY, CDC6, SEMA3B, TUBG1, KIAA0513, CSTF2, CPD, TNFRSF9, IGF2BP3, NCAPH, YWHAE, SBF1, PVRL2, ORC1L, EIF1AX, ZWINT, GDPD5, GRM1, ARHGDIG, MCM4, LLGL2, BUB1B, GALK1, ACTN2, RBCK1, DDX10, C9orf127, PTTG1, ACTG2, CLIC1, RNF185, CCNA2, RBM34, LTBP2, SORT1, ZIC2, CDH13, GPC3, ST6GALNAC, CYP21A2, AKR1B10, CEBPA, PRKCG, HIST1H2AG, UBE2C, CCNB2, CST6, NAT9, MYH6, HOXD4, CAMK1, CNTNAP2, MUTYH, SFTPA2, ALK, CDC2, CD34, NFKBIB, TP53I11, CHST1, NOTCH3, HMGB2, FOLR1, BLVRA, LGALS3, RFC4, MKRN3, CHAF1A, KPNA2, ITGA2, EPHA4, CELSR2, SLC16A3, GNAZ, KCTD2, MAPK3, TPSD1, GJA5, RRP1B, BAP1, CAD, HIST1H2BJ, PPP1R12B, REG3A, ESM1, LYPD3, TRIP13, GML, CKS1B, ROBO1, ORC6L, FGF9, SYN2, MYCN, ELAVL3, AFP, CCL20, CBX5, LEF1, SERPINB2, IRF5, ISG20, BCAT1, SLC6A12, REG1A, C4orf8, TPST2, S100P, NR5A1, MAP2K6, CHL1, HIST1H2AD, MMP9, CORO2A, SQLE, PHLDA2, CSPG4, CCNB1, CRYGA, STX1A, UNG, SULT1C2, MYOM1, MAOA, ZNF43, FKBP1B, HOXA5, GAS7, CYP51A1, EYA1, PRSS1, CHRNA4, MAN2A1, OCRL, ALDH3A1, F13A1, VCAN, STMN1, FLJ33790, EFNA1, PLA2G1B, KCNQ3, PAGE4, TCAP, CDKN2C, NQO1, CYP17A1, KIAA0101, TSHB, KLRC3, DKK1, ESPL1, MRPL49, NKX2‐5, PTHR2, NTN1, CTHRC1, TBL3, MMP11, PROK1, TSR1, ADAMDEC1, MS4A2, SCGB1D2, PCOLCE2, NUP62, NINJ2, LAPTM4B, DMN, RABGGTA, LOX, SOX2, SSTR5, NRXN3, CDKN3 | NEK2, NUSAP1, DLG7, LCN2, SFRP4, RRM2, HIST1H3H, TAGLN2, MYBL2, TK1, PRCC, E2F1, BRCA1, CENPF, SCML2, BIRC5, TOP2A, SPINK1, MDK, GNG10, FOXM1, RAB3B, ECT2, KIAA0859, PIGC, SLC4A3, TTK, CDC20, GPSM2, PLK4, TSTA3, MAPT, VDR, MCM2, CIAO1, XRCC2, MELK, MAP4K4, CCNE2, MAP2, AMELY, CDC6, SEMA3B, TUBG1, KIAA0513, CSTF2, CPD, TNFRSF9, IGF2BP3, NCAPH, YWHAE, SBF1, PVRL2, ORC1L, EIF1AX, GDPD5, GRM1, ARHGDIG, MCM4, LLGL2, BUB1B, GALK1, ACTN2, RBCK1, DDX10, C9orf127, PTTG1, ACTG2, CLIC1, RNF185, CCNA2, RBM34, LTBP2, SORT1, CDH13, GPC3, ST6GALNAC, CYP21A2, AKR1B10, PRKCG, HIST1H2AG, UBE2C, CCNB2, CST6, NAT9, MYH6, HOXD4, CAMK1, CNTNAP2, MUTYH, PSMC3IP, ALK, CDC2, CD34, NFKBIB, TP53I11, NOTCH3, HMGB2, FOLR1, BLVRA, LGALS3, RFC4, MKRN3, CHAF1A, KPNA2, ITGA2, EPHA4, CELSR2, SLC16A3, GNAZ, KCTD2, MAPK3, TPSD1, GJA5, RRP1B, CAD, HIST1H2BJ, PPP1R12B, REG3A, ESM1, WDR1, LYPD3, TRIP13, GML, CKS1B, ROBO1, ORC6L, SYN2, MYCN, ELAVL3, AFP, CCL20, CBX5, LEF1, SERPINB2, IRF5, ISG20, BCAT1, PRKG2, REG1A, C4orf8, TPST2, S100P, NR5A1, MAP2K6, CHL1, HIST1H2AD, MMP9, CORO2A, PTHLH, SQLE, PHLDA2, CSPG4, CCNB1, CRYGA, STX1A, UNG, SULT1C2, MYOM1, MAOA, FKBP1B, HOXA5, GAS7, CYP51A1, EYA1, PRSS1, CHRNA4, KATNB1, MAN2A1, OCRL, ALDH3A1, F13A1, VCAN, STMN1, FLJ33790, EFNA1, PLA2G1B, KCNQ3, PAGE4, TCAP, CDKN2C, NQO1, CYP17A1, KIAA0101, TSHB, KLRC3, DKK1, ESPL1, MRPL49, RIMS3, NKX2‐ 5, PTHR2, NTN1, CTHRC1, TBL3, MMP11, PROK1, TSR1, ADAMDEC1, MS4A2, SCGB1D2, PCOLCE2, NUP62, NINJ2, LAPTM4B, DMN, RABGGTA, LOX, SOX2, SSTR5, NRXN3, CDKN3 |
Discussion
Our aim here was to compare and analyse novel CDKN3 cell cycle networks between non‐malignancy associated hepatitis/cirrhosis and HCC transformation for potential novel markers for prognosis and therapy of HCC. On the basis of our previous published novel molecular network constructions and functional comparison from different databases presented in our papers (18, 19, 20, 21, 22, 23, 24, 25, 26, 27), we constructed and analysed significant higher expression gene CDKN3 activated and inhibited cell cycle network from 25 HCC versus 25 non‐malignancy associated hepatitis/cirrhosis patients (viral infection HCV or HBV) in GEO Dataset GSE10140‐10141 by a combination of gene regulatory network inference methods based on linear programming and decomposition procedure with the CapitalBio MAS 3.0 software, based on integration of public databases including Gene Ontology, KEGG, BioCarta, GenMapp, Intact, UniGene, OMIM, and more. We identified some same and other different novel activated and inhibited upstream and downstream genes of CDKN3’s cell cycle module between non‐malignancy associated hepatitis/cirrhosis and HCC, on the condition that our CDKN3 network covered CDKN3 pathways and matched cell cycle enrichment analysis by the CapitalBio MAS 3.0 software, from published data.
To confirm our prediction for covering published data, we setup CDKN3 interaction and pathways in HCC using CapitalBio MAS 3.0 software for standard comparison with our CDKN3 network. We identified CDKN3 interaction and pathways from our total established network by inputting 225 significant high expression genes (fold change ≥2) to the CapitalBio MAS 3.0 software based on integration of public databases including Gene Ontology, KEGG, BioCarta, GenMapp, Intact, UniGene, OMIM, and more. CDKN3 interaction molecules and genes included CDC2, CDK3, CDK2, CEBPA, CDC25A, CDKN3, MS4A3, CDC28_YEAST. CDKN3 pathway molecules consisted of SBF1, EYA1, MCM2, MCM7, UBE2C, CHAF1A, CKS1B, SPINK1, MAPT, CYP21A2, REG3A, REG1A, STX1A, PRSS1, PLA2G1B, CYP17A1, TSHB, RIMS3, CCNE2, TNFRSF9, BAP1, GML, PTHLH, SSTR5, TTK, CIAO1, DDX11, BRCA1, BUB1B, CCNA2, CDC6, CDKN2C. By comparison of similarities, we observed that our high expression molecules in HCC did not contain CDKN3 interaction proteins, whereas they completely covered CDKN3 pathway proteins (28).
To establish candidate novel genes concerned CDKN3 upstream and downstream network covering CDKN3 pathways, we assayed GRNInfer in several thresholds 1, 0.5, 0.1, 0.001, 0.0001, 0.000001, 0.0000001, 0.00000001, 0.000000001. Finally, we selected threshold 0.000000001 and set up candidate genes of the CDKN3 network between non‐malignancy associated hepatitis/cirrhosis and HCC by GRNInfer (Table 2). Our GRNInfer finding was verified covering CDKN3 pathways using the CapitalBio MAS 3.0 software from published data.
To construct novel CDKN3 cell cycle modules between non‐malignancy associated hepatitis/cirrhosis and HCC respectively by GRNInfer, our CDKN3 network needed to match cell cycle enrichment analysis. We identified cell cycle enrichment from our total established enrichment results by inputting 225 significant high expression genes (fold change ≥2) to the CapitalBio MAS 3.0 software based on integration of public databases including Gene Ontology, KEGG, BioCarta, GenMapp, Intact, UniGene, OMIM, and more. We data‐mined molecules and genes of cell cycle enrichment including NEK2, NUSAP1, CDKN3, DLGAP5, BIRC5, CDC20, MCM2, MCM7, CCNE2, CDC6, ZWINT, LLGL2, PTTG1, CCNA2, UBE2C, CCNB2, CDC2, CHAF1A, MAPK3, CKS1B, DDX11, CCNB1, KATNB1, CDKN2C. On the basis of cell cycle enrichment, we predicted novel candidate activated and inhibited upstream and downstream networks of CDKN3 cell cycle components between non‐malignancy associated hepatitis/cirrhosis and HCC by GRNInfer separately, as shown in 1, 2. Study into ion binding relationship in tumour cells is presented previously in many papers (28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46). However, implications concerning CDKN3 cell cycle networks between non‐malignancy associated hepatitis/cirrhosis and HCC transformation remained to be elucidated. We further compared and interpreted candidate molecules of CDKN3 cell cycle modules between non‐malignancy associated hepatitis/cirrhosis and HCC transformation considering activation and inhibition relationships (29).
Figure 1.
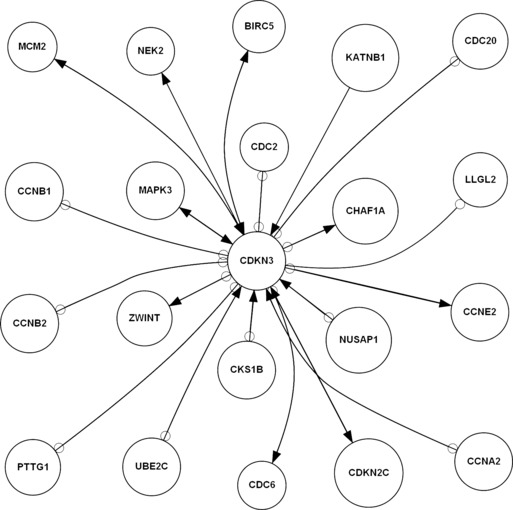
Candidate activated and inhibited upstream and downstream network of CDKN3 in the cell cycle in non‐malignancy associated hepatitis/cirrhosis by GRNInfer. Arrowhead represents activation relationship, empty circle represents inhibition relationship.
Figure 2.
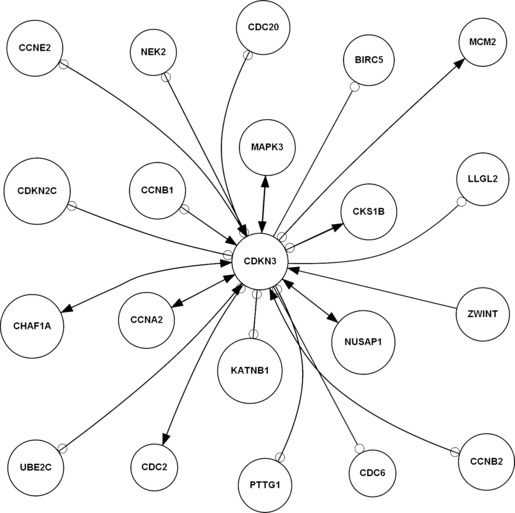
Candidate activated and inhibited upstream and downstream network of CDKN3 in the cell cycle in HCC by GRNInfer. Arrowhead represents activation relationship, empty circle represents inhibition relationship.
First, we identified the same novel molecules of CDKN3 cell cycle modules in both non‐malignancy associated hepatitis/cirrhosis and HCC. The same upstream NUSAP1, CCNA2, UBE2C, MAPK3 activated CDKN3, and CDC20, PTTG1, CDC2 inhibited CDKN3. The same downstream CDKN3 activated MCM2, CHAF1A, MAPK3 and inhibited CDC20, LLGL2, PTTG1, UBE2C, CCNB2, CCNB1 in both non‐malignancy associated hepatitis/cirrhosis and HCC. To interpret molecular mechanisms of CDKN3 cell cycle modules between non‐malignancy associated hepatitis/cirrhosis and HCC transformation, we analysed GO of the same activated and inhibited molecules. For example, activated MCM2 between non‐malignancy associated hepatitis/cirrhosis and an HCC biological process is relevant to DNA replication, DNA unwinding during replication, DNA replication initiation, nucleosome assembly, transcription, regulation of transcription, DNA‐dependency and cell cycle progression. Inhibited CDC20 between non‐malignancy associated hepatitis/cirrhosis and HCC biological processes is involved in ubiquitin‐dependent protein catabolism, cell cycle progression, mitosis, anaphase‐promoting complex‐dependent proteasomal ubiquitin‐dependent protein catabolism, cell division, negative regulation of ubiquitin ligase activity during mitotic cell cycle and positive regulation of ubiquitin ligase activity during mitosis of the cell cycle. Compared to the same and different activated and inhibited CDKN3 networks with GO analysis between non‐malignancy associated hepatitis/cirrhosis and HCC, our results suggested that the same molecular network of CDKN3 cell cycle modules maybe stronger DNA replication and weaker ubiquitin‐dependent protein catabolism, as common characteristics in both non‐malignancy associated hepatitis/cirrhosis and HCC.
Secondly, we identified the different novel molecules of CDKN3 cell cycle modules between non‐malignancy associated hepatitis/cirrhosis compared to HCC. The different upstream BIRC5, MCM2, CKS1B, KATNB1, CDKN2C activated CDKN3, and CCNE2, CDC6, ZWINT, CCNB2, CHAF1A, CCNB1 inhibited CDKN3; different downstream CDKN3 activated NEK2, BIRC5, CCNE2, CDC6, ZWINT, CDKN2C and inhibited NUSAP1, CCNA2, CDC2, CKS1B in non‐malignancy associated hepatitis/cirrhosis. To further interpret molecular mechanisms of CDKN3 in cell cycle modules of non‐malignancy associated hepatitis/cirrhosis, we analysed GO of the different activated and inhibited molecules from non‐malignancy associated hepatitis/cirrhosis. For example, activated NEK2 in the non‐malignancy associated hepatitis/cirrhosis biological process is relevant to cell cycle progression, regulation of mitosis, meiosis, protein amino acid autophosphorylation, centrosome separation and cell division. Inhibited CCNA2 in the HCC biological process is involved in cell cycle progression, mitosis, mitotic G2 checkpoint, positive regulation of transcription, cell division. Compared to the same and different activated & inhibited CDKN3 networks with GO analysis, our results demonstrated different molecular networks of CDKN3 cell cycle modules, stronger cell division and the weaker mitotic G2 checkpoint as the only characteristics in non‐malignancy associated hepatitis/cirrhosis.
Thirdly, we identified the different novel molecules of CDKN3 cell cycle modules in HCC compared to non‐malignancy associated hepatitis/cirrhosis. The different upstream NEK2, CCNE2, ZWINT, CCNB2, CHAF1A, CCNB1 activated CDKN3, and MCM2, CKS1B, KATNB1, CDKN2C inhibited CDKN3; different downstream CDKN3 activated NUSAP1, CCNA2, CDC2, CKS1B and inhibited NEK2, BIRC5, CCNE2, CDC6, KATNB1, CDKN2C in HCC. To interpret further molecular mechanisms of the CDKN3 cell cycle module in HCC, we analysed GO of the different activated and inhibited molecules from HCC. For example, activated NUSAP1 in the HCC biological process is relevant to cytokinesis after mitosis, cell cycle progression, mitotic chromosome condensation, establishment of mitotic spindle localization, positive regulation of mitosis and cell division progression. Inhibited BIRC5 in the HCC biological process is involved in G2/M transition of during the cell cycle, cytokinesis, apoptosis, anti‐apoptosis, cell cycle progression, mitosis, protein complex localization, positive regulation of exit from mitosis, spindle checkpoint, negative regulation of caspase activity, positive regulation of progression through mitosis of the cell cycle and establishment of chromosome localization. Compared to the same and different activated and inhibited CDKN3 networks with GO analysis, our results indicate different molecular network of CDKN3 cell cycle modules, stronger ell cycle progression and weaker cytokinesis, and as a result formation of multinucleate cells in HCC.
In conclusion, our high expression molecules of HCC did not contain CDKN3 interaction proteins, whereas they completely covered CDKN3 pathway proteins by comparison of similarities. We identified same and different novel activated and inhibited upstream and downstream genes related to CDKN3 in the cell cycle module between non‐malignancy associated hepatitis/cirrhosis and HCC, on condition that our CDKN3 network covered CDKN3 pathways and matched cell cycle enrichment analysis by the CapitalBio MAS 3.0 software from published data. Comparing same and different activated & inhibited CDKN3 networks with GO analysis between non‐malignancy associated hepatitis/cirrhosis and HCC, our results suggested CDKN3 cell cycle networks (1) with stronger DNA replication and weaker ubiquitin‐dependent protein catabolism as common characteristics in both non‐malignancy associated hepatitis/cirrhosis and HCC; (2) with stronger cell division and the weaker mitotic G2 checkpoint in non‐malignancy associated hepatitis/cirrhosis and (3) with stronger cell cycle and weaker cytokinesis ‐ and as a result formation of multinucleate cells in HCC. Thus, it is most useful to identify CDKN3 cell cycle networks for understanding of molecular mechanisms between non‐malignancy associated hepatitis/cirrhosis and HCC transformation.
Acknowledgements
This work was supported by the National Natural Science Foundation in China (No.60871100). The Returned Overseas Chinese Scholars for Scientific research Foundation of State Education Ministry. Significant Science and Technology Project for new transgenic biological Species (2009ZX08012‐001B). State Key Lab of Pattern Recognition Open Foundation.
All authors contributed equally to this study.
References
- 1. Dai Y, Grant S (2006) CDK inhibitor targets: a hit or miss proposition?: cyclin‐dependent kinase inhibitors kill tumor cells by downregulation of anti‐apoptotic proteins. Cancer Biol. Ther. 5, 171–173. [DOI] [PubMed] [Google Scholar]
- 2. Doganay L, Altaner S, Bilgi S, Kaya E, Ekuklu G, Kutlu K (2003) Expression of the cyclin‐dependent kinase inhibitor p27 in transitional cell bladder cancers: is it a good predictor for tumor behavior? Int. Urol. Nephrol. 35, 181–188. [DOI] [PubMed] [Google Scholar]
- 3. Evangelou K, Bramis J, Peros I, Zacharatos P, Dasiou‐Plakida D, Kalogeropoulos N et al. (2004) Electron microscopy evidence that cytoplasmic localization of the p16(INK4A) “nuclear” cyclin‐dependent kinase inhibitor (CKI) in tumor cells is specific and not an artifact. A study in non‐small cell lung carcinomas. Biotech. Histochem. 79, 5–10. [DOI] [PubMed] [Google Scholar]
- 4. Gong Z, Fu J (2001) [Cyclin‐dependent kinase 4 inhibitor a and tumor]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 18, 219–221. [PubMed] [Google Scholar]
- 5. Iurisci I, Filipski E, Reinhardt J, Bach S, Gianella‐Borradori A, Iacobelli S et al. (2006) Improved tumor control through circadian clock induction by Seliciclib, a cyclin‐dependent kinase inhibitor. Cancer Res. 66, 10720–10728. [DOI] [PubMed] [Google Scholar]
- 6. Koutsodontis G, Tentes I, Papakosta P, Moustakas A, Kardassis D (2001) Sp1 plays a critical role in the transcriptional activation of the human cyclin‐dependent kinase inhibitor p21(WAF1/Cip1) gene by the p53 tumor suppressor protein. J. Biol. Chem. 276, 29116–29125. [DOI] [PubMed] [Google Scholar]
- 7. Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J et al. (2003) The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin‐dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell. Biol. 23, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le XF, Claret FX, Lammayot A, Tian L, Deshpande D, LaPushin R et al. (2003) The role of cyclin‐dependent kinase inhibitor p27Kip1 in anti‐HER2 antibody‐induced G1 cell cycle arrest and tumor growth inhibition. J. Biol. Chem. 278, 23441–23450. [DOI] [PubMed] [Google Scholar]
- 9. Li G, Hundemer M, Wolfrum S, Ho AD, Goldschmidt H, Witzens‐Harig M (2006) Identification and characterization of HLA‐class‐I‐restricted T‐cell epitopes in the putative tumor‐associated antigens P21‐activated serin kinase 2 (PAK2) and cyclin‐dependent kinase inhibitor 1A (CDKN1A). Ann. Hematol. 85, 583–590. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Poi MJ, Qin D, Selby TL, Byeon IJ, Tsai MD (2000). Tumor suppressor INK4: quantitative structure‐function analyses of p18INK4C as an inhibitor of cyclin‐dependent kinase 4. Biochemistry 39, 649–657. [DOI] [PubMed] [Google Scholar]
- 11. Li X, Tanaka K, Nakatani F, Matsunobu T, Sakimura R, Hanada M et al. (2005) Transactivation of cyclin E gene by EWS‐Fli1 and antitumor effects of cyclin dependent kinase inhibitor on Ewing’s family tumor cells. Int. J. Cancer 116, 385–394. [DOI] [PubMed] [Google Scholar]
- 12. Li Y, Tanaka K, Li X, Okada T, Nakamura T, Takasaki M et al. (2007) Cyclin‐dependent kinase inhibitor, flavopiridol, induces apoptosis and inhibits tumor growth in drug‐resistant osteosarcoma and Ewing’s family tumor cells. Int. J. Cancer 121, 1212–1218. [DOI] [PubMed] [Google Scholar]
- 13. Maziere C, Marcheux V, Louandre C, Maziere JC (2002) Oxidized low density lipoprotein induces the cyclin‐dependent kinase inhibitor p21(waf1) and the tumor suppressor Rb. Biochem. Biophys. Res. Commun. 293, 1327–1332. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Joshi T, Zhang XS, Xu D, Chen L (2006) Inferring gene regulatory networks from multiple microarray datasets. Bioinformatics 22, 2413–2420. [DOI] [PubMed] [Google Scholar]
- 15. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A et al. (2008) Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 359, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Storey JD (2002) A direct approach to false discovery rates. J. Roy. Stat. Soc., Ser. B 64, 479–498. [Google Scholar]
- 17. Leon LG, Donadel OJ, Tonn CE, Padron JM (2010) ip1 in anti‐HER2 antibody‐induced G1 cell cycle arrest and tumor growth inhibition. J. Biol. Chem. 278, 23441–23450. [DOI] [PubMed] [Google Scholar]
- 18. Huang J, Wang L, Jiang M, Zheng X (2010) Interferon α‐inducible protein 27 computational network construction and comparison between the frontal cortex of HIV encephalitis (HIVE) and HIVE‐control patients. Open Genomics J. 3, 1–8. [Google Scholar]
- 19. Huang JX, Wang L, Jiang MH (2010) TNFRSF11B computational development network construction and analysis between frontal cortex of HIV encephalitis (HIVE) and HIVE‐control patients. J. Inflamm. (Lond.) 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun Y, Wang L, Jiang M, Huang J, Liu Z, Wolfl S (2009) Secreted phosphoprotein 1 upstream invasive network construction and analysis of lung adenocarcinoma compared with human normal adjacent tissues by integrative biocomputation. Cell Biochem. Biophys. 56, 59–71. [DOI] [PubMed] [Google Scholar]
- 21. Sun Y, Wang L, Lui L (2008) Integrative decomposition procedure and Kappa statistics set up ATF2 ion binding module in malignant pleural mesothelioma (MPM). Front. Electrical Electronic Eng. China 3, 381–387. [Google Scholar]
- 22. Wang L, Huang J, Jiang M (2010) CREB5 Computational Regulation Network Construction and Analysis Between Frontal Cortex of HIV Encephalitis (HIVE) and HIVE‐Control Patients. Cell Biochem. Biophys. 7, 50. [DOI] [PubMed] [Google Scholar]
- 23. Wang L, Huang J, Jiang M (2010) RRM2 computational phosphoprotein network construction and analysis between no‐tumor hepatitis/cirrhotic liver tissues and human hepatocellular carcinoma (HCC). Cell Physiol. Biochem. 26, 303–310. [DOI] [PubMed] [Google Scholar]
- 24. Wang L, Huang J, Jiang M, Sun L (2010) MYBPC1 computational phosphoprotein network construction and analysis between frontal cortex of HIV encephalitis (HIVE) and HIVE‐control patients. Cell. Mol. Neurobiol., 1573–6830 (Electronic), 0272–4340 (Linking). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang L, Huang J, Jiang M, Zheng X (2010) AFP computational secreted network construction and analysis between human hepatocellular carcinoma (HCC) and no‐tumor hepatitis/cirrhotic liver tissues. Tumour Biol. 31, 417–425. [DOI] [PubMed] [Google Scholar]
- 26. Wang L, Sun Y, Jiang M, Zhang S, Wolfl S (2009) FOS proliferating network construction in early colorectal cancer (CRC) based on integrative significant function cluster and inferring analysis. Cancer Invest. 27, 816–824. [DOI] [PubMed] [Google Scholar]
- 27. Wang L, Sun Y, Jiang M, Zheng X (2009) Integrative decomposition procedure and Kappa statistics for the distinguished single molecular network construction and analysis. J. Biomed. Biotechnol. 3, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leon LG, Donadel OJ, Tonn CE, Padron JM (2009) Tessaric acid derivatives induce G2/M cell cycle arrest in human solid tumor cell lines. Bioorg. Med. Chem. 17, 6251–6256. [DOI] [PubMed] [Google Scholar]
- 29. Li D, Zhu J, Zhou Y, Liu X (2009) [Influence of DNAzymes against cyclin D1 in tumor cell cycle]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 26, 374–378. [PubMed] [Google Scholar]
- 30. Liang QC, Xiong H, Zhao ZW, Jia D, Li WX, Qin HZ et al. (2009) Inhibition of transcription factor STAT5b suppresses proliferation, induces G1 cell cycle arrest and reduces tumor cell invasion in human glioblastoma multiforme cells. Cancer Lett. 273, 164–171. [DOI] [PubMed] [Google Scholar]
- 31. Lu X, Qian J, Yu Y, Yang H, Li J (2009) Expression of the tumor suppressor ARHI inhibits the growth of pancreatic cancer cells by inducing G1 cell cycle arrest. Oncol. Rep. 22, 635–640. [PubMed] [Google Scholar]
- 32. Ma S, Yang Y, Wang C, Hui N, Gu L, Zhong H et al. (2009) Endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest and apoptosis through down‐regulation of the phosphatidylinositide 3‐kinase/Akt/HDM2 pathway. J. Biol. Chem. 284, 24773–24782. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V et al. (2009) BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 30, 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pecherzewska R, Nawrot B (2009) [FHIT – tumor suppressor protein involved in induction of apoptosis and cell cycle regulation]. Postepy Biochem. 55, 66–75. [PubMed] [Google Scholar]
- 35. Ritta M, De Andrea M, Mondini M, Mazibrada J, Giordano C, Pecorari G et al. (2009) Cell cycle and viral and immunologic profiles of head and neck squamous cell carcinoma as predictable variables of tumor progression. Head Neck 31, 318–327. [DOI] [PubMed] [Google Scholar]
- 36. Scoumanne A, Zhang J, Chen X (2009) PRMT5 is required for cell‐cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skladanowski A, Bozko P, Sabisz M (2009) DNA structure and integrity checkpoints during the cell cycle and their role in drug targeting and sensitivity of tumor cells to anticancer treatment. Chem. Rev. 109, 2951–2973. [DOI] [PubMed] [Google Scholar]
- 38. Stewart CJ, Crook ML, Leung YC, Platten M (2009) Expression of cell cycle regulatory proteins in endometrial adenocarcinoma: variations in conventional tumor areas and in microcystic, elongated and fragmented glands. Mod. Pathol. 22, 725–733. [DOI] [PubMed] [Google Scholar]
- 39. Teyssot ML, Jarrousse AS, Chevry A, De Haze A, Beaudoin C, Manin M et al. (2009) Toxicity of copper(I)‐NHC complexes against human tumor cells: induction of cell cycle arrest, apoptosis, and DNA cleavage. Chemistry 15, 314–318. [DOI] [PubMed] [Google Scholar]
- 40. Tu S, Wai‐Yin Sun R, Lin MC, Tao Cui J, Zou B, Gu Q et al. (2009) Gold (III) porphyrin complexes induce apoptosis and cell cycle arrest and inhibit tumor growth in colon cancer. Cancer 115, 4459–4469. [DOI] [PubMed] [Google Scholar]
- 41. Varamini P, Soltani M, Ghaderi A (2009) Cell cycle analysis and cytotoxic potential of Ruta graveolens against human tumor cell lines. Neoplasma 56, 490–493. [DOI] [PubMed] [Google Scholar]
- 42. Wrona IE, Lowe JT, Turbyville TJ, Johnson TR, Beignet J, Beutler JA et al. (2009) Synthesis of a 35‐member stereoisomer library of bistramide A: evaluation of effects on actin state, cell cycle and tumor cell growth. J. Org. Chem. 74, 1897–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xie YF, Sheng W, Xiang J, Zhang H, Ye Z, Yang J (2009) Adenovirus‐mediated ING4 expression suppresses pancreatic carcinoma cell growth via induction of cell‐cycle alteration, apoptosis, and inhibition of tumor angiogenesis. Cancer Biother. Radiopharm. 24, 261–269. [DOI] [PubMed] [Google Scholar]
- 44. Xiong H, Su WY, Liang QC, Zhang ZG, Chen HM, Du W et al. (2009) Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Invest. 89, 717–725. [DOI] [PubMed] [Google Scholar]
- 45. Xu L, Niesen MI, Blanck G (2009) Linkage of a tumor immune function and cell cycle de‐regulation via a gene regulatory network subcircuit. Mol. Immunol. 46, 569–575. [DOI] [PubMed] [Google Scholar]
- 46. Zolochevska O, Figueiredo ML (2009) Expression of cell cycle regulator cdk2ap1 suppresses tumor cell phenotype by non‐cell‐autonomous mechanisms. Oral Oncol. 45, e106–e112. [DOI] [PMC free article] [PubMed] [Google Scholar]