

Abstract
Abstract. The use of non‐steroidal anti‐inflammatory drugs has proved of great interest in the prevention and treatment of colorectal cancer, although their precise mechanisms of action remain unclear. Overexpression of cyclooxygenase‐2 (COX‐2) and subsequent prostaglandin production promote metastasis and have been shown to increase cell motility in vitro. Objective: We have aimed to elucidate whether specific inhibition of COX‐2 with NS‐398 (NS‐398 is a selective inhibitor of COX‐2) would be able to inhibit motility of colorectal cancer cells and whether this was modulated through epidermal growth factor receptor (EGFR) transactivation. Materials and Methods: A transwell filter assay was used to study cell motility. Expression of COX‐2, EGFR phosphorylation and prostaglandin E2 (PGE2) receptors were assessed by Western blot analysis and reverse transcriptase‐polymerase chain reaction. PGE2 concentrations after NS‐398 treatment were estimated by enzyme immunoassay. Results: Treatment with NS‐398 significantly reduced PGE2 levels and reduced cell migration in the HT29 and HCA7 colorectal carcinoma cell lines and this effect was rescued by addition of PGE2. Furthermore, specific inhibition of COX‐2 with NS‐398 reduced EGFR phosphorylation in colorectal cancer cells. Direct inhibition of EGFR activity with AG1478 reduced PGE2‐stimulated motility, clearly demonstrating that PGE2 acts via the EGFR‐signalling pathway. The novel combination of NS‐398 and AG1478 dramatically reduced migration of colorectal cancer cells. Conclusion: The data presented indicate that the use of NS‐398 in chemoprevention and adjuvant therapy for colorectal cancer may work in part, through the inhibition of cell motility. Furthermore, our data suggest that the combined use of non‐steroidal anti‐inflammatory drugs with EGFR antagonists could be explored further for future use in the clinic.
INTRODUCTION
Colorectal carcinoma is the second highest cause of cancer mortality in the Western world. It is frequently the process of metastasis that increases the risk of death in patients with this disease. Therefore, identification of drugs that can target this process is of particular importance. There is great interest in the use of non‐steroidal anti‐inflammatory drugs (NSAIDs) for the prevention and adjuvant therapy of colorectal cancer. Evidence from both human and animal experiments and from epidemiological studies has shown that NSAIDs have potent antitumour effects (Chen et al. 2001). One property shared by all these drugs is their ability to inhibit cyclooxygenase (COX), the rate‐limiting step in conversion of arachidonic acid to prostaglandins (PGs). Studies have confirmed the presence of two forms of COX of which COX‐2 is frequently overexpressed in colonic adenomas and carcinomas (Bamba et al. 1999; Zhang & Sun 2002). Specific inhibitors of COX‐2 have been shown to induce apoptosis in tumour cells in vivo and in vitro and to reduce tumour growth in animal models and in humans (Elder et al. 1997; Sheng et al. 1997; Sawaoka et al. 1998). For example, administration of the selective COX‐2 inhibitor celecoxib significantly reduced incidence of colonic tumours in rats by 53–78% (Reddy et al. 2000). The principal PG generated in colorectal carcinomas and adenomas appears to be prostaglandin E2 (PGE2), and its levels are elevated when compared to normal intestinal tissue (Rigas et al. 1993; Pugh & Thomas 1994; Adam et al. 2001). PGE2 has been shown to stimulate growth, modulate apoptosis and enhance cell motility in colon carcinoma cell lines in vitro (Qiao et al. 1995; 1998, 2001; Pai et al. 2002). The EP4 receptor mediates the effect of PGE2 on cell motility (Sheng et al. 2001), and a recent study by Pai et al. (2002), reported that PGE2 is also able to stimulate motility through transactivation of the epidermal growth factor receptor (EGFR). This mechanism presents a novel target for NSAIDs in the treatment of colorectal cancer and raises the possibility of increasing treatment efficacy through combining COX‐2 inhibition with direct antagonism of the EGFR.
Although PGE2 has been shown to stimulate migration in colorectal cancer cells, no previous study has addressed whether the use of NSAIDs can inhibit motility and whether COX‐2 inhibition directly affects the transactivation of the EGFR. Selective COX‐2 inhibitors may effectively inhibit both COX‐2 and EGF signalling pathways and thus effectively reduce the metastatic potential of colorectal tumours.
Using a mouse model of colorectal cancer, Torrance et al. (2000) evaluated the effects of combining the NSAID sulindac (which inhibits both COX‐1 and COX‐2) with a novel EGFR kinase inhibitor (EKB‐569). The APC+/MIN mice, which normally develop multiple intestinal polyps due to a mutation in the APC tumour suppressor gene, showed dramatic reduction in polyp occurrence when treated with the combination of drugs, with half the mice developing no polyps at all (Torrance et al. 2000). These data illustrate the benefit of targeting both pathways, but this approach has yet to be taken using human cells or using a COX‐2 selective inhibitor.
The aim of the study presented here is to determine whether the selective COX‐2 inhibitor NS‐398 reduces colorectal cancer cell motility and whether this occurs through modulation of EGFR activation. The combination of COX‐2 inhibition with the selective EGFR antagonist AG1478 was also tested to see if this combination of approaches had therapeutic potential, by modulating cell migration.
MATERIALS AND METHODS
Cell culture
HCA7 cells were established from a moderately well differentiated mucinous carcinoma of the colon (Kirkland 1985) and were a kind gift from Dr Sue Kirkland (London, UK). HCA7‐col29 was subcloned from the parental line (Marsh et al. 1993) and will be referred to as HCA7. HT29 and HCT116 human colorectal carcinoma cell lines were obtained from the American Type Culture Collection, Rockville, MD, USA. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% foetal calf serum at 37 °C, in a humidified atmosphere of 5% CO2.
Transwell filter migration assay
Cell migration assays were carried out using a transwell filter migration assay as previously described (Efstathiou et al. 1999); they were treated with NS‐398 (Sigma, St. Louis, MO, USA) in serum‐free and calcium free medium for 24 h. NS‐398 was dissolved in dimethyl sulfoxide at stock solution of 30 mm, with each control and treated flask receiving the same amount of dimethyl sulfoxide. Insert filter (Falcon, Bedford, MA, USA), 8 µm in pore size was coated with 10 µg/mL Vitrogen type I collagen (Cohesion, Palo Alto, CA, USA); 1 × 105 cells were placed in each insert filter. After an incubation period of 24 h at 37 °C, cells were removed from the upper filter surface with a cotton swab. The filters were fixed and stained with haematoxylin. Cells on the lower filter surface were considered migratory and were counted, 10 high power fields, at ×40 magnification. The effect of NS‐398 on migratory abilities of the cells was examined at basal conditions. The outcome of PGE2 (Sigma) on NS‐398 inhibition of migration was studied by re‐adding PGE2 during the migration assay. The role of tyrphostin (AG1478) (Sigma) on cell motility was investigated by pre‐treatment of the cells and by adding AG1478 during the migration assay. Independent experiments were carried out, and the data are expressed as the mean ± SE of assays performed in triplicate.
Assessment of apoptosis
Cell death by apoptosis was determined by Annexin V binding in combination with 7‐amino‐actinomycin D uptake. We have previously used this method to assess apoptosis induction in colorectal cancer cell lines (Buda et al. 2003), but briefly, samples collected from treated and untreated cells were incubated with Annexin V conjugated with phycoerythrin and 7‐amino‐actinomycin D and results were analysed by FACSscan.
Determination of PGE2 production
PGE2 was measured in the culture media taken from cells, by competitive enzyme immunoassay, according to the manufacturer's protocol (Cayman Chemical Co., Ann Arbor, MI, USA). Briefly, 2 × 105 cells were seeded and grown in T25 flasks up to 70–80% confluency. Then, they were treated with 10 µm NS‐398 for 24 h. The harvested medium was centrifuged at 500 g for 5 min to remove floating cells and the supernatant was frozen at –70 °C until used for the PGE2 assay.
Reverse transcriptase‐polymerase chain reaction
RNA was extracted from 8 × 106 cells using the RNeasy mini kit (Qiagen GmbH, Hilden, Germany) with ‘on‐column’ DNase treatment. Single‐stranded cDNA was synthesized from 10 µg RNA in a 50‐µL volume containing 1 µg oligo‐dT primer, 400 U Moloney murine leukaemia virus reverse transcriptase, 80 U rRNasin (Promega Corporation, Madison, WI, USA) and 0.2 µm each dNTP in appropriate buffer. Polymerase chain reactions (PCR) (25 µL) contained 2 µL cDNA solution, 12.5 µL 2× PCR Master Mix (Promega Corporation) and 1 µm primers. PCR was carried out for 35 cycles for each primer and conditions were: 94 °C for 1 min, 61 °C for 1 min and 72 °C for 2 min. Control PCR was performed directly on RNA without the step of cDNA synthesis. PCR fragments were sequenced to confirm their identity. PCR primer sequences used were: EP1 receptor: forward, 5′‐GGCGGGCGAGGCGACCACA‐3′; reverse, 5′‐GGACCCAGGCCGATGAAGCACCAC‐3′ (product = 549 bp); EP2 receptor: forward, 5′‐CCAGGTAAAGGCCGGGAGAGGAG‐3′; reverse, 5′‐GTCATGGCGAAAGCGAAGTAGGTG‐3′ (product = 401 bp); EP3 receptor: forward, 5′‐CGGGGCTACGGAGGGGATGC‐3; reverse, 5′‐ATGGCGCTGGCGATGAACAACGAG‐3′ (product = 440 bp); EP4 receptor: forward, 5′‐TCGCGCAAGGAGCAGAAGGAGACG‐3′; reverse, 5′‐GGACGGTGGCGAGAATGAGGAAGG‐3′ (product = 469 bp).
Western blotting
Samples of 2 × 106 cells were prepared for Western blotting as described previously by Palmer et al. (1997). Briefly, proteins were resolved on 7.5% polyacrylamide gels and were transferred to Immobilon‐P polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). COX‐2 protein was detected using monoclonal anti‐COX‐2 at 250 µg/mL (Cayman Chemical Co.) and tyrosine phosphorylated EGFR was detected by a specific monoclonal antibody raised against the tyrosine phosphorylated EGFR, used at 1 µg/mL (Chemicon International, Temecula, CA, USA). Total EGFR protein levels were evaluated using mouse monoclonal antibody (BD Bioscience, San Diego, CA, USA) used at 0.25 µg/mL. Blots were subsequently probed with anti‐α‐tubulin (Sigma) to show equal sample loading.
Statistical analysis
Statistical analysis of the data on cell migration was performed using Student's t‐test. Differences were considered significant when P‐values were < 0.05.
RESULTS
NS‐398 reduces migration of COX‐2‐positive colon cancer cells and this effect can be reversed by addition of PGE2
To define the model system, COX‐2 expression by the cell lines studied was evaluated by Western blotting. HT29 and HCA7 cell lines showed expression of COX‐2 protein (Fig. 1a). COX‐2 expression was higher in HCA7 cells compared to HT29s due to increased COX‐2 mRNA stability in HCA7 (Shao et al. 2000). In HCT116 cells, COX‐2 protein was virtually undetectable (Fig. 1a). These data concur with findings reported elsewhere (Agarwal et al. 2003).
Figure 1.
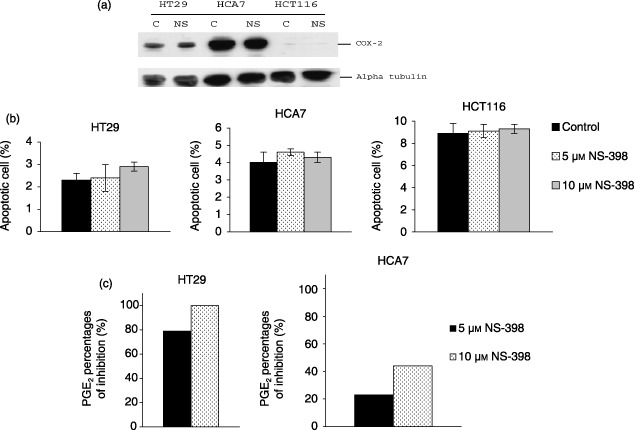
(a) Effect of NS‐398 (10 µm) treatment for 24 h on COX‐2 expression in HT29, HCA7 and HCT116 cell lines. Lysates of 2 × 106 cells from untreated (C) and NS‐398 treated (NS) cultures were resolved by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis and were probed with anti‐COX‐2 monoclonal antibody (72 kDa). Protein levels were determined by Western immunoblotting. Blots were re‐probed with anti‐α‐tubulin to confirm equal loading. (b) Effect of NS‐398 (5 and 10 µm) on apoptosis in colorectal cancer cell lines. Results expressed as percentage of apoptotic cells (annexin V, positive/7‐amino‐actinomycin, negative). NS‐398 treatment did not increase the percentage of apoptotic cells. Values are expressed as mean (SEM) from three different experiments. (c) Inhibition of PGE2 secretion by NS‐398 treatment. HT29, HCA7 and HCT116 colonic carcinoma cell lines were treated with solvent, control and 5 or 10 µm of NS‐398 for 24 h. NS‐398 significantly reduced PGE2 levels in HT29 and HCA7 cells. Data shown are results of duplicate measurements.
The effect of NS‐398 on COX‐2 protein expression is still not clearly defined, although some researchers report that doses of NS‐398 in excess of 20 µm elevate COX‐2 expression in colon cancer cells (Elder et al. 2000, 2002). Here, levels of COX‐2 protein were evaluated following 10 µm NS‐398 treatment in HT29, HCA7 and HCT116 cells by Western blotting (Fig. 1a). In this study, control and NS‐398 treated cells showed unchanged total levels of COX‐2 protein.
It has been previously shown that doses of NS‐398 in excess of 20 µm induce apoptosis of colorectal cancer cells (Elder et al. 2002). In the current study, NS‐398 was used at 5 and 10 µm. Previous work has shown that 10 µm NS‐398 does not inhibit the growth of colorectal cancer cells (Crew et al. 2000). In order to show that the effects on cell migration were not due to altered cell survival, apoptosis was studied by Annexin V binding. Treatment with 5 or 10 µm of NS‐398 did not significantly increase the percentage of apoptotic cells compared to controls, in all cell lines tested (Fig. 1b).
Because the biological effects of COX‐2 appear to be mediated by prostaglandins, we estimated PGE2 levels in the cells after treatment with NS‐398. HT29 cells secreted much less PGE2 than HCA7s, whereas PGE2 was undetectable in HCT116 cells (data not shown). NS‐398 treatment significantly decreases PGE2 production in COX‐2‐positive colorectal cancer cells (Fig. 1c) (Elder et al. 2002) and treatment of colorectal cancer cells with 10 µm NS‐398 functionally inhibits COX‐2 without altering its expression or inducing cell death.
Treatment with 5 or 10 µm of NS‐398 for 24 h reduced basal migration of COX‐2‐positive HT29 and HCA7 human colorectal carcinoma cells compared to controls (Fig. 2a). HCA7 cells (with the highest level of COX‐2 expression), showed the highest level of basal migration. NS‐398 did not affect cell migration in COX‐2‐negative HCT116 cells (Fig. 2a).
Figure 2.
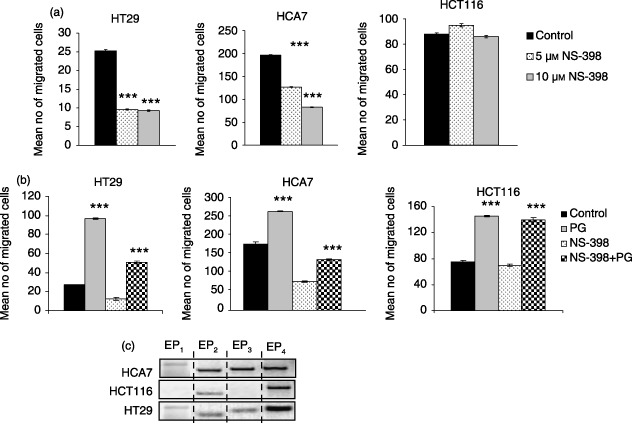
(a) Effects of NS‐398 on cell migration of HT29, HCA7 and HCT116 human colonic adenocarcinoma cell lines. Cells were treated with 5 and 10 µm NS‐398 for 24 h. 1 × 105 cells were resuspended in serum‐free and calcium‐free (SFCF) medium and were added to each insert filter. SFCF medium was placed in wells. After 24 h, the number of migrated cells in 10 high power fields was counted. Data represent mean ± SE of three independent experiments done in triplicates. ***P < 0.001 compared to control. (b) Effects of PGE2 on motility of colon cancer cells. (a) PGE2 stimulated cell migration in these human colonic carcinoma cells. 1 × 105 cells resuspended in SFCF were seeded into the insert filter, and the assay was carried out for 24 h with either vehicle or 1 µm PGE2 as attractant. (b) PGE2 partially reversed effect of NS‐398 on cell migration. 1 µm PGE2 was added to each insert containing 1 × 105 NS‐398‐treated cells. SFCF was placed in the wells. After 24 h the number of migrated cells in 10 high power fields was counted. Data represent mean ± SE of three independent experiments performed in triplicates. ***P < 0.001 control versus PGE2 and NS‐398 versus NS‐398+PGE2. (c) EP receptor expression in colon cancer cells. EP receptor mRNA expression in human HT29, HCA7 and HCT116 human colonic adenocarcinoma cells detected by RT‐PCR. No bands were observed in RNA samples that had not undergone reverse transcription.
In order to confirm that the effect of NS‐398 on cell migration was due to functional inhibition of COX‐2, PGE2 was added following pre‐treatment with 10 µm NS‐398. Exogenous addition of PGE2 without NS‐398 treatment increased motility of both COX‐2‐positive (HT29 and HCA7) and COX‐2‐negative (HCT116) cell lines (Fig. 2b). In HT29 cells, PGE2 addition restored migration to greater than control levels, whereas for HCA7 cells, addition of 1 µm PGE2 was unable to completely reverse the effect of NS‐398 (Fig. 2b). These findings suggest that inhibition of cell migration by NS‐398 is related, at least in part, to a decrease in PGE2 production.
Combined treatment of COX‐2‐negative HCT116 cells with NS‐398 and PGE2 increased cell migration above the basal level, to the same extent, as treatment with PGE2 alone. This response of these cells suggests that although not producing prostaglandins, these cells retain the ability to respond to this type of stimulus. For this reason, expression of PGE2 receptors was analysed in the three cells lines by RT‐PCR using gene‐specific primers (Fig. 2c). All PCR products were sequenced to confirm their identity.
Each of the three carcinoma cell lines expressed EP receptors. Whereas the COX‐2‐positive cell lines (HCA7 and HT29) expressed transcripts for all four EP receptor subtypes, the COX‐2‐negative cell line (HCT116) expressed EP2 and EP4 transcripts only.
NS‐398 reduces prostaglandin‐stimulated phosphorylation of EGFR in colorectal cancer cells
Previous studies have demonstrated that PGE2 can transactivate the EGFR in colorectal cancer cells (Pai et al. 2002; Shao et al. 2004). Furthermore, Buchanan et al. (2003), showed that this prostaglandin‐induced signalling through EGFR enhanced cell motility in the LS174T cell line. The use of COX‐2 inhibition as an adjuvant therapy could reduce tumour metastasis through affecting EGFR activity. To investigate this possibility, the effects of NS‐398 on EGFR phosphorylation were assessed using a phospho‐specific antibody (Campos‐Gonzalez & Glenney 1991) in the colorectal cancer cell lines used above. NS‐398 treatment reduced the level of phosphorylated EGFR in the COX‐2‐positive cell lines compared to controls (Fig. 3). Analysis using an antibody for total EGFR showed that this effect of NS‐398 was not due to modulation of EGFR expression levels (Fig. 3). Unsurprisingly, NS‐398 did not affect EGFR phosphorylation in the COX‐2‐negative HCT116 cell line as these cells do not produce PGE2. This finding serves as further evidence that NS‐398 is having COX‐2‐specific effects at the dose used. Furthermore, treatment of all three cell lines with exogenous PGE2 resulted in elevation of phospho‐EGFR; this effect was less pronounced in HCA7 cells due to high endogenous PGE2 levels. Treatment with a combination of NS‐398 and PGE2 showed lesser induction in the COX‐2‐positive cell lines (due to inhibition of endogenous prostaglandin production) but not in the COX‐2‐negative HCT116 cells.
Figure 3.
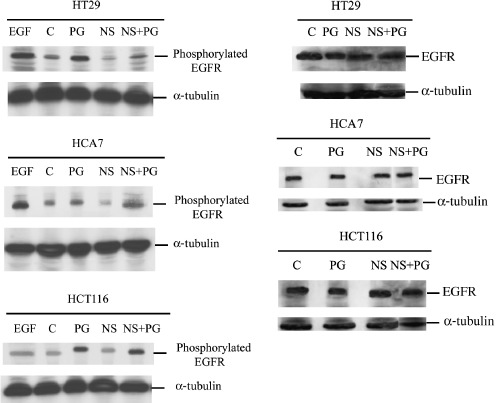
Effect of PGE2 (1 µm) and NS398 (10 µm) on EGFR activation and EGFR total protein levels. Lysates of 2 × 106 cells from untreated (C), PGE2 (PG)‐, NS‐398 (NS)‐ and PGE2+ NS‐398 (NS+PG)‐treated 24 h samples were prepared. EGF (0.6 µg/mL) was used as control for the EGFR activated form. Samples were resolved by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis, probed with anti‐EGF receptor (activated) monoclonal antibody and anti‐EGF receptor monoclonal antibody. Protein levels were determined by Western immunoblotting (185 kDa). Equal loading is shown by repeat probing with anti‐α‐tubulin.
Combination of the EGFR antagonist AG1478 with NS‐398 dramatically reduces motility of colorectal cancer cells
The use of combinations of therapeutic drugs in the treatment of cancer is of great interest, as it can allow administration of lower doses of drug and therefore reduction of potentially adverse side effects, and can also be used to target multiple pathways. Drugs that target cellular motility could have antimetastatic effects when used in an adjuvant setting.
The data presented above show a potent antimigratory effect of the COX‐2 inhibitor NS‐398 in colon cancer cells, which may be mediated through modulation of EGFR activity. EGFR antagonists are in widespread clinical use and therefore the effects of combining a COX‐2 inhibitor with the EGFR antagonist were tested using the in vitro model of colorectal cancer cell motility employed above (Fig. 4).
Figure 4.
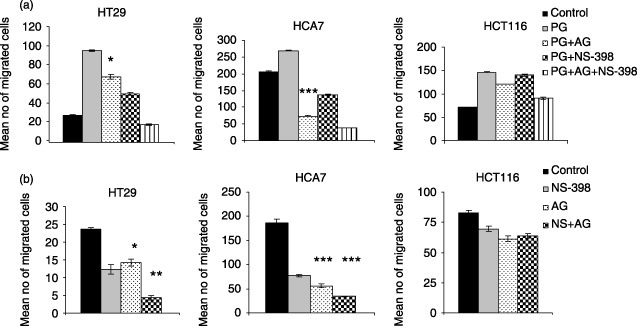
Effect of AG1478 on cell migration. Cells were treated with 1 µm AG1478 for 30 min 1 × 105 control or AG1478 treated cells were added to each insert filter. (a) 1 µm PGE2 was used as the attractant. Effects of PGE2 on NS398‐ and AG1478‐reduced cell migration were studied by placing 1 × 105 cells treated with both 10 µm NS‐398 (24 h) and 1 µm AG1478 (30 min) in each insert filter. 1 µm PGE2 and 1 µm AG1478 was added to the insert filter during the assay. (b) 1 × 105 control, 10 µm NS‐398 (24 h) treated or both NS‐398 and 1 µm AG1478 (30 min) cells were added to each insert filter. After 24 h, the number of migrated cells in 10 high power fields was counted. Data represent mean ± SE of three independent experiments performed in triplicates. *P < 0.05; **P < 0.01; ***P < 0.001 PG versus PG+AG; control versus AG and NS‐398 versus NS‐398+ AG.
In order to assess functionality of the EGFR pathway in these cells, and activity of the antagonist, treatments were carried out with exogenous EGF and AG1478 (a selective EGFR tyrosine kinase inhibitor) (Patrik & Hochegger 1999) alone, and in combination. EGF treatment stimulated migration in HT29 and HCA7 cells, but not in HCT116. Effects of EGF could be abrogated in the responsive cell lines by the addition of AG1478, but the drug had no effect on the unresponsive HCT116 cells suggesting that the consequences of AG1478 treatment were EGFR‐dependent (data not shown).
AG1478 was able to reduce PGE2‐stimulated motility in all three cell lines, and this was significant in HT29 and HCA7 cells. This finding is in concurrence with previous work by Buchanan et al. (2003) in the colon cancer cell line LS174T. These data further support the hypothesis that PGE2 is working through EGFR to stimulate motility in HT29 and HCA7 cells, but to a lesser extent in HCT116 cells.
The novel combination of AG1478 with NS‐398 reduced cell migration in HT29 and HCA7 cells and this was significantly greater than for NS‐398 treatment alone. In the absence of exogenous PGE2, this combination of potential therapeutic approaches almost completely abolished cell motility in HT29 cells.
DISCUSSION
COX‐2 inhibitors have been shown to have diverse antitumour effects and are of interest in the prevention and treatment of colorectal cancer. However, the precise mechanisms by which COX‐2 inhibitors exert their antitumour action are not completely understood. Furthermore, there are some adverse side effects associated with long‐term NSAID use (Mulcahy & O'Donoghue 2002) and thus there is need to define their mechanism of action in order to target biological processes more specifically in the tumour, and to increase drug efficacy by using novel combinations of therapeutics.
By the time patients present at the clinic with colorectal cancer, their disease is often at an advanced stage and it is frequently the metastases and not the primary tumour that kills the patient. Therefore, there is considerable interest in targeting the processes involved in metastasis, for adjuvant therapy. Aberrant cell motility is necessary for tumour cell invasion and metastasis. Previous studies have shown that COX‐2 overexpression and subsequent PGE2 production can promote cell motility in colorectal cancer cell lines CaCo2 and LS174T (Tsujii et al. 1997; Sheng et al. 2001), respectively, and invasiveness in the SW420 and LoVo lines (Pai et al. 2003). Here, in this study we have investigated the effect of the selective COX‐2 inhibitor, NS‐398 on cell motility, using a transwell filter assay.
We demonstrated that treatment with 10 µm of NS‐398 significantly reduced PGE2 secretion and reduced cell migration of the COX‐2‐positive colorectal cancer cells. Treatment with this dose of NS‐398 did not affect cell survival as assessed by Annexin V binding, showing that the result on migration was not due to reduced cell viability. Elder et al. (2000) have shown that NS‐398 at higher concentrations (20–75 µm) induces COX‐2 expression in HT29 cells. The inhibitory effect on motility by the low‐dose (10 µm) NS‐398 used in this study was not associated with changes in COX‐2 protein levels. This finding is significant as the induction of COX‐2 expression by its inhibitor could result in chemoresistance, and also it demonstrates that lower doses of this drug can still have potent antitumour consequences.
The ability of exogenous PGE2 to reverse the characteristic of cell motility demonstrates the specificity of drug action. Furthermore, the lack of effect of NS‐398 on the COX‐2‐negative cell line HCT116 emphasizes the lack of ‘off‐target’ result at the dose used. Higher doses of NSAIDs are frequently associated with COX‐2‐independent effects although these are not well defined as yet (Williams et al. 2000).
Analysis of expression of EP receptors in the cell lines used revealed that although COX‐2 negative, the HCT116 cells express EP2 and EP4 PGE2 receptors. Sheng et al. (2001) have shown EP4 to mediate PG‐induced migration in LS174T colon cancer cells. This finding explains the mechanism by which HCT116 respond to exogenous PGE2 addition and has implications for this type of therapeutic approach in COX‐2 negative tumours. Although the epithelial component of a tumour may be COX‐2‐negative, the tumour cells may still respond to stromally produced prostaglandins in a paracrine manner and therefore the patient may still benefit from adjuvant therapy.
Recent work by other investigators has shown that PGE2 induces phosphorylation of EGFR (Pai et al. 2002; Shao et al. 2004), and subsequent cell migration of the colorectal cancer cell line LS174T (Buchanan et al. 2003). Whereas blocking PGE2 synthesis or the EP1 receptor inhibited EGF‐induced EGFR phosphorylation in human cholangiocarcinoma cell line CCLP1 (Han & Wu 2005). We have demonstrated that treatment with exogenous PGE2 induces phosphorylation of the EGFR in both COX‐2‐positive HT29 and HCA7 cells and in COX‐2‐negative HCT116 cells. The HCT116 result is not unexpected as we have shown expression of EP receptors in these cells.
We wished to ascertain whether inhibition of cell motility by NS‐398 was modulated through altering transactivation of EGFR. NS‐398 reduces basal EGFR phosphorylation in the COX‐2‐positive lines. This is due to inhibition of endogenous prostaglandin production, illustrated by the fact that NS‐398 did not affect EGFR activity in HCT116 cells and further demonstrating the specificity of NS‐398. Effects of PGE2 and NS‐398 were also shown not be due to modulation of EGFR expression levels. These findings highlight EGFR as a possible novel target for therapeutic use of NSAIDs.
A combinatorial approach to the use of anticancer drugs can allow use of lower, therefore less toxic, doses of each drug. Novel combinations of therapies can also produce synergistic effects. As we have shown, NS‐398 inhibits the motility of colorectal cancer cells and this inhibition appears to work, at least in part, through the modulation of EGFR activity; we tried a novel combination of NS‐398 and the selective antagonist AG1478.
Combination of these two drugs produced a striking inhibition of cell migration in the COX‐2‐positive cells HT29 and HCA7. Exogenous PGE2 addition was only modestly able to rescue this effect as the inclusion of AG1478 had apparently abrogated its key route for promoting motility. This combinatorial approach was most dramatic in HT29 cells, where migration was almost entirely blocked. Data for HCA7 cells indicate that this cell line has a high dependence on EGFR signalling for cell motility, illustrated by its sensitivity to AG1478 and the inability of PGE2 to reverse this effect.
Cell migration in the COX‐2‐negative HCT116 cells appears to be relatively independent of EGFR signalling, suggesting that other mechanisms drive motility in these cells. Although they respond to PGE2 treatment, it appears that very little of the PGE2‐induced effect is due to any detectable induction of EGFR phosphorylation. HCT116 cells express EP receptors, but the mechanisms by which cell migration can be induced by these, other than through EGFR, remain unclear; this cell line presents itself as an ideal model for the study of these mechanisms.
In conclusion, the data presented here highlight another mechanism of in vivo action for COX‐2 inhibitors in cell motility and that significant effects can be achieved with lower doses of the drug than necessary for the induction of apoptosis. This is significant following recent findings regarding potential side effects of long‐term NSAID use (Warner & Mitchell 2004). Furthermore, the novel combination of a NSAID with an EGFR antagonist has been demonstrated to have dramatic effect on cell motility in vitro and may prove an extremely effective strategy in the clinic. This type of approach could greatly affect the metastatic potential of tumour cells and therefore greatly improve disease outcome for patients with colorectal cancer.
REFERENCES
- Adam L, Mazumdar A, Sharma T, Jones TR, Kumar R (2001) A three‐dimensional and temporo‐spatial model to study invasiveness of cancer cells by heregulin and prostaglandin E2 . Cancer Res. 61, 81–87. [PubMed] [Google Scholar]
- Agarwal B, Swaroop P, Protiva P, Raj SV, Shirin H, Holt PR (2003) Cox‐2 is needed but not sufficient for apoptosis induced by Cox‐2 selective inhibitors in colon cancer cells. Apoptosis 8, 649–654. [DOI] [PubMed] [Google Scholar]
- Bamba H, Ota S, Kato A, Adachi A, Itoyama S, Matsuzaki F (1999) High expression of cyclooxygenase‐2 in macrophages of human colonic adenoma. Int. J. Cancer 83, 470–475. [DOI] [PubMed] [Google Scholar]
- Buchanan FG, Wang D, Bargiacchi F, Dubois RN (2003) Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J. Biol. Chem. 278, 35451–35457. [DOI] [PubMed] [Google Scholar]
- Buda A, Qualtrough D, Jepson MA, Martines D, Paraskeva C, Pignatelli M (2003) Butyrate downregulates α2β1 integrin: a possible role in the induction of apoptosis in colorectal cancer cell lines. Gut 52, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos‐Gonzalez R, Glenney JR Jr (1991) Immunodetection of the ligand‐activated receptor for epidermal growth factor. Growth Factors 4, 305–316. [DOI] [PubMed] [Google Scholar]
- Chen WS, Wei SJ, Liu JM, Hsiao M, Kou‐Lin J, Yang WK (2001) Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase‐2 (COX‐2) expression and inhibited by a COX‐2 selective inhibitor, etodolac. Int. J. Cancer 91, 894–899. [DOI] [PubMed] [Google Scholar]
- Crew TE, Elder DJE, Paraskeva C (2000) A cyclooxygenase‐2 (COX‐2) selective non‐steroidal anti‐inflammatory drug enhances the growth inhibitory effect of butyrate in colorectal carcinoma cells expressing COX‐2 protein: regulation of COX‐2 by butyrate. Carcinogenesis 21, 69–77. [DOI] [PubMed] [Google Scholar]
- Efstathiou JA, Liu D, Wheeler JM, Kim HC, Beck NE, Ilyas M, Karayiannakis AJ, Mortensen NJ, Kmiot W, Playford RJ, Pignatelli M, Bodmer WF (1999) Mutated epithelial cadherin is associated with increased tumorigenicity and loss of adhesion and of responsiveness to the motogenic trefoil factor 2 in colon carcinoma cells. Proc. Natl Acad. Sci. USA 96, 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder DJ, Halton DE, Crew TE, Paraskeva C (2000) Apoptosis induction and cyclooxygenase‐2 regulation in human colorectal adenoma and carcinoma cell lines by the cyclooxygenase‐2‐selective non‐steroidal anti‐inflammatory drug NS‐398. Int. J. Cancer 86, 553–560. [DOI] [PubMed] [Google Scholar]
- Elder DJE, Halton DE, Hague A, Paraskeva C (1997) Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase‐2 (COX‐2) selective nonsteroidal anti‐inflammatory drug: Independence from COX‐2 protein expression. Clin. Cancer Res. 3, 1679–1683. [PubMed] [Google Scholar]
- Elder DJ, Halton DE, Playle LC, Paraskeva C (2002) The MEK/ERK pathway mediates COX‐2‐selective NSAID‐induced apoptosis and induced COX‐2 protein expression in colorectal carcinoma cells. Int. J. Cancer 99, 323–327. [DOI] [PubMed] [Google Scholar]
- Han C, Wu T (2005) Cyclooxygenase‐2‐derived prostaglandin E2 promotes human cholangiocarcinoma cell growth and invasion through EP1 receptor‐mediated activation of the epidermal growth factor receptor and Akt. J. Biol. Chem. 280, 24053–24063. [DOI] [PubMed] [Google Scholar]
- Kirkland SC (1985) Dome formation by a human colonic adenocarcinoma cell line (HCA‐7). Cancer Res. 45, 3790–3795. [PubMed] [Google Scholar]
- Marsh KA, Stamp GWH, Kirkland SC (1993) Isolation and characterisation of multiple cell types from a single human colonic carcinoma: tumorigenicity of these cell types in a xenograft system. J. Pathol. 170, 441–450. [DOI] [PubMed] [Google Scholar]
- Mulcahy HE, O'Donoghue D (2002) Nonsteroidal anti‐inflammatory drugs and their colonic effects: more interesting than irritating? Eur. J. Gastroenterol. Hepatol. 14, 1177–1178. [DOI] [PubMed] [Google Scholar]
- Pai R, Nakamura T, Moon WS, Tarnawski AS (2003) Prostaglandins promote colon cancer cell invasion; signaling by cross‐talk between two distinct growth factor receptors. FASEB J. 17, 1640–1647. [DOI] [PubMed] [Google Scholar]
- Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS (2002) Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 8, 289–293. [DOI] [PubMed] [Google Scholar]
- Palmer DG, Paraskeva C, William AC (1997) Modulation of p53 expression in cultured colonic adenoma cell lines by the naturally occuring lumenal factors butyrate and deoxycholate. Int. J. Cancer 73, 702–706. [DOI] [PubMed] [Google Scholar]
- Patrik G, Hochegger K (1999) Inhibition of epidermal‐growth‐factor‐dependent signalling by tyrphostins A25 and AG1478 blocks growth and induces apoptosis in colorectal tumor cells in vitro . J. Cancer Res. Clin. Oncol. 125, 379–388. [DOI] [PubMed] [Google Scholar]
- Pugh S, Thomas GAO (1994) Patients with adenomatous potyps and carcinomas have increased colonic mucosal prostaglandin E2 . Gut 35, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, Shiff SJ, Rigas B (1995) Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo . Biochim. Biophys. Acta 1258, 215–223. [DOI] [PubMed] [Google Scholar]
- Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV (2000) Chemoprevention of colon cancer by specific cyclooxygenase‐2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 60, 293–297. [PubMed] [Google Scholar]
- Rigas B, Goldman IS, Levine L (1993) Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 122, 518–523. [PubMed] [Google Scholar]
- Sawaoka H, Kawano S, Tsuji S, Tsujii M, Gunawan ES, Takei Y, Nagano K, Hori M (1998) Cyclooxygenase‐2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am. J. Physiol. 274, G1061–G1067. [DOI] [PubMed] [Google Scholar]
- Shao J, Evers BM, Sheng H (2004) Prostaglandin E2 synergistically enhances receptor tyrosine kinase‐dependent signaling system in colon cancer cells. J. Biol. Chem. 279, 14287–14293. [DOI] [PubMed] [Google Scholar]
- Shao J, Sheng H, Inoue H, Morrow JD, Dubois RN (2000) Regulation of constitutive cyclooxygenase‐2 expression in colon carcinoma cells. J. Biol. Chem. 275, 33951–33956. [DOI] [PubMed] [Google Scholar]
- Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J (1997) Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase‐2. J. Clin. Invest. 99, 2254–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Shao J, Morrow JD, Beauchamp RD, Dubois RN (1998) Modulation of apoptosis and Bcl‐2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 58, 362–366. [PubMed] [Google Scholar]
- Sheng H, Shao J, Washington MK, Dubois RN (2001) Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J. Biol. Chem. 276, 18075–18081. [DOI] [PubMed] [Google Scholar]
- Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM (2000) Combinatorial chemoprevention of intestinal neoplasia. Nat. Med. 6, 1024–1928. [DOI] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, Dubois RN (1997) Cyclooxygenase‐2 expression in human colon cancer cells increases metastatic potential. Proc. Natl Acad. Sci. USA 94, 3336–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TD, Mitchell JA (2004) Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 18, 790–804. [DOI] [PubMed] [Google Scholar]
- Williams CS, Watson AJ, Sheng H, Helou R, Shao J, Dubois RN (2000) Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res. 60, 6045–6051. [PubMed] [Google Scholar]
- Zhang H, Sun XF (2002) Overexpression of cyclooxygenase‐2 correlates with advanced stages of colorectal cancer. Am. J. Gastroenterol. 97, 1037–1041. [DOI] [PubMed] [Google Scholar]