

Abstract
Abstract. Objectives: We have evaluated the physiological roles of transforming growth factor‐β1 (TGF‐β1) on differentiation, migration, proliferation and anti‐apoptosis characteristics of cultured spinal cord‐derived neural progenitor cells. Methods: We have used neural progenitor cells that had been isolated and cultured from mouse spinal cord tissue, and we also assessed the relevant reaction mechanisms using an activin‐like kinase (ALK)‐specific inhibitory system including an inhibitory RNA, and found that it involved potential signalling molecules such as phosphatidylinositol‐3‐OH kinase (PI3K)/Akt and mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated kinase (ERK1/2). Results and Conclusions: Transforming growth factor‐β1‐mediated cell population growth was activated after treatment and was also effectively blocked by an ALK41517‐synthetic inhibitor (4‐(5‐benzo(1,3) dioxol‐5‐yl‐4‐pyridine‐2‐yl‐1H‐imidazole‐2‐yl) benzamide (SB431542) and ALK siRNA, thereby indicating the involvement of SMAD2 in the TGF‐β1‐mediated growth and migration of these neural progenitors cells (NPC). In the present study, TGF‐β1 actively induced NPC migration in vitro. Furthermore, TGF‐β1 demonstrated extreme anti‐apoptotic behaviour against hydrogen peroxide‐mediated apoptotic cell death. At low dosages, TGF‐β1 enhanced (by approximately 76%) cell survival against hydrogen peroxide treatment via inactivation of caspase‐3 and ‐9. TGF‐β1‐treated NPCs down‐regulated Bax expression and cytochrome c release; in addition, the cells showed up‐regulated Bcl‐2 and thioredoxin reductase 1. They also had increased p38, Akt and ERK1/2 phosphorylation, showing the involvement of both the PI3K/Akt and MAPK/ERK1/2 pathways in the neuroprotective effects of TGF‐β1. Interestingly, these effects operate on specific subtypes of cells, including neurones, neural progenitor cells and astrocytes in cultured spinal cord tissue‐derived cells. Lesion sites of spinal cord‐overexpressing TGF‐β1‐mediated prevention of cell death, cell growth and migration enhancement activity have been introduced as a possible new basis for therapeutic strategy in treatment of neurodegenerative disorders, including spinal cord injuries.
INTRODUCTION
Stem cells are characterized by their capacity for self‐renewal and differentiation into distinct cell types that perform specialized functions. Since their discovery, neural stem/progenitor cells have evoked great interest due to their potential to differentiate into neural cell lineages, thereby providing a renewable resource for cell‐based therapies for the treatment of neurological diseases. Multiple signalling networks are known to orchestrate the self‐renewal and differentiation of neural stem/progenitor cells, including that of the transforming growth factor‐β (TGF‐β) family of proteins (Bakin et al. 2000, 2002; Massague & Wotton 2000; Bhowmick et al. 2001). Among these, TGF‐β1 has been shown to exert tissue‐specific stimulatory or inhibitory effects on a variety of cellular functions, including differentiation, proliferation and apoptosis (Mattson et al. 1997; Flanders et al. 1998; Krieglstein et al. 2002). For example, TGF‐β1 has been shown to induce autonomic gangliogenesis and proliferation of olfactory neurones during development (Gangemi et al. 2004). Moreover, several groups have shown that TGF‐β1 exerts a neuroprotective effect after cerebral injuries (Henrich‐Noack et al. 1996; Ruocco et al. 1999; Pang et al. 2001; Tikka et al. 2001) and on cultured neurones exposed to a variety of stress inducers (Prehn et al. 1996; Ren & Flanders 1996). The neuroprotective mechanism of TGF‐β1 has been linked to its capacity to induce the expression of plasminogen activator inhibitor‐1 (Buisson et al. 1998; Docagne et al. 2002), Bcl‐2 and Bcl‐xl (Prehn et al. 1994), as well as to regulate Ca2+ homeostasis and inhibit caspase‐3 activation. In this study, we have also demonstrated that TGF‐β1 inactivates the pro‐apoptotic protein Bax via activation of the mitogen‐activated protein kinase (MAPK) pathway. Therefore, although the Small Mothers Against Decapentaplegic (SMAD) was the first signalling pathway demonstrated to mediate the effects of TGF‐β1, a growing number of studies have indicated that this cytokine may also stimulate MAPK and phosphatidylinositol‐3‐OH kinase (PI3k)/Akt pathways (Wyss‐Coray et al. 1995; Yrjanheikki et al. 1998). Here, we have assessed whether MAPK, PI3K/Akt or SMAD activation might contribute to the anti‐apoptotic effects of TGF‐β1 on spinal cord‐derived neural progenitor cells in a model of spinal cord injury.
MATERIALS AND METHODS
Animals and spinal cord injury induction and histological analysis
Adult female Wistar rats weighting 260 g or at 6 weeks of age were used in our experiments. Animal care was in compliance with Korean regulations on protection of animals used for experimental and other scientific purposes. They were subjected to traumatic injury followed by a modified protocol as described in detail in Kang et al. (2006). Briefly, rats were anaesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg). Spinous processes of thoracics 9 and 10 were removed with rongeurs and a laminectomy was performed using a dental drill and rongeurs to expose the dorsal spinal cord. Dorsal and ventral columns, which include the dorsal and ventral corticospinal tract, were cut by lowering microscissors attached to a stereotaxic arm to a depth of 3.5 mm below the dorsal surface of the spinal cord and cutting twice. Experimental trials were first performed to ascertain that this surgical procedure produced a consistent total transection of the corticospinal tract on both sides. The animals were sacrificed after being anaesthetized with sodium pentobarbital (60 mg/kg, intraperitoneally) and their spinal cords were fixed by transcardial perfusion with 0.1 M PBS (phosphate‐buffered saline) followed by fixation in 4% paraformaldehyde in 0.1 m phosphate buffer. Fixed spinal cords were equilibrated in 30% sucrose for 48 h and then frozen blocks were cut into 10 µm coronal sections on a cryotome. Intact control spinal cords and one sciatic nerve were also processed in the same manner. For analysis of protein up‐regulation in injured spinal cords, tissues at the centre of the lesion were incubated overnight at 4 °C with primary antibodies against anti‐TGF‐β1 (1 : 400, Sigma, St. Louis, MO, USA). After extensive washing with PBS, tissue sections were incubated for 30 min with fluorescein isothiocyanate‐secondary antibodies (1 : 250, Molecular Probe, Eugene, OR, USA); cell nuclei were labelled with Topro‐3 (Molecular Probe). Results were analysed by confocal microscopy (Leica Microsystems, Exton, PA, USA) using a Leica TCS sp2 laser scanning microscope, equipped with three lasers. Immunohistochemical procedures were repeated at least three times.
Isolation of adult spinal cord‐derived neural progenitors cells
Anaesthetized rats were sacrificed by decapitation and then the region of the complete cervical enlargement (spinal cord level cervical 3 through T1) was dissected. After removal of the dura, tissue was minced, washed in sterile PBS and incubated for 30 min at 37 °C in Hank's balanced salt solution containing 0.125% trypsin and 0.01% DNase (Sigma, St. Louis, MO, USA). Cells were then plated at a density of 5 × 105 in 10 cm dishes and were cultured in neurobasal (NB) medium containing B27 supplement, basic fibroblastic growth factor (20 ng/mL) and epidermal growth factor (20 ng/mL). Cells were either grown as neurospheres in uncoated plates or as a monolayer on poly‐D‐lysine coated plates.
Transforming growth factor‐β1 treatment and H2O2 exposure
Spinal cord‐derived neural progenitors cells (NPC) were seeded on 10 cm dishes at a density of 5 × 105 and cultured in NB medium for 8 h in a humidified chamber with 5% CO2. They were then pre‐treated with TGF‐β1 (10 ng/mL) for 8 h followed by H2O2 (0.3 mm) for a further 6 h. Optimum concentrations of TGF‐β1 and H2O2 were selected based on results obtained from cytotoxicity studies using a broad concentration range for each reagent. Dilutions of H2O2 were made from a 30% stock solution freshly prepared before each experiment. We observed that toxicity of H2O2 was reduced if H2O2 exposures were not performed within 5 min of dilution. This is likely due to the highly reactive nature of hydrogen peroxide and its short half‐life in dilute solutions.
Analysis of cell viability
Viability of spinal cord‐derived NPCs was assessed by counting the number of live cells after staining with trypan blue. Apoptosis was evaluated using the deoxynucleotidyl transferase (TdT) in situ apoptosis detector kit (Roche, Indianapolis, IN, USA) according to the manufacturer's specifications. After fixation, cultured cells fixed in 4% paraformaldehyde were incubated in TUNEL (TdT‐mediated dUTP‐X nick end labelling) reaction mixture containing TdT buffer with TdT and biotinylated dUTP, incubated in a humidified atmosphere at 37 °C for 90 min, and then washed with PBS. Cells were incubated at room temperature for 30 min with antihorseradish peroxidase‐conjugated antibody, and signals were visualized with diaminobenzidine. The results were analysed under a fluorescence microscope (Leica Microsystems), and TUNEL‐positive apoptotic cells were quantified by counting positively stained cells. Three digital microscopic images at a magnification ×100 were randomly captured at areas where positive cells were abundant around the lesion site, for each section. The number of positively stained cells in the three images was averaged. Three sections at each of the two areas were examined by an observer blind to the treatment, and the mean value of cell counting was used. The result was expressed as relative percentage of positive cells per field of view. For the assay of caspase‐3, ‐8 and ‐9 activities, 10 µg of protein in 50 µL total volume was mixed with 50 µL of equilibrated Caspase‐Glo 3/7, 8 or 9 reagents (Promega, Madison, WI, USA). After incubating at room temperature for 1 h, luminescence was measured using a TD‐20/20 luminometer (Turner Designs, Sunnyvale, CA, USA). Blank values were subtracted and fold increases in activity were calculated based on activity measured from untreated cells. Each sample was measured in triplicate. Each assay was performed in triplicate and each experiment was repeated at least three times. Raw data from each experiment were analysed using analysis of variance either with the Fisher or the t‐test.
5‐Bromo‐2′‐deoxyuridine incorporation
The proliferative potential of spinal cord‐derived NPCs was assessed by measuring their uptake of 5‐bromo‐2′‐deoxyuridine (BrdUrd) using a commercial kit (BD Pharmingen, San Diego, CA, USA). Briefly, cells were incubated for 24 h in growth media supplemented with 10 µm BrdUrd (Sigma, Deisenhofen, Germany). 72 h after TGF‐β1 stimulation (10 ng/mL), NPCs were fixed using 4% paraformaldehyde for 15 min and then were immunostained using an anti‐BrdUrd antibody according to the manufacturer's instructions. Fluorescence was monitored using an inverse confocal laser scanning microscope (LSM 5, Pascal, Carl Zeiss, Jena, Germany). Relative cell proliferation was calculated from fluorescence of five fields of view (n = 4 for each condition). Differences in relative cell proliferation were assessed by two‐way anova followed by a post hoc t‐test. Differences between two conditions at P ≤ 0.05 and P < 0.001 were considered statistically significant.
Cell migration assay
Cells were transferred to culture dishes containing serum‐free growth medium, which consisted of NB medium with B27 supplement and TGF‐β1 (10 ng/mL). They were grown as neurospheres in Petri dishes, or poly‐D‐lysine coated culture dishes. Cultured neurospheres, transferred onto transwell membranes (Costar, Cambridge, MA, USA; 8 µm pore size) were coated on both sides with laminin, and were placed on 6‐well plates. Below the membrane, TGF‐β1 (10 ng/mL) containing NB/B27 media was added to each well. To the upper chamber, 200–300 µL of NPC suspension were added and were incubated for 2 h at 37 °C in NB/B27 medium, and the plate was incubated overnight at 37 °C in a CO2 incubator. Cells on the lower surface were air dried and counterstained with Harris’ haematoxylin for 20 min and were washed. Stained inserts were placed on object slides and the numbers of cells on lower surfaces were assessed at ×200 using an inverted bright field microscope. Ten × 20 fields per insert were counted. Spontaneous migration (expressed as a percentage of cells per field), towards the bottom, was assessed.
Immunostaining
To determine expression of neural markers, differentiated spinal cord‐derived NPCs were fixed with 4% paraformaldehyde solution for 30 min at room temperature. After extensive washing with PBS, cells were blocked at room temperature for 30 min with 1% normal goat serum. Then, they were incubated with primary antibodies against nestin (1 : 200, Sigma, St. Louis, MO, USA), TuJ (1 : 250, Sigma, St. Louis, MO, USA) and glial fibrillary acidic protein (GFAP) (1 : 1500, DAKO, Carpinteria, CA, USA). After further extensive washing in PBS, they were incubated for 30 min with FITC or Texas Red‐conjugated secondary antibodies (1 : 250, Molecular Probes). Cells were labelled with 4′,6‐diamidino‐2‐phenylindoline (Vector Laboratories, Peterborough, UK) and were analysed using a fluorescence microscope (Leica Microsystems).
RNA extraction and semiquantitative real‐time polymerase chain reaction
Total cellular RNA was extracted from NPCs with Trizol (Life Technologies, Frederick, MA, USA) that were reverse transcribed into first strand cDNA, using an oligo‐dT primer. cDNA was then amplified by polymerase chain reaction (PCR) with 20 pM of specific primers for 35 cycles (94 °C, 1 min; 55 °C, 1 min; 72 °C, 1 min). Results of PCR reactions were electrophoresed through a 1.5% agarose gel. Alternatively, real‐time PCR was performed using an ABI7700 Prism Sequence Detection System with the SYBR green kit (Applied Biosystems, Foster, CA, USA). Primer sequences were designed using Primer Express software (PE‐Applied Biosystems, Warrington, UK) and gene sequences were obtained from the GeneBank database.
Western blot analysis
Spinal cord‐derived NPCs exposed to TGF‐β1 and/or H2O2 for the indicated times were lysed in 500 µL of lysis buffer (20 mm Tris‐HCl [pH 7.5], 150 mm NaCl, 1 mm ethylenediaminetetraacetic acid, 1% Triton X‐100, 2.5 mm sodium pyrophosphate, 1 mm ethyleneglycoltetraacetic acid, 1 mm glycerophosphate, 1 mm Na3VO4 and 1 mm phenylmethylsulphonyl fluoride), clarified by centrifugation at 15 000 g for 10 min, and total protein content was determined using a Bio‐Rad protein assay kit (Milan, Italy). For Western blot, equal amounts (40 µg) of protein extract in lysis buffer were subjected to 10% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis analysis and were transferred to a nitrocellulose membrane. Anti‐GFAP (1 : 3000, DAKO), antinestin (1 : 500, Sigma, St. Louis, MO, USA), anti‐TuJ (1 : 300, Sigma, St. Louis, MO, USA), anti‐SMAD1/2 (1 : 1000, Cell Signalling, Beverly, MA, USA), anti‐pERK (1 : 1000, Cell Signalling), anti‐Bcl‐2 (1 : 1000, Cell Signalling), anti‐Bax (1 : 1000, Cell Signalling), anti‐p38 (1 : 1000, Cell Signalling), anti‐Akt (1 : 1000, Cell Signalling), anti‐GSKβ1 (1 : 1000, Cell Signalling), anticytochrome c (1 : 1000, Cell Signalling) and anti‐β‐actin (1 : 500, Sigma, St. Louis, MO, USA) antibodies were incubated with membranes. Relative band intensities were determined by Quality‐one 1‐D analysis software (Bio‐Rad, Hercules, CA, USA). Anti‐thioredoxin reductase 1 antibody was kindly provided by Dr. SR Lee, Chonnam National University, Jeollanam‐do, South Korea.
Neural progenitors cell growth inhibition by activin‐like kinase siRNA
Two complementary hairpin siRNA template oligonucleotides containing 21 target sequences of mouse activin‐like kinase were used for transient transfection with LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA) using 50 nm siRNA; the amount of siRNA had been optimized as per the manufacturer's instructions. Three separate activin‐like kinase (ALK) siRNAs (Silencer® predesigned siRNA, Ambion, Austin, TX, USA), and scrambled siRNA with the same nucleotide content were tested. When compared with unrelated control siRNA and scrambled siRNA, the ALK siRNA resulted in an 80–95% decrease in ALK1 mRNA and protein levels as determined by real‐time PCR. The siRNA that provided most efficient inhibition (90–95%) was used for experiments. For detection of growth inhibition of NPCs by ALK siRNA, we transfected siRNA into cultured NPCs and then counted dye‐excluding viable cells for 6 days.
Statistical analysis
All data are presented as mean ± SEM from five or more independent experiments. The statistical significance of difference between groups was calculated by using Student's two tailed t‐test.
RESULTS
Transforming growth factor‐β1‐induced cell proliferation involves activation of SMAD2 and runx, AP2 and forkhead boxes
Exposure of adult spinal cord‐derived NPCs to varying concentrations of TGF‐β1 (0, 2, 5 and 10 ng/mL) over a time course of 6 days generated a dose‐dependent increase in cell proliferation, as measured by BrdUrd uptake (Fig. 1a,b). This effect of TGF‐β1 on cell population growth was inhibited in a dose‐dependent manner by the addition of ALK41517 synthetic inhibitor SB431542 and by ALK siRNA (Fig. 1c–f). Western blot analysis showed that both extracellular‐regulated kinase (ERK1/2) and SMAD2 were activated in spinal cord‐derived NPCs by TGF‐β1 treatment, and that the level of activation was detectable after only 15 min of exposure (Fig. 1c). By way of contrast, Western blot using a phospho‐specific antibody against pJNK failed to reveal activation of this kinase by TGF‐β1, at all examined time points. When the cells were cultured in TGF‐β1 in the presence of the SMAD2 inhibitor SB431542, activation of SMAD2 and ERK1/2 were inhibited in a dose‐dependent manner (Fig. 1f). Finally, TGF‐β1 treatment also induced expression of runx2 and activating protein transcription factor 2 (AP2) in spinal cord‐derived NPCs, but did not significantly alter the levels of fox mRNA expression (Fig. 1d).
Figure 1.
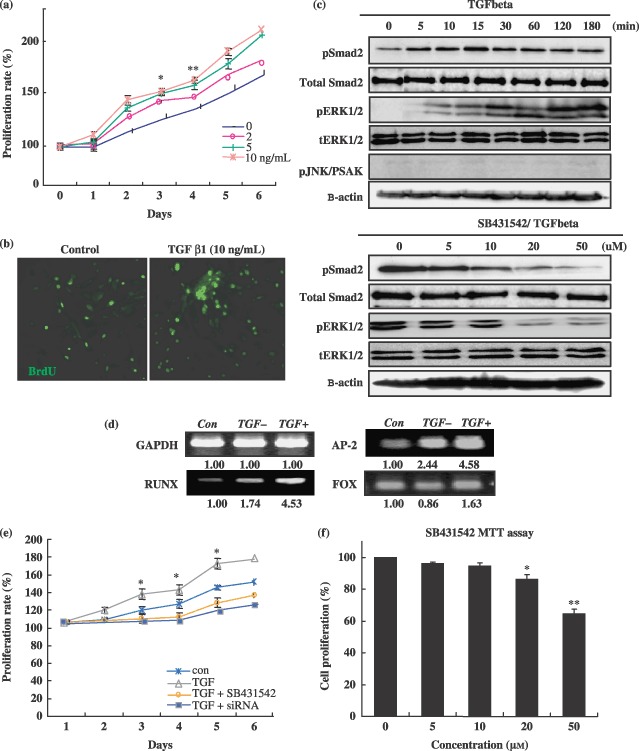
Effects of transforming growth factor‐β1 (TGF‐β1) on neural progenitor cell population growth. Cultured neural progenitors cells (NPC) were treated with TGF‐β1 alone or with TGF‐β1 inhibitor SB431542 pre‐treatment. (a) To determine TGF‐β1's effect on cell proliferation, visual cell counts were conducted in conjunction with trypan blue exclusion methods and MTT assays. (b) The proliferation potential of spinal cord‐derived NPCs was assessed by measuring their uptake of 5‐bromo‐2′‐deoxyuridine (BrdUrd) with a commercial kit. In all proliferation assays, triplicate wells were used for each condition, and each experiment was repeated at least three times. (c–f) Transforming growth factor β1‐mediated activation of SMAD2 and proliferation‐related transcript expression during proliferation of neural progenitor cells. In order to block SMAD2 activation, SB431542 at various concentrations was added to the cells 2 h prior to TGF‐β1 treatment. Control cultures received vehicle treatment only. Cells were harvested 3 h after addition of TGF‐β1 for Western blot and RT‐PCR of SMAD2 and proliferation‐related genes. Data are expressed as the means ± standard deviation (*P < 0.001; **P < 0.05).
In an effort to determine whether TGF‐β1 exerted specific effects on distinct subpopulations of progenitor cells, populations exposed to TGF‐β1 in the presence of BrdUrd were simultaneously stained with antibodies against BrdUrd and specific neural markers. BrdUrd incorporation was more than 2.5 times higher in the TGF‐β1‐treated cells than in the untreated controls (data not shown). Moreover, cells induced to proliferate in response to TGF‐β1, as shown by BrdUrd incorporation, co‐expressed the neural stem cell marker nestin or the oligodendrocyte marker A2B5 (Fig. 2). Thus, TGF‐β1 significantly (*P < 0.001, **P ≤ 0.05) activated proliferation of spinal cord‐derived NPCs.
Figure 2.
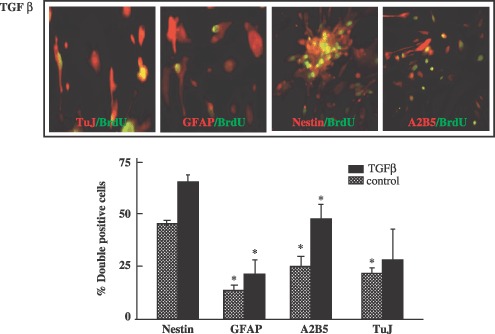
Transforming growth factor‐β1 effect on cell proliferation, cultured neural progenitors cell (NPC) subtypes after BrdU incorporation. For BrdUrd assays, 10 µm BrdUrd was added to the NPC cultures daily. 72 h after TGF‐β1 stimulation (10 ng/mL), the cultured cells were fixed with 3.7% PFA for 60 min at 4 °C and were stained with BrdUrd primary antibody. Incorporation of BrdUrd into newly synthesized DNA in the NPCs was detected via BrdUrd‐dependent fluorescence enhancement of ToPro‐3. Fluorescence was monitored using an inverse confocal laser scanning microscope. Relative cell proliferation was calculated from fluorescence of five fields of view (n = 4 for each condition) as follows, relative percentage of cell proliferation. Differences in relative cell proliferation were evaluated via anova followed by a post hoc t‐test (*P < 0.001).
Transforming growth factor‐β1 induces neural progenitor cell migration
In addition to its effects on cell population growth, TGF‐β1 was also shown to induce migration of spinal cord‐derived NPCs. In these studies, neurospheres derived from the progenitor populations were dissociated and were plated into transwell inserts. Inserts were then transferred to multiwell plates and were cultured for 16 h in media supplemented with varying TGF‐β1 concentrations (Fig. 3). As is shown in Fig. 4b, TGF‐β1 (2 and 10 ng/mL) actively stimulated migration of spinal cord‐derived NPCs in a dose‐dependent fashion, and this effect was inhibited by the ALK‐specific inhibitor, SB431542. Thus, it was shown that TGF‐β1 stimulates chemotaxis of spinal cord‐derived NPCs in vitro.
Figure 3.
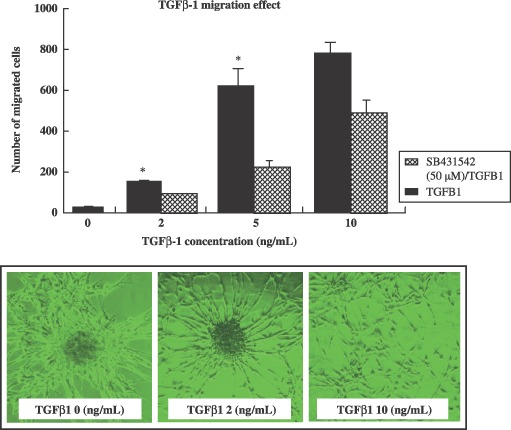
The effect of transforming growth factor‐β1 (TGF‐β1) on cultured neural progenitor cell migration. Cells were transferred to culture dishes containing serum‐free growth medium, which consisted of NB medium with B27 supplement and TGF‐β1 (10 ng/mL) with or without ALK inhibitor (SB431542) pre‐treatment. Cells were grown as neurospheres in Petri dishes, or in poly‐D‐lysine coated culture dishes. Cultured neurospheres, transferred to transwell membranes (Costar, 8 µm pore size) were coated on both sides with laminin, and were placed in 6‐well plates. Below the membrane, TGF‐β1 containing NB medium with B27 supplement was added to each well. To the upper chamber, NPC cells were incubated overnight in a 37 °C CO2 incubator. Cells on the lower surface were air‐dried and counterstained with Harris’ haematoxylin for 20 min, then washed. The stained inserts were placed on object slides, and numbers of cells on lower surfaces were assessed at ×200 using an inverted bright field microscope. Migration is expressed as a percentage (of cells per field) of spontaneous migration towards the cell bottom. Data are expressed as the means ± standard deviation (*P < 0.001).
Figure 4.
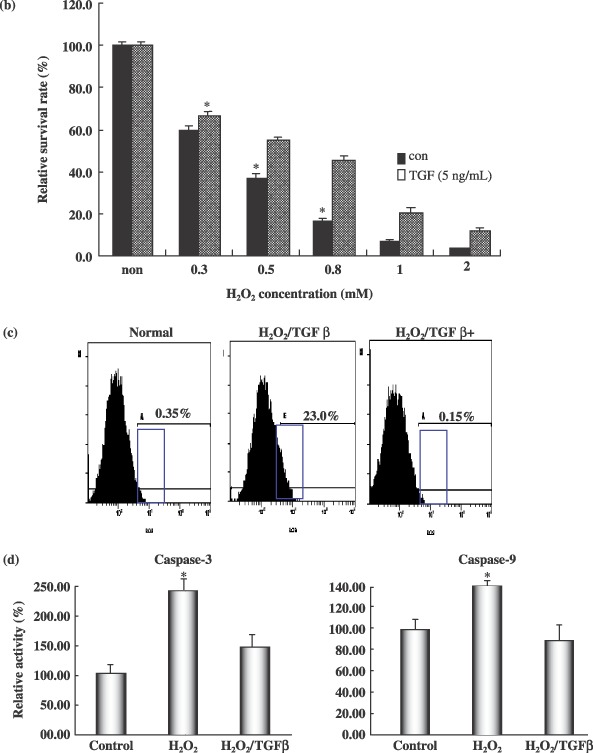
Transforming growth factor‐β1 reduced apoptotic cell death and affected H2O2‐induced apoptotic features in cultured neural progenitor cells. (a) Cultured neural progenitors cells (NPC) were treated with various concentrations of H2O2 alone or with various concentrations of TGF‐β1 pre‐treatment. Cell viability was assessed by TUNEL staining. TUNEL‐positive apoptotic cells were quantified by counting the positively stained cells. (b) Transforming growth factor‐β1 effect on cell survival at the various concentrations of hydrogen peroxide. Visual cell counts were conducted in conjunction with trypan blue exclusion methods and MTT assays. In all viability assays, triplicate wells were employed for each condition, and each experiment was repeated at least three times. Raw data from each experiment were analysed using analysis of variance with Fisher's or t‐tests. Each of the samples was measured in triplicate. (c) For caspase‐3 and ‐9 activity assays, 10 µg of protein in a total volume of 50 µL was mixed with 50 µL of equilibrated Caspase‐Glo 3/7, 8 or 9 reagents (Promega). After 1 h of incubation at room temperature, luminescence was measured using a TD‐20/20 luminometer (Turner Designs). Blank values were subtracted and fold‐increases in activity were calculated on the basis of activity measured for untreated cells. Each sample was measured in triplicate. Data are expressed as relative activity (%) against protein‐free blank reaction mixtures. (d) Intracellular reactive oxygen species (ROS) was assessed using the fluorescent probe DCFDA. For fluorescent microscopic visualization, the NPCs were plated in glass‐bottomed dishes and were treated with 50 µm tBHP, alone or with SS‐31, for 6 h. Cells were then washed and loaded with 10 µm of DCFDA for 30 min at 37 °C, and imaged via fluorescent microscopy (ex/em = 495/525 nm). For quantitative assessments of ROS production, NPC cells in 6‐well plates were washed with HBSS and loaded with 10 µm of DCFDA for 30 min at 37 °C. Cells were washed three times with HBSS and exposed to 0.3 mm of H2O2, alone or with TGF‐β1 (10 ng/mL). DCF oxidation was monitored in real time with a microplate spectrofluorometer (Molecular Devices) using ex/em wavelengths of 485/530 nm. Data are expressed as means ± standard deviations (*P < 0.001).
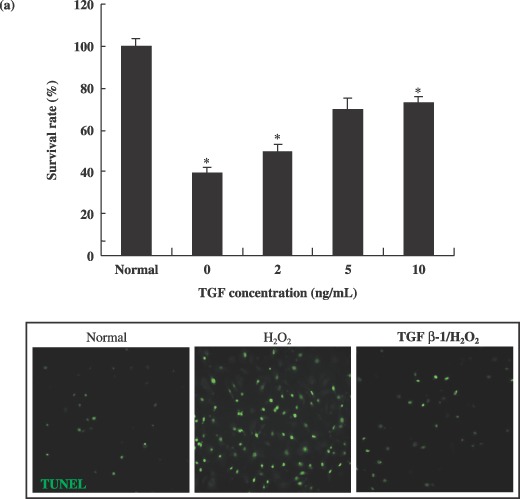
Transforming growth factor‐β1 protects neural progenitor cell from H2O2‐induced apoptotic cell death
Exposure of spinal cord‐derived NPCs to increasing hydrogen peroxide concentrations resulted in a progressive reduction in cell survival, such that concentrations in excess of 0.3 mm generated a statistically significant (P < 0.001) reduction in the numbers of viable cells (Fig. 4a,b). TUNEL assays confirmed that a majority of H2O2‐exposed cells underwent apoptotically mediated cell death (Fig. 4a). Typically, apoptosis of spinal cord‐derived NPCs was evident 2 days after H2O2 exposure. However, pre‐treatment of cells with 5 ng/mL of TGF‐β1 resulted in a dramatic reduction in the extent of apoptosis, leading to a significant increase in cell survival after exposure to H2O2 (P < 0.001) (Fig. 4a). Sixty percent of NPCs pre‐treated with 10 ng/mL of TGF‐β1 survived after exposure to H2O2 (Fig. 4a). In addition, NPCs were capable of growth in a TGF‐β1‐containing medium at a concentration of 10 ng/mL after being subjected to treatment with 0.3 mm H2O2 (Fig. 4b). TGF‐β1 at concentrations of 5 and 10 ng/mL facilitated survival and growth of these spinal cord‐derived NPCs over a 5‐day period. Neural progenitor cells grew well from the 2nd to the 5th day, with 5 and 10 ng/mL of TGF‐β1 being the optimal pre‐treatment concentration (Fig. 4a).
Transforming growth factor‐β1 inhibits H2O2‐induced apoptotic features in cultured neural progenitor cells
In order to characterize the mechanism by which TGF‐β1 blocked apoptosis of spinal cord‐derived NPCs, we assessed levels of reactive oxygen species (ROS) in cells after H2O2 exposure. As is shown in Fig. 4d, spinal cord‐derived NPCs loaded with membrane permeable molecule 2′‐7′‐dichlorofluorescein‐diacetate (DCFDA), showed marked increases in intracellular fluorescence following H2O2 exposure. However, H2O2‐induced increases in intracellular fluorescence were essentially abolished by pre‐treatment of cells with 10 ng/mL of TGF‐β1 (Fig. 4d). TGF‐β1 itself did not alter ROS production, but TGF‐β1 treatment indirectly induced a reduction in ROS generation, from 23% to 0.15% (Fig. 4d). H2O2 has also been demonstrated to induce caspase activity in cells (Liu et al. 2000). Consistent with this finding, we determined that activities of caspase‐3 and ‐9 were increased approximately 2.5‐ and 1.4‐fold, respectively, in spinal cord‐derived NPCs following H2O2 exposure. By way of contrast, caspase‐3 and ‐9 activities were induced only by 1.5‐ and 0.8‐fold, respectively, as the result of H2O2 exposure in NPCs pre‐treated with TGF‐β1, which was significantly lower than that measured in those that did not receive TGF‐β1 pre‐treatment (P < 0.001) (Fig. 4c). Taken together, these findings show that TGF‐β1 inhibits H2O2‐induced increases in ROS and caspase activity in spinal cord‐derived neural progenitor cells. We also attempted to determine whether TGF‐β1 could inhibit H2O2‐induced increases in cytochrome c release from mitochondria of exposed NPCs. The results of Western blot analysis verified that TGF‐β1 inhibited expression of other signal proteins, p38 and c‐Jun–NH2‐terminal kinase (SAPK/JNK) in H2O2‐exposed cells. As a result, TGF‐β1 treatment enhanced down‐regulated expression of p38 (Fig. 5a). Furthermore, TGF‐β1 pre‐treatment induces thioredoxin reductase 1 (cytosolic) expression after treatment with H2O2 (Fig. 5a). However, H2O2 induced expression of the pro‐apoptotic protein Bax in the NPCs, but TGF‐β1 pre‐treatment attenuated these effects (Fig. 5). Moreover, pre‐treatment with TGF‐β1 also induced expression of specific cell survival factors in NPCs, including Bcl‐2 and Akt (Fig. 5a). Taken together, these findings show that TGF‐β1 promotes survival via prevention of H2O2‐induced activation of the apoptotic pathway in the cells. In addition, TuJ‐positive neurones and the GFAP‐expressing subpopulation generally survived after TGF‐β1/H2O2 treatment (Fig. 5b). We also attempted to determine whether the MAPK/ERK1/2 and PI3k/Akt pathways were involved in induction of cell survival by TGF‐β1. As is shown in Fig. 6a, TGF‐β1 significantly (P < 0.001) increased cell survival by activation of the PI3K/Akt signalling pathway and by in‐activation of Bax pro‐apoptotic protein. The PI3K inhibitor LY294002 induced a significant reduction in cell survival after H2O2 treatment in NPC cell cultures (Fig. 6b) (P < 0.001). In addition, levels of pAkt and cell survival were detected in cell cultures exposed to LY294002 in order to clarify whether TGF‐β1‐mediated cell survival was dependent on activation of the PI3k/Akt‐signalling pathways. TGF‐β1 (10 ng/mL) increased cell survival and pAkt levels beginning 15 h after H2O2 treatment (Fig. 6). TGF‐β1‐mediated pAkt activation was profoundly reduced by LY294002 (Fig. 6a). Our results indicated that TGF‐β1 attenuated considerably mitochondria‐ and Akt/PI3K‐mediated apoptotic cell death signal pathways, and ultimately negatively regulated neuronal cell death in cultured spinal cord‐derived NPC cells (5, 6).
Figure 5.
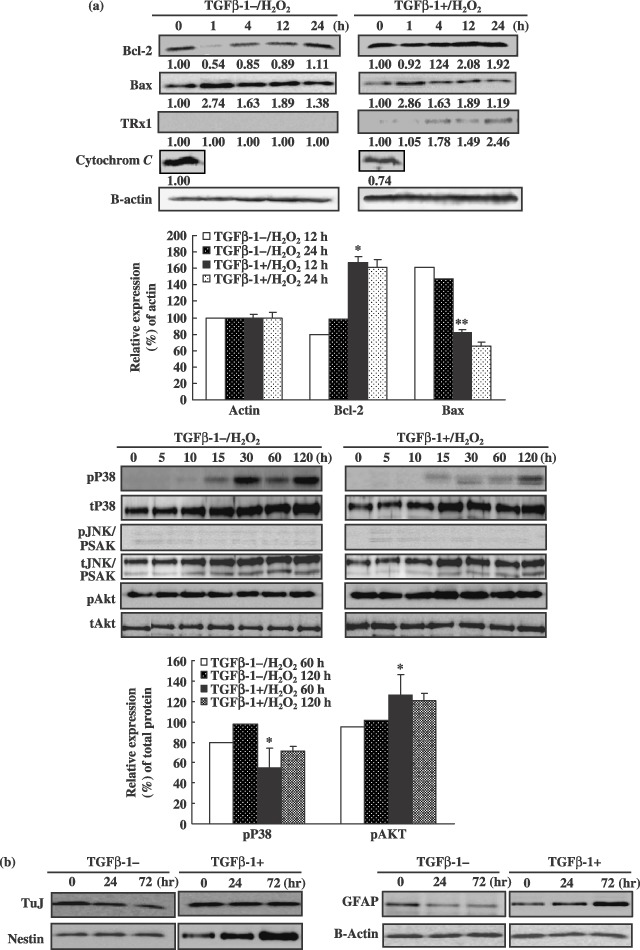
Differential expressions of apoptotic, survival and proliferation signal proteins and neural marker expression in the neural progenitor cells after transforming growth factor‐β1 (TGF‐β1) treatment. (a) Western blots and densitogram showing expression levels of p38, pJNK/SAPK and Akt over a time course of H2O2 exposure or cytochrome c, Bax and Bcl‐2 in neural progenitors cells (NPC) with or without TGF‐β1 pre‐treatment. Data are expressed as the means ± standard deviations (*P < 0.05; **P < 0.001). (b) To confirm differentially expressed neural markers with or without TGF‐β1 treatment, Western blots of cultured NPCs cells were analysed.
Figure 6.
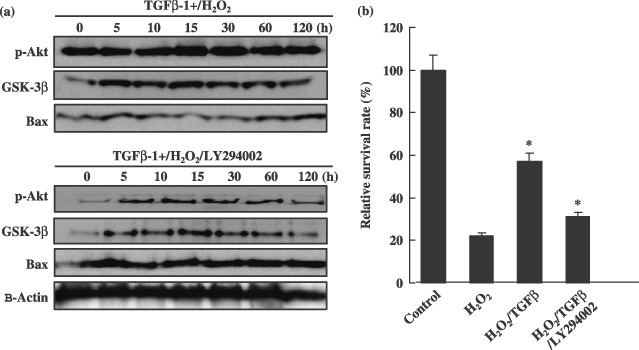
Transforming growth factor‐β1 induced cell survival via activation of the PI3K/Akt‐signalling pathway and inactivation of the Bax pro‐apoptotic protein. (a) Immunoanalysis of Akt, GSK‐3β and Bax expression after TGF‐β1/H2O2 or TGF‐β1/H2O2/LY294002 (10 µm) combined treatment. Confirmation of PI3kinase activity inhibition and differential expression of signal proteins were conducted via Western blot analysis of one of its downstream targets, Akt, including apoptotic cell death‐related proteins. (b) The PI3kinase inhibitor, LY294002, effectively mediated cell death and death signal protein expression in cultured NPC cells. After TGF‐β1/H2O2 or TGF‐β1/H2O2/LY294002 treatment (10 µm), cells were cultured for 3 days. After incubation, numbers of viable cells were measured by trypan blue exclusion. Data are plotted as means ± standard deviations (*P < 0.05).
DISCUSSION
Stem cells utilize multiple signalling networks to orchestrate their differentiation into functional neuronal, haematopoietic, mesenchymal and epithelial lineages, among other functions. Signalling gradients activated by the TGF‐β1‐related bone morphogenetic proteins often result in selection of a defined differentiation pathway. TGF‐β family signalling then further drives differentiation along the lineage, and maintains the differentiated phenotypes of epithelial, mesenchymal and other cell types via interactions with other growth factors that help promote the expansion of the cell population. During development, TGF‐β promotes differentiation and lineage expansion of established progenitors, for example, by inducing autonomic gangliogenesis or olfactory neurone proliferation (Gangemi et al. 2004). A reduction in cerebellar Purkinje cells in Smad4 −/– mice and proliferation of precursor cells in the developing cortex of mice lacking the SMAD adaptor protein embryonic liver fodrin (ELF), has previously been reported (Tang et al. 2003). In addition, astroglial overproduction of TGF‐β1 has been shown to exacerbate inflammatory central nervous system diseases in transgenic mice (Wrana et al. 1992) and also rat spinal cord injury lesion side (Supplementary Fig. 1). By way of contrast, the overexpression or injection of TGF‐β1 into the central nervous system has resulted in reduction in microglial cell activation and reduced induction of pro‐inflammatory chemokines after severe hypoxic–ischaemic injury (Gross et al. 1993; McNeill et al. 1994; Henrich‐Noack et al. 1996). Thus, effects of TGF‐β on inflammatory cells after injury are both complex and contextual. TGF‐β conducts signalling via a complex of two type II and two type I transmembrane serine/threonine kinase receptors (Attisano et al. 1993; Letterio & Roberts 1998; Wakefield & Roberts 2002). Ligand–receptor interaction then results in phosphorylation of SMAD2 and SMAD3 by the type I receptor (Heldin et al. 1997; Derynck & Zhang 2003). These receptor‐activated SMADs subsequently form a complex with the pathway‐independent SMAD4 (Lagna et al. 1996), finally translocating to the nucleus, in which they regulate transcription of specific genes (Massague & Wotton 2000). SMADs interact with a diverse array of transcription factors to induce TGF‐β‐regulated transcription (Ten & Hill 2004; Feng & Derynck 2005). However, although the SMAD proteins are crucial to many of the functions of TGF‐β, TGF‐β utilizes several different pathways for signalling within the cells, including SMAD‐independent pathways (Massague 2003). Although we now appreciate the central role of TGF‐β in mediation of the response to central nervous system injury, very little is known regarding in vivo signalling pathways that regulate central nervous system response to TGF‐β1 after injury.
The present study elucidates an additional novel mechanism underlying the neuroprotective effect of TGF‐β1 against apoptotic insult. We hypothesized that Akt/PI3K is a target of TGF‐β1 against apoptotic insult. Our biochemical studies proved that treatment of cultured NPCs with TGF‐β1 limited apoptosis, as shown by decreased expression of caspase‐3 and ‐9, together with a reduction in the number of TUNEL‐positive cells (Fig. 4a,c). Caspases are known to be important intracellular signalling molecules that promote apoptosis after spinal cord injury. Moreover, among members of the caspase family, caspase‐3 is regarded as the principal effector of apoptosis (Armstrond et al. 1997). Caspase‐3 is known to cleave poly(ADP‐ribose)‐polymerase (PARP) directly, which blocks DNA repair, but also preserves cellular pools of ATP that may be necessary for execution of the cell death programme (Burkle 2001). PARP activity is believed to contribute to neuronal cell death under a variety of neurological conditions (Skaper 2003), including traumatic brain injury (LaPlaca et al. 2001) and spinal cord injury (Scott et al. 1999). This is the result of energy failure or modification of the activity of various proteins by poly(ADP‐ribosylation) (Burkle 2001). At present, it remains unclear whether TGF‐β1 exerts its inhibitory effect on apoptosis at the level of cell surface receptor activation, cell signalling or transcriptional activation (Fig. 5). However, the results of our in vitro biochemical studies showed that TGF‐β1 promotes discernable changes in expressed levels of survival and proliferation‐promoting signalling molecules, including Bcl‐2, ERK1/2, p38 and pAkt in NPCs (Fig. 5). Other studies have shown that alterations in these signalling pathways have neuroprotective effects in brain injury models (Yrjanheikki et al. 1998; Ten & Hill 2004), and furthermore, it is well established that JAK/STAT3 and PLC/Raf/ERK1/2 signalling is induced by cytokines or other factors that promote cell survival and inhibit apoptosis (Roux & Blenis 2004). Phospho‐SAPK/JNK and p38 MAPK are stress‐induced signalling pathway molecules that may be involved in cytokine production and cell survival, differentiation, growth and apoptosis (Kyriakis & Avruch 2001). In conclusion, in this study we have evaluated cell population growth, migration and neuroprotective effects of TGF‐β1. Studies conducted in vitro have indicated that TGF‐β1 protects spinal cord‐derived NPCs against H2O2‐mediated apoptosis by down‐regulating the expression of a variety of apoptotic mediators, including caspase‐3 and Bax, as well as inducing expression of survival factors, most notably Bcl‐2. Additionally, TGF‐β1 effectively induced proliferation of NPCs via activation of the SMAD1 molecule in vitro. Our findings show that overexpressed TGF‐β1 in injury lesions plays an important role in mediation of the regeneration response to central nervous system injuries in vivo, which in turn regulates the response of the central nervous system to TGF‐β1 after injury.
Supporting information
Figure S1. Immunostaining with anti‐TGF‐&bgr;1 antibody at the lesion site of a spinal cord. One week after injury, TGF‐&bgr; antigen expression increased slightly in the lesion sites of spinal cord injury animals compared with that of normal animals. Normal rat spinal cord tissue expresses almost no TGF‐&bgr;1 antigen. For analysis of TGF‐&bgr;1 overexpression in lesion sites of injured spinal cords, tissues at the centre of the lesion were incubated with primary antibodies against anti‐TGF‐&bgr;1 (1 : 300). After extensive washing with phosphate‐buffered saline, these tissue sections were incubated for 30 min with fluorescein isothiocyanate‐secondary antibodies (1 : 250). Cell nuclei were labelled with Topro‐3 (Molecular Probe). Results were analysed using confocal microscopy. Numbers of TGF‐&bgr;1‐positive cells were compared between the two experimental groups.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
ACKNOWLEDGEMENTS
This work was supported by the 21st Century Frontier/Stem Cell Research Committee (SC3130).
REFERENCES
- Armstrond RC, Aja TJ, Hoand KD (1997) Activation of the CED3/ICE‐related protease‐CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J. Neurosci. 17, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L, Carcamo J, Ventura F, Weis FM, Massague J, Wrana JL (1993) Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 75, 671–680. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL (2000) Phosphatidylinositol 3‐kinase function is required for transforming growth factor β‐mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275, 36803–36810. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL (2002) p38 mitogen‐activated protein kinase is required for TGF‐β‐mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A (2001) Transforming growth factor β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA‐dependent mechanism. Mol. Biol. Cell 12, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson A, Nicole O, Docagne F, Sartelet H, Mackenzie ET, Vivien D (1998) Up‐regulation of a serine protease inhibitor in astrocytes mediates the neuroprotective activity of transforming growth factor β1. FASEB J. 12, 1683–1691. [PubMed] [Google Scholar]
- Burkle A (2001) Physiology and pathophysiology of poly (ADP‐ribosyl)ation. BioEssays 23, 795–806. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE (2003) Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 425, 577–584. [DOI] [PubMed] [Google Scholar]
- Docagne F, Nicole O, Gabriel C, Fernandez‐Monreal M, Lesne S, Ali C, Plawinski L, Carmeliet P, MacKenzie ET, Buisson A, Vivien D (2002) Smad3‐dependent induction of PAl‐1 in astrocytes mediates neuroprotective activity of TGF‐β1 against NMDA‐induced necrosis. Mol. Cell. Neurosci. 21, 634–644. [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R (2005) Specificity and versatihity in TGF‐β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF (1998) Transforming growth factor betas in neurodegenerative diseases. Prog. Neurobiol. 54, 71–85. [DOI] [PubMed] [Google Scholar]
- Gangemi RM, Perera M, Corte G (2004) Regulatory genes controlling cell fate choice in embryonic and adult neural stem cells. J. Neurochem. 89, 286–306. [DOI] [PubMed] [Google Scholar]
- Gross CE, Bednar MM, Howard DB, Sporn MB (1993) Transforming growth factor‐beta I reduces infarct size after experimental cerebral ischemia in a rabbit model. Stroke 24, 558–562. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, Ten Dijke P (1997) TGF‐beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471. [DOI] [PubMed] [Google Scholar]
- Henrich‐Noack P, Prehn JH, Krieglstein J (1996) TGF‐beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose–response relationship and potential neuroprotective mechanisms. Stroke 27, 1609–1614. [DOI] [PubMed] [Google Scholar]
- Kang SK, So HW, Moon YS, Kim CH (2006) Proteomic analysis of injured spinal cord tissue protein using 2 D‐E and MALDI‐TOF Mass. Proteomics 6, 2797–2812. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Strelau J, Shober A, Sullivan A, Unsicker K (2002) TGF‐β and the regulation of neuron survival and death. J. Physiol. (Paris) 96, 25–30. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (2001) Mammalian mitogen‐activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–886. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati‐Brivanlou A, Massague J (1996) Partnership between DPC4 and SMAD proteins in TGF‐beta signalling pathways. Nature 383, 832–836. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R (2001) Pharmacologic inhibition of poly (ADP‐b ribose) polymerase is neuroprotective following traumatic brain injury in rats. J. Neurotrauma 18, 369–376. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB (1998) Regulation of immune responses by TGF‐β. Annu. Rev. Immunol. 16, 137–161. [DOI] [PubMed] [Google Scholar]
- Liu D, Ling X, Wen J, Liu J (2000) The role of reactive nitrogen species in secondary spinal cord injury: formation of nitric oxide, peroxynitrite, and nitrated protein. J. Neurochem. 75, 2144–2154. [DOI] [PubMed] [Google Scholar]
- Massague J (2003) Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 17, 2993–2997. [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D (2000) Transcriptional control by the TGF‐beta/Smad signaling system. EMBO J. 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Furnkawa K, Bruce AJ, Wyss‐Coray T, Mark RJ, Mucke L (1997) Cellular signalling role of TGF‐β, TNFalpha and beta APP in brain injury responses and Alzheimer's disease. Brain Res. Rev. 23, 47–61. [DOI] [PubMed] [Google Scholar]
- McNeill H, Williams C, Guan J, Dragunow M, Lawlor P, Sirimanne E, Nikolics K, Gluckman P (1994) Neuronal rescue with transforming growth factor‐beta 1 after hypoxic‐ischaemic brain injury. NeuroReport 5, 901–904. [DOI] [PubMed] [Google Scholar]
- Pang L, Wen Y, Che XM, Roessler BJ, Betz AL, Yang GY (2001) Reduction of inflammatory response in the mouse brain with adenoviral‐mediated transforming growth factor‐β1 expression. Stroke 32, 544–552. [DOI] [PubMed] [Google Scholar]
- Prehn JHM, Bindokas VP, Jordan J, Galindo MF, Ghadge GD, Roos FP, Bosie LH, Tompson CB, Krajewski S, Reed JC, Miller RJ (1996) Protective effect of transforming growth factor‐β1 on β‐amyloid neurotoxicity in rat hippocampal neurons. Mol. Pharmacol. 49, 319–328. [PubMed] [Google Scholar]
- Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski D, Reed JC, Miller RJ (1994) Regulation of neuronal Bcl‐2 protein expression and calcium homeostasis by transforming growth factor beta confers wide‐ranging protection on rat hippocampal neurons. Proc. Natl. Acad. Sci. USA 91, 12599–12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren RF, Flanders KC (1996) Transforming growth factor‐β protects primary rat hippocampal neuronal cultures from degeneration induced by β‐amyloid peptide. Brain Res. 732, 16–24. [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J (2004) ERK and p38 MAPK‐activated protein kinases with diverse biological function. Microbiol. Mol. Biol. Rev. 68, 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A (1999) A transforming growth factor‐beta antagonist unmasks the ischemic brain injury. J. Cereb. Blood Flow Metab. 19, 1345–1353. [DOI] [PubMed] [Google Scholar]
- Scott GS, Jakeman LB, Stokes BT, Szabo C (1999) Peroxynitrite production and activation of poly (adenosine diphosphate‐ribose) synthetase in spinal cord injury. Ann. Neurol. 45, 120–124. [PubMed] [Google Scholar]
- Skaper SD (2003) Poly (ADP‐Ribose) polymerase‐1 in acute neuronal death and inflammation: a strategy for neuroprotection. Ann. N.Y. Acad. Sci. 993, 217–228. [DOI] [PubMed] [Google Scholar]
- Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L (2003) Disruption of transforming growth factor‐β signaling in ELF β‐spectrin‐deficient mice. Science 24, 574–577. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Hill CS (2004) New insights into TGF‐β SMAD signaling. Trend Biochem. Sci. 29, 265–273. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. 21, 2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield LM, Roberts AB (2002) TGF‐β signaling: positive and negative effects on tumorigenesis. Curr. Opin. Genet. Dev. 12, 22–29. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J (1992) TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L (1995) Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor‐beta 1. Am. J. Pathol. 147, 53–67. [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. USA 95, 15769–15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunostaining with anti‐TGF‐&bgr;1 antibody at the lesion site of a spinal cord. One week after injury, TGF‐&bgr; antigen expression increased slightly in the lesion sites of spinal cord injury animals compared with that of normal animals. Normal rat spinal cord tissue expresses almost no TGF‐&bgr;1 antigen. For analysis of TGF‐&bgr;1 overexpression in lesion sites of injured spinal cords, tissues at the centre of the lesion were incubated with primary antibodies against anti‐TGF‐&bgr;1 (1 : 300). After extensive washing with phosphate‐buffered saline, these tissue sections were incubated for 30 min with fluorescein isothiocyanate‐secondary antibodies (1 : 250). Cell nuclei were labelled with Topro‐3 (Molecular Probe). Results were analysed using confocal microscopy. Numbers of TGF‐&bgr;1‐positive cells were compared between the two experimental groups.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item