

Abstract
Cell cycle progression is controlled by both extracellular and intracellular signalling molecules. It has been generally believed that cdc2/CDK1 only control G2‐M transition in mammalian and many other higher eukaryotic cells. Accumulating evidence shows that cdc2 not only promotes G2‐M transition but is also capable of regulating G1 progress and G1‐S transition via association with multiple interphase cyclins; cdc2 activity can be inhibited by p21 and p27, two traditional G1 CDK inhibitors. In addition, cdc2‐cyclin B controls pronuclear union in interphase fertilized eggs. These data suggest that cdc2 may be a pluripotent CDK. Although mechanisms responsible for the multiple functions of cdc2 remain to be further investigated, interactions of cdc2 with pRb and with several important transcription factors may provide a clue to the pluripotent role of cdc2.
Introduction
The critical role of cdc2 (CDK1) in cell cycle control has been well documented. In fission yeast, both G2‐M and G1‐S transitions are triggered by activity of a single protein kinase, cdc2 (or cdc28p in budding yeast). In higher eukaryotes, multiple cyclin‐dependent kinases (CDKs; more than 11) have been identified, and these play different roles in the cell cycle. In mammalian cells, it has long been believed that CDK2, CDK4 and CDK6 drive cells through interphase, whereas cdc2 has been primarily implicated only in G2‐M transition, mainly in association with cyclin B. However, recent studies suggest that cdc2 is able to drive G1/S transition (1, 2). This new concept challenges the traditional model and suggests that cdc2 may be a pluripotent protein kinase acting globally in cell cycle control. In this mini‐review, we will first briefly discuss cell cycle control and the role of cdc2 in G2‐M transition. Then, we will focus on recent progress of cdc2 in G1‐S transition and its interactions with several important transcription factors. Both terms (cdc2 and CDK1) will be used throughout this mini‐review depending on usage described in the original publications.
Overview of cell cycle control
In mammalian cells, cell cycle progression is regulated by a group of CDKs and their regulatory subunits in sequential order: cyclin D‐CDK4/CDK6 and cyclin E‐CDK2 complexes act on G1 and G1‐S transition, respectively, followed by cyclin A‐CDK2 on S and cyclin B‐cdc2 at G2‐M transition. G1 cyclin‐CDK complexes have been reported to modify pRb by phosphorylation, thereby promoting cell cycle progression towards DNA replication. Dephosphorylation of pRb negatively regulates cell cycle progression through interactions with the E2F family of transcription factors. In contrast, phosphorylation of pRb results in loss of its replication suppression property (3, 4). However, in some types of cells, pRb may be not a major player in cell cycle control (5, 6).
Cell cycle progression controlled by cyclin‐dependent kinases is counterbalanced by CDK inhibitors (CDKIs). There are two families of such inhibitors, one of which is INK4A‐D, including p16INK4A, p15INK4B, p18 INK4C and p19INK4D. The INK4 inhibitors negatively regulate cyclin D‐CDK4/6 kinase activity. The second family of CDKIs, termed Cip/Kip, includes p21Cip, p27kip1 and p57kip2, which are efficient inhibitors of cyclin E‐CDK2 and cyclin D‐CDK4/CDK6. CDK regulation by CDK inhibitors is an important step in linking anti‐mitogenic signals to cell cycle progression.
Identification of G2 protein kinase(s) is directly related to discovery of maturation‐promoting factor (MPF) (7). For several years in the late 1960s, it had been suspected that some cytoplasmic factors might regulate nuclear activity during cell division. However, evidence for the existence of cytoplasmic factors that initiate M phase was not clear until 1971 during which year, Masui and Markert published their discovery of MPF (now termed M‐phase promoting factor). In their studies, protein factors in cytoplasm from mature unfertilized eggs could promote oocytes to be released from G2 and proceed through the first meiotic division. Subsequently, it was found that similar mitotic cell extracts from all eukaryotic cells tested including starfish, frog, sea urchin, yeast and human can function as MPF, demonstrating that MPF not only promotes meiosis but also triggers mitosis. Meanwhile, CDK, the protein that controls cell division, was first identified in yeast (8, 9) then in a broad range of cell types. By the late 1980s, several key experiments led to realization that cdc2 gene product, cdc2, controls entry into mitosis and is a catalytic subunit of MPF (10, 11). In 1987, the human gene that encodes the protein corresponding to cdc2 of Xenopus MPF was already found to induce cell division in cdc2‐deficient yeast mutants (12).
cdc2 interacts primarily with cyclin B or homologous B type cyclins to regulate G2‐M transition (13, 14). Expression of cyclin B has periodic behaviour, which is parallel to expression of MPF activity (15). During interphase, concentration of cyclin B gradually increases following G1, S and G2, and reaches a critical threshold at the end of G2, which promotes activation of cdc2 and triggers onset of mitosis. Activity of cdc2 is also regulated by phosphorylation and dephosphorylation and changes in subcellular localization (16, 17). Before reaching their threshold, cdc2‐cyclin B complexes are kept inactive through phosphorylation at Thr14 and Tyr15 of cdc2 by Myt1 and Wee1 (18, 19). Myt1 is a cell membrane‐associated protein kinase that is able to bind and phosphorylate cdc2 at both Thr14 and Tyr15, preventing its nuclear translocation. Different from Myt1, Wee1 suppresses cdc2 kinase activity by phosphorylation at Tyr15 in the nucleus. By the end of G2, Myt1 and Wee1 are inactivated and a specific dual‐phosphatase, cdc25, is activated. Activated cdc25 dephosphorylates two residues (Thr14 and Tyr15) in cdc2, leading to activation of cdc2 (20). cdc25 has three isoforms: cdc25A, cdc25B and cdc25C. cdc25A is thought to regulate G1‐S transition, cdc25B both G1‐S and G2‐M transition, and cdc25c only G2‐M transition (21). However, one recent report reveals that overexpression of cdc25A and cdc25B, but not cdc25c, promotes activation of cdc2 (22). Both Wee1 and cdc25 are regulated by Chk1 and 14‐3‐3 via phosphorylation during interphase (23, 24, 25).
Cdc2 drives G1‐S transition
As described above, cyclin D‐CDK4/CDK6 complexes are thought to be essential for G1 progress, whereas cyclin E‐CDK2 and cyclin A‐CDK2 regulate G1‐S transition and S phase progress, respectively. However, several recent reports have challenged this traditional model. One group has generated two strains of mice, one of which lacks CDK4 expression and the other expresses a CDK4 molecule with an inactivating mutation. Although loss of CDK4 causes insulin‐deficient diabetes and partial sterility, embryonic fibroblasts proliferate normally and the mice lacking CDK4 are viable (26). Consistent with this report, CDK4 deficiency does not affect normal keratinocyte proliferation (27). In a further report, disruption of CDK4 delayed cell cycle entry of mouse fibroblasts; however, CDK4−/− and p27−/− cells showed partial recovery of G0‐S transition (28), suggesting that delay of cell cycle entry was not simply caused by deletion of CDK4. A more recent study, published in 2004, presented evidence that mouse embryos defective in CDK4 and CDK6 initially displayed normal organogenesis and most cell types proliferated normally, although the mice died during late stages of embryonic development due to severe anaemia. Additional supporting evidence is that quiescent CDK4/CDK6‐null cells were able to enter S phase in response to serum stimulation (29). These data demonstrated that CDK4 and CDK6 are not as critical for cell cycle progression as had previously been believed. As D‐type cyclins are the regulatory subunits of CDK4 and CDK6, ‘normal’ cell cycle progression in CDK4−/− and CDK6−/− cells may be due to presence of D type‐cyclins, although these cyclins, in theory, would not be able to compensate for CDK4 or CDK6. To test this possibility, mice lacking all D‐cyclins were generated and cell proliferation and mouse development were investigated. As expected, D‐type cyclin (D1, D2 and D3)‐deficient fibroblasts of these mice still proliferated almost normally, although with increased requirement for mitogenic stimulation (30).
A further important protein kinase involved in interphase progression in mammalian cells is CDK2, which targets many substrates that are important in DNA replication and transcription (31, 32, 33). Is it possible that CDK2 regulates the entire cell cycle and/or compensates for CDK4 or CDK6 in the absence of these molecules? Recently, two groups have reported their findings. Both studies generated CDK2 knockout mice to determine whether deletion of CDK2 could prevent cell progression. Surprisingly, CDK2 knockout mice survived and developed normally (34, 35). Moreover, mouse embryonic fibroblasts (MEFs) lacking CDK2 proliferated normally in culture and re‐entered the cell cycle without significant delay after stimulation with serum. Cyclin E is the major regulatory subunit of CDK2. Two research teams have tested requirement of cyclin E in mouse development. They found that cyclin E1‐ or cyclin E2‐deficient animals developed normally (36, 37), but double‐knock out cyclin E1−/− and cyclin E2−/− genotypes were embryonically lethal (36). One possible explanation for results caused by E1 and E2 double‐knock outs is that cyclin Es, as regulatory subunits, not only associate with CDK2, but also bind to other CDK(s), whose functions are critical or important for cell cycle progression.
All data from gene knockout mice described above have demonstrated that deletion of interphase CDKs is not lethal to the mice and that their cells still proliferated in a ‘normal’ way, suggesting that some other molecules may compensate for CDK2, CDK4 and CDK6. By generating CDK2−/− p27−/− double knockout mice, Aleem et al. detected high numbers of cells in S and M phases in thymus and spleen, parallel to high levels of cyclin E activity (2). As cyclin E is a regulatory subunit of CDK2 and CDK2 is deleted here, high cyclin E activity must relate to (an)other associated protein kinase(s). Subsequently, this group demonstrated that in CDK2−/− cells, cyclin E was associated with cdc2 as an active complex. If cdc2 in wild type and CDK2−/− mice was silenced by shRNA, cells progression through S phase was slow and cell proliferation was significantly reduced, suggesting that cdc2 was able to drive G1‐S transition in the presence or absence of CDK2. However, deletion of CDK2 greatly enhanced efficiency of cdc2 in promoting G1‐S transition. Consistent with this observation, Satyanarayana et al. observed that CDK1 was translocated to the nucleus in cdk2−/− MEFs after serum stimulation, while wild type cells expressing cdk2 had delayed and weak nuclear translocation of CDK1 (38). The role of cdc2 in interphase progression was confirmed by application of Roscovitine, a highly potent inhibitor of CDK (especially cdc2 and CDK2) activity (39). Addition of Roscovitine caused significant growth inhibition of cells of several human cancer cell lines in culture (40). In human HeLa cells, CDK1 was shown to be required for establishment of G1 phase, as cells expressing mutant CDK1 (CDK1AF) were able to enter and exit M phase, but were not able to carry out cytokinesis or karyokinesis compared to wild type cells (41). In fibroblasts, it has been shown that cdc2 mRNA is inducible in response to fresh culture medium; during quiescence, cdc2 mRNA is almost undetectable. Stimulation of cells with medium induces cdc2 expression, beginning at G1‐S transition and reaching maximum levels during late S and G2 phases (42). Supplementary to these findings, experiments with TGFβ (43) and Cks1 (44) also support a pluripotent role for CDK1 in the presence of interphase CDKs; TGFβ is well known as a G1 protein kinase inhibitor. We have found, in our previous studies on human myeloid leukaemia cells, that TGFβ not only inhibits several G1 checkpoint kinases but also strongly downregulates expression of cdc2 without causing accumulation of the cells in G2‐M. As cdc2 is associated with pRb during S phase, our data suggest that cdc2 may participate in G1‐S regulation (43). Cks1 is a small protein component of CDK complexes, which regulates CDK1 activity. Deletion of Cks1 by siRNA in MCF‐derived cells blocks oestrogen‐ and further growth factor‐induced signalling pathways, slowed progression of cells through G1‐S and blocked their entry into M phase. Protein analysis demonstrated that deletion of Cks1 causes significant reduction of CDK1 and accumulation of hypophosphorylated pRb (44). More surprising results came from Santamaria’s group’s studies in which cdc2 was shown to execute all the events that are required to drive cell division, suggesting that cdc2 alone is sufficient to drive mammalian cell cycle progression (1). First, they demonstrated that CDK1 was able to interact with D‐type cyclins in lysates extracted from embryos lacking CDK4 and it interacted with cyclin E in embryos lacking CDK2 and CDK4. CDK1‐cylin D and CDK1‐cyclin E complexes were able to phosphorylate pRb proteins in vitro. Second, although knockdown of CDK1 had no effect on interphase progression of primary MEFs induced by CDK4 and CDK2, CDK1 deletion completely abrogated S phase entry in embryonic cells lacking CDK4 (−/−),CDK6 (−/−) and CDK2 (−/−). Finally, by generating mutant mice at cdc2a, the locus encoding CDK1, they demonstrated that CDK1 is essential for early stages of embryonic development. Additional information on cdc2 came from a report published in 2008 according to which CDK1‐cyclin B controls pronuclear union by regulating formation of sperm astral microtubules in interphase in fertilized eggs of starfish (45). Briefly, these data suggest that CDK1 is a pluripotent CDK that is able to promote cells’ entry into S phase, as well as G2‐M transition, not only in somatic cells but also in gonad cells. The pluripotent role of cdc2 is shown in Fig. 1.
Figure 1.
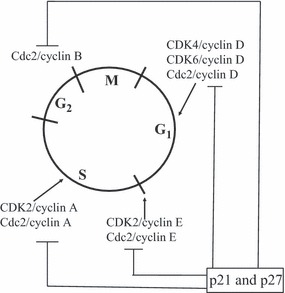
Cell cycle control by cdks and cdk inhibitors: cdc2 versus CDK2 and CDK4/6.
Interactions between cdc2 and cell cycle regulatory molecules/transcription factors
pRb and E2F
If cdc2 is a pluripotent CDK, what are the fundamental mechanisms responsible for the multiple functions of cdc2 described above? Interactions of cdc2 with several key molecules that control cell growth may provide a clue. It is well known that Rb is a tumour suppressor gene. The gene product, pRb, has the ability to suppress cell proliferation, which is regulated by G1 cyclin‐CDK complexes (3, 46, 47). Subsequently, studies of several groups have demonstrated that not only G1 CDKs but also cdc2 have ability to phosphorylate pRb in various species including humans (43, 48, 49, 50). As early as 1998, Taieb et al. observed, by microinjection of human Rb gene into Xenopus oocytes, elevated activities of cdc2‐cyclin B and MAPK, but not cyclin D‐CDK4/6 complexes, accompanying pRb phosphorylation. As inactivation or overexpression of cyclin D‐CDK4/6 complexes does not affect Rb kinase activities, elevated cdc2 activity might be responsible for pRb phosphorylation (48). Soon, physical association of pRb with cdc2 and its phosphorylation by cdc2 in human cells have been observed (49). By using human myeloid leukaemia cell lines as research tools for cell cycle study, we have detected that in proliferating human myeloid leukaemia cells, there was a significant formation of cdc2‐pRb complexes. Immunoprecipitates of cdc2 extracted from these cells had significant kinase activity upon pRb phosphorylation and inhibition of cdc2 contributes to transforming growth factor‐beta‐induced G1‐arrest (43). pRb may also directly or indirectly regulate cdc2 activity, as increased CDK1 (cdc2) activity has been detected in Rb‐deficient mouse fibroblasts (51).
It has been reported that dephosphorylated pRb suppresses cell replication partly by turning off transcription of genes required for cell cycle progression, through interaction with E2Fs (50). The pRb family has three members: pRb, p107 and p130, which are collectively called ‘pocket proteins’. Each member can bind E2Fs and inhibit E2F activity. In contrast, free E2Fs bind to target DNA and activate DNA transcription. A number of reports have revealed that cdc2 is one of E2F’s targets. In quiescent human fibroblasts, p130‐E2F4 complexes were found to bind to the cdc2 promoter, resulting in inhibition of cdc2 transcription (42). The binding site was detected at the R box of the cdc2 promoter, which is located downstream of AP1 or SP1 sites (52). In contrast, E2F1, E2F2 and E2F3 bind to positive‐acting site in the cdc2 promoter and induces cdc2 expression (53). Thus, cdc2‐pRb‐E2F forms a positive feedback loop, which may amplify cdc2‐induced proliferation of cells (Fig. 2), whereas inhibition of cdc2 gene may contribute to pocket protein‐E2F complex‐induced replication inhibition.
Figure 2.
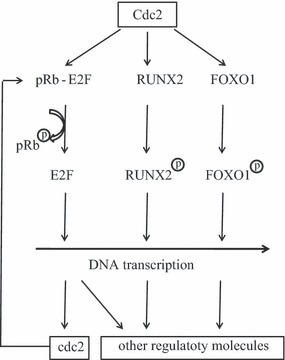
DNA transcription induced by Cdc2.
p21cip and p27kip1
As a negative regulator of cell cycle progression, p21cip is thought to bind and inhibit CDK2/cyclin E and/or CDK4/cyclin D complexes, thereby arresting cells in G1 phase. Induction of p21cip is regulated by tumour suppressor gene, p53, in response to DNA damage. If cdc2 drives interphase progression, one would expect that p21cip might be able to bind and inhibit cdc2, as it does with other G1 CDKs, in response to p53 activation. In 2001, one group found that overexpression of p53 suppressed cell proliferation and cdc2 activity in TR9‐7 cells; in contrast, deletion of p21cip substantially impaired ability of p53 to repress the cdc2 promoter, suggesting that p21cip is required for p53‐induced inhibition of cdc2 activity (52). As there was no evidence for direct interaction between p21cip and cdc2, the reasonable explanation for p21cip‐induced inhibition of cdc2 is that overexpression of p53 activates transcription of p21cip (p53‐p21cip pathway), which in turn inhibits interphase CDK activity. As a result, CDKs lose ability to phosphorylate pRb family proteins. In the dephosporylated form, p130‐E2F4 complexes bind to the cdc2 promoter and inhibit cdc2 transcription. Apparently, the explanation described above is indirect and crucial information is missing. Direct physical association between cdc2 and p21cip has been reported recently. In CDK2−/− MEFs, but not in wild‐type cells, both p21cip and CDK1 were predominantly found in the nucleolus at about the same time, after cells were irradiated and serum stimulated. Immunoprecipitation revealed elevated levels of p21cip‐CDK1 complexes between 6 and 24 h in cells lacking CDK2 after stimulation (38). As time points typically represented G1 progression and G1/S transition after receiving a stimulating signal, this suggests that formation of a p21cip‐cdc2 complex is responsible for cell arrest at G1/S transition in the absence of cdk2.
p27 kip1 was first identified as an inhibitor of cyclin E‐CDK2. Subsequently, it was found that p27 kip1 can target cyclin‐CDK4 complexes. Since then, p27 kip1 as an interphase CDK inhibitor has been widely accepted; however, some recent progress in the field has challenged this model. In work performed by Martin et al., forced expression of p27kip1 and p21cip inhibited replication of mouse fibroblasts expressing CDK2 and cells without CDK2. In contrast, p27 kip1 and p21cip double‐knockouts caused similar proliferation in both CDK+/+ and CDK−/− cells. In agreement with these results, mice lacking p27kip1 have about the same size and body weight in presence or absence of CDK2. As p27kip1 and p21cip had no any significant effect on expression or activity of CDK4, the data described above suggest that CDK2 is not a primary target of p27kip1 and p21cip and these inhibitor‐induced cell cycle arrests could be a result of their interaction with molecules other than cdk2 or CDK4 (54). In the same year, another group reported that cdc2 was able to interact directly with p27kip1 (2). In CDK2−/− MEFs p27kip1 was found to bind and inhibit cdc2 activity. In contrast, deletion of p27 kip1 significantly upregulated cdc2 activity and promoted G1‐S transition. Clearly, these new findings advanced our knowledge on the roles of CDK inhibitors and cdc2 in cell cycle control. Most likely, p27kip1‐ or p21cip‐induced growth inhibition, which has been well reported in the literature, is partially due to negative regulation of cdc2 activity (Fig. 1).
FOXO and RUNX
Forkhead box O (FOXO) is a group of transcription factors that belong to the FOX superfamily (55, 56). These transcription factors possess tumour suppressor functions by regulating expression of genes involved in cell death and proliferation. For example, upregulation of FOXO activates proapoptotic genes encoding for Fas ligand, Bim and TRAIL (57, 58, 59). FOXO proteins also arrest cells in G1 by upregulating CDK inhibitors, p27 (60, 61), or p130‐E2F4 complexes (62). The FOXO subfamily includes FOXO1, FOXO3a, FOXO4 and FOX6 in humans. The role of FOXO1 in regulating cell proliferation and cell cycle regulation has received particular attention recently (63, 64). Both CDK1‐induced apoptosis and proliferation through interaction with FOXO1 have been reported. In neurons, CDK1 phosphorylates FOXO1 at Ser249 in vitro and in vivo. Phosphorylated FOXO1 disrupts FOXO1 binding to 14‐3‐3 proteins and thereby causes nuclear accumulation of FOXO1, followed by activation of FOXO1‐dependent transcription and cell death (65). In prostate cancer (Pca) cells, experiments performed by Liu et al. have shown that ectopically expressed CDK1 forms a complex with FOXO1 and inhibits FOXO1‐induced apoptosis (66). As CDK1 and cyclin B1 are often overexpressed in human cancers, the authors believed that their findings suggested that aberrant activation of CDK1 may contribute to tumourigenesis by promoting cell proliferation via phosphorylation of FOXO1.
The RUNX transcriptional regulators have been reported to be essential for haematopoiesis, bone formation and gastric development. The family has three members, RUNX1, RUNX2 and RUNX3. Both RUNX1 and RUNX2 are able to promote bone marrow cell proliferation and cell cycle progression by up‐regulation of D‐type cyclins and down‐regulation of CDK inhibitor, p21 (67, 68, 69). One of the mechanisms responsible for RUNX2‐mediated cell proliferation involves interaction between RUNX2 and cdc2. Qian et al. reported that in proliferating endothelial cells, RUNX2 DNA binding activity is high and RUNX2 is associated with cyclin B1. In culture, cdc2 inhibitor, Roscovitine, dose‐dependently inhibits RUNX2 DNA binding activity. In vitro protein kinase assay has shown that cdc2 phosphorylated RUNX2 at the Ser451 residue (69). Thus, phosphorylation of RUNX2 by cdc2 is linked to RUNX2‐mediated cell cycle progression in bone marrow endothelial cells.
Conclusion
Taken together, recent findings have clearly demonstrated that cdc2, a traditional G2‐M regulator, not only promotes G2/M transition but also regulates interphase progression and some other biological processes in mammalian and other higher eukaryotic cells. In the absence of interphase CDKs, cdc2 is able to associate itself with various interphase cyclins. Multiple functions of cdc2 may link to its interactions with the pRb‐E2F and several other transcription factors (Fig. 2). Although much has been learnt from these new findings, many fundamental questions remain for future studies. First, as most of the multiple functions of cdc2 have been detected in the absence of interphase CDKs by approaches of gene deletion or gene knock‐mice, the pluripotent role of cdc2 and its regulation in the presence of interphase CDKs, that is, in intact cells, are less clear. Although interactions between cdc2 and interphase cyclins are required for the role of cdc2 in driving G1‐S transition, it is not ruled out that cdc2 may regulate interphase progression via association with some yet unidentified cyclins. Second, the loops and pathways for interactions between cdc2 and the multiple transcription factors described in this review are far from understood. If cdc2 is indeed a pluripotent CDK, this knowledge may have a profound impact on our understanding of tumourigenesis and therapeutic applications.
Acknowledgements
We apologize to those authors whose work was not cited due to space limitations. We thank Shakima St. Clair and Djenny Pierre for their effort in the preparation of the references.
References
- 1. Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K et al. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815. [DOI] [PubMed] [Google Scholar]
- 2. Aleem E, Kiyokawa H, Kaldis P (2005) Cdc2‐cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 7, 831–836. [DOI] [PubMed] [Google Scholar]
- 3. Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 9, 585–593. [PubMed] [Google Scholar]
- 4. Wu L, Timmers C, Maiti B, Saabedra HI, Sang L, Chong GT et al. (2001) The E2F1‐3 transcription factors are essential for cellular proliferation. Nature 414, 457–462. [DOI] [PubMed] [Google Scholar]
- 5. Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpoles S, Watson RJ et al. (2002) E2F mediates cell cycle‐dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 16, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi Y, Rayman JB, Dynlacht DB (2000) Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816. [PMC free article] [PubMed] [Google Scholar]
- 7. Masui Y, Markert CL (1971) Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 177, 129–145. [DOI] [PubMed] [Google Scholar]
- 8. Nurse P, Thuriaux P (1980) Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics 96, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reed SI, Ferguson J, Groppe JC (1982) Preliminary characterization of the transcriptional and translational products of the Saccharomyces cerevisiae cell division cycle gene CDC28 . Mol. Cell. Biol. 2, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lohka MJ, Hayer MK, Maller JL (1988) Purification of maturation‐promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 85, 3009–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunphy WG, Brizuela L, Beach D, Newport J (1988) The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell 54, 423–431. [DOI] [PubMed] [Google Scholar]
- 12. Lee M, Nurse P (1987) Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2+ . Nature 335, 251–254. [DOI] [PubMed] [Google Scholar]
- 13. Sherr CJ (1994) G1 phase progression: cycling on cue. Cell 18, 551–555. [DOI] [PubMed] [Google Scholar]
- 14. Malumbres M, Barbacid M (2005) Mammalian cyclin‐dependent kinases. Trends Biochem. Sci. 30, 630–641. [DOI] [PubMed] [Google Scholar]
- 15. Dorée M, Peaucellier G, Picard A (1983) Activity of the maturation‐promoting factor and the extent of protein phosphorylation oscillate simultaneously during meiotic maturation of starfish oocytes. Dev. Biol. 99, 489–501. [DOI] [PubMed] [Google Scholar]
- 16. Smits VA, Medema RH (2001) Checking out the G(2)/M transition. Biochim. Biophys. Acta 1519, 1–12. [DOI] [PubMed] [Google Scholar]
- 17. Takizawa CG, Morgan DO (2000) Control of mitosis by changes in the subcellular location of cyclin‐B1‐cdk1 and cdc25c. Curr. Opin. Cell Biol. 12, 658–665. [DOI] [PubMed] [Google Scholar]
- 18. Mueller PR, Coleman TR, Kumagai A, Dunphy WG (1995) Myt1: a membrane‐associated inhibitory kinase that phosphorylates cdc2 on both threonine‐14 and tyrosine‐15. Science 270, 86–90. [DOI] [PubMed] [Google Scholar]
- 19. Featherstone C, Russell P (1991) Fission yeast p107wee 1 mitotic inhibitor is a tyrosine/serine kinase. Nature 349, 808–811. [DOI] [PubMed] [Google Scholar]
- 20. Donzelli M, Draetta GF (2003) Regulating mammalian checkpoints through cdc25 inactivation. EMBO Rep. 4, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nilsson I, Hoffmann I (2000) Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 4, 107–114. [DOI] [PubMed] [Google Scholar]
- 22. Timofeev O, Cizmecioglu O, Settele F, Kempf T, Hoffmann I (2010) Cdc25 phosphatases are required for timely assembly of CDK1‐cyclin B at the G2/M transition. J. Biol. Chem. 285, 16978–16990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Graves PR, Lovly CM, Uy GL, Piwnica‐Worms H (2001) Localization of human cdc25C is regulated both by nuclear export and 14‐3‐3 protein binding. Oncogene 20, 1839–1851. [DOI] [PubMed] [Google Scholar]
- 24. Kumagai A, Dunphy WG (1999) Binding of 14‐3‐3 proteins and nuclear export control the intracellular localization of the mitotic inducer cdc25. Genes Dev. 13, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee J, Kumagai A, Dunphy WG (2001) Positive regulation of Wee1 by Chk1 and 14‐3‐3 proteins. Mol. Biol. Cell 12, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP et al. (1999) Loss of Cdk4 expression causes insulin‐deficient diabetes and Cdk4 activation results in beta‐islet cell hyperplasia. Nat. Genet. 1, 44–52. [DOI] [PubMed] [Google Scholar]
- 27. Rodriguez‐Puebla ML, de Marval PL, LaCava M, Moons DS, Kiyokawa H, Conti CJ (2002) Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am. J. Pathol. 161, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A et al. (1999) Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity. Mol. Cell. Biol. 19, 7011–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S et al. (2004) Mammalian cells cycle without the D‐type cyclin‐dependent kinases Cdk4 and Cdk6. Cell 118, 493–504. [DOI] [PubMed] [Google Scholar]
- 30. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E et al. (2004) Mouse development and cell proliferation in the absence of D‐cyclins. Cell 118, 477–491. [DOI] [PubMed] [Google Scholar]
- 31. Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK et al. (2000) Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103, 127–140. [DOI] [PubMed] [Google Scholar]
- 32. Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD et al. (2000) Cell cycle‐regulated phosphorylation of p220(NPAT) by cyclinE/cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 14, 2298–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA et al. (2000) NPAT links cyclin E‐Cdk2 to the regulation of replication‐dependent histone gene transcription. Genes Dev. 14, 2283–2287. [PMC free article] [PubMed] [Google Scholar]
- 34. Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P (2003) Cdk2 knockout mice are viable. Curr. Biol. 13, 1775–1785. [DOI] [PubMed] [Google Scholar]
- 35. Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R et al. (2003) Cyclin‐dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31. [DOI] [PubMed] [Google Scholar]
- 36. Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S et al. (2003) Cyclin E ablation in the mouse. Cell 114, 431–443. [DOI] [PubMed] [Google Scholar]
- 37. Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z et al. (2003) Cyclins E1 and E2are required for endoreplication in placental trophoblast giant cells. EMBO J. 22, 4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satyanarayana A, Hilton MB, Kaldis P (2008) p21 inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol. Biol. Cell 19, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N et al. (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin‐dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243(1–2), 527–536. [DOI] [PubMed] [Google Scholar]
- 40. Goke R, Barth P, Schmidt A, Samans B, Lankat‐Buttgereit B (2004) Programmed cell death protein 4 suppresses cdk1/cdc2 via induction of p21. Am. J. Physiol. Cell Physiol. 287, c1541–c1546. [DOI] [PubMed] [Google Scholar]
- 41. Pomerening J, Ubersax J, Ferrell J Jr (2008) Rapid cycling and precocious termination of G1 phase in cells expressing CDK1AF. Mol. Biol. Cell 19, 3426–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tommasi S, Pfeifer GP (1995) In vivo structure of the human cdc2 promoter: release of a p130‐E2F‐4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol. Cell. Biol. 12, 6901–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu X, Cui D, Moscinski L, Zhang X, Maccachero V, Zuckerman KS (2007) TGFβ regulates the expression and activities of G2 checkpoint kinases in human myeloid leukemia cells. Cytokine 37, 155–162. [DOI] [PubMed] [Google Scholar]
- 44. Westbrook L, Manuvakhova M, Kern FG, Estes NR II, Ramanathan HN, Thottassery JV (2007) Cks1 regulates cdk1 expression: a novel role during mitotic entry in breast cancer cells. Cancer Res. 67, 11393–11401. [DOI] [PubMed] [Google Scholar]
- 45. Tachibana K, Hara M, Hattori Y, Kishimoto T (2008) Cyclin B‐cdk1 controls pronuclear union in interphase. Curr. Biol. 18, 1308–1313. [DOI] [PubMed] [Google Scholar]
- 46. Dyson N (1998) The regulation of E2F by pRB‐family proteins. Genes Dev. 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 47. Nevins JR (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703. [DOI] [PubMed] [Google Scholar]
- 48. Taieb F, Karaiskou A, Rime H, Jessus C (1998) Human retinoblastoma protein (Rb) is phosphorylated by cdc2 kinase and MAP kinase in Xenopus maturing oocytes. FEBS Lett. 425, 465–471. [DOI] [PubMed] [Google Scholar]
- 49. Choi KS, Eom YW, Kang Y, Ha MJ, Rhee H, Yoon JW et al. (1999) Cdc2 and cdk2 kinase activated by transforming growth factor‐β1 trigger apoptosis through the phosphorylation of retinoblastoma protein in FaO hepatoma cells. J. Biol. Chem. 274, 31775–31783. [DOI] [PubMed] [Google Scholar]
- 50. Frolov MV, Dyson NJ (2004) Molecular mechanisms of E2F‐dependent activation and pRB‐mediated repression. J. Cell Sci. 117, 2173–2181. [DOI] [PubMed] [Google Scholar]
- 51. Li W, Kotoshiba S, Berthet C, Hilton MB, Kaldis P (2009) Rb/Cdk2/Cdk4 triple mutant mice elicit an alternative mechanism for regulation of the G1/S transition. Proc. Natl. Acad. Sci. USA 106, 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taylor WR, Schonthal AH, Galante J, Stark GR (2001) p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J. Biol. Chem. 276, 1998–2006. [DOI] [PubMed] [Google Scholar]
- 53. Zhu W, Giangrande PH, Nevins JR (2004) E2Fs link the control of G1/S and G2/M transcription. EMBO J. 23, 4615–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martín A, Odajima J, Hunt SL, Dubus P, Ortega S, Malumbres M et al. (2005) Cdk2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1). Cancer Cell 7, 591–598. [DOI] [PubMed] [Google Scholar]
- 55. Tuteja G, Kaestner KH (2007a) SnapShot: forkhead transcription factors I. Cell 130, 1160. [DOI] [PubMed] [Google Scholar]
- 56. Tuteja G, Kaestner KH (2007b) SnapShot: forkhead transcription factors II. Cell 131, 192. [DOI] [PubMed] [Google Scholar]
- 57. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- 58. Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ (2000) Expression of the pro‐apoptotic Bcl‐2 family member Bim is regulated by the forkhead transcription factor FKHR‐L1. Curr. Biol. 10, 1201–1204. [DOI] [PubMed] [Google Scholar]
- 59. Modur V, Nagarajan R, Evers BM, Milbrandt J (2002) FOXO proteins regulate tumor necrosis factor‐related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J. Biol. Chem. 277, 47928–47937. [DOI] [PubMed] [Google Scholar]
- 60. Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787. [DOI] [PubMed] [Google Scholar]
- 61. Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR (2000) Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 20, 8969–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ et al. (2002) Control of cell cycle exit and entry by protein kinase B‐regulated forkhead transcription factors. Mol. Cell. Biol. 22, 2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sakamaki J, Daitoku H, Yoshimochi K, Miwa M, Fukamizu A (2009) Regulation of FOXO1‐mediated transcription and cell proliferation by PARP‐1. Biochem. Biophys. Res. Commun. 382, 497–502. [DOI] [PubMed] [Google Scholar]
- 64. Ai J, Jingjing D, Xiaoyan L, Mianzhi C, Qiutan Y, Huan S et al. (2010) Overexpression of FoxO1 causes proliferation of cultured pancreatic β cells exposed to low nutrition. Biochemistry 49, 218–225. [DOI] [PubMed] [Google Scholar]
- 65. Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y et al. (2008) Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 319, 1665–1668. [DOI] [PubMed] [Google Scholar]
- 66. Liu P, Kao TP, Huang H (2008) CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 27, 4733–4744. [DOI] [PubMed] [Google Scholar]
- 67. Sun L, Vitolo MI, Qiao M, Anglin IE, Passaniti A (2004) Regulation of TGFb1‐mediated growth inhibition and apoptosis by RUNX2 isoforms in endothelial cells. Oncogene 23, 4722–4734. [DOI] [PubMed] [Google Scholar]
- 68. Westendorf JJ, Zaidi SK, Cascino JE, Kahler R, van Wijnen AJ, Lian JB et al. (2002) Runx2 (Cbfa1, AML‐3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22, 7982–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A (2006) Cell cycle‐dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J. Biol. Chem. 281, 7118–7128. [DOI] [PubMed] [Google Scholar]