

Abstract
Objectives
To characterize potency of menstrual blood‐derived stem cells (MenSCs) for future cell therapies, we examined differentiation potential of MenSCs into adipocytes.
Materials and methods
Differentiation potential of MenSCs in comparison to bone marrow stem cells (BMSCs) was assessed in conventional culture medium. Differentiation potential of MenSCs into adipocytes was improved using different combinations of growth factors and hormones.
Results
First, we demonstrated that MenSCs preserve their appearance and karyotypic stability during passages. Although these cells express mesenchymal stem cells markers, they cannot simply be classified as mesenchymal stem cells due to expression of embryonic stem cells marker, OCT‐4. Oil red O staining showed that differentiated MenSCs in conventional medium with/without retinoic acid (protocols 1 and 2) did not attain adipocyte characteristics, whereas differentiated BMSCs in conventional medium accumulated oil vacuoles typically. Nevertheless, real‐time RT‐PCR results showed that LPL gene expression was up‐regulated in both protocols 1 and 2, whereas LEPR was up‐regulated only in protocol 2 (fortified with retinoic acid). Surprisingly, protocol 3 (including rosiglitazone) had odd influence on mRNA expression of all genes (LEPR, LPL and PPAR‐γ). Oil red O staining confirmed fat‐producing ability of MenSCs under protocol 3.
Conclusions
Presented data suggest an efficient differentiation protocol for in vitro production of MenSC‐derived adipocytes. These cells are suggested to be an apt alternative to BMSCs for future stem cell therapy of soft tissue injuries.
Introduction
A large percentage of plastic and reconstructive surgical procedures are performed each year to restore soft tissue defects resulting from serious burns, tumour resection, and genetic and congenital defects 1. Strategies to restore soft tissue injuries consist of use of adipose tissue as an implant and filler 2. However, grafting adipose tissue has problems, including tissue resorption, immunological rejection and more 3. Due to these limitations, in recent decades, characteristics of stem cells such as self‐renewal potential and trans‐differentiation into other lineages (including adipocytes) have impelled scientists to take advantage of stem cell therapy as a novel therapeutic approach for soft tissue healing 4. Nevertheless, the main challenge in everyday clinical practice is to achieve high cell density from an easily accessible and safe stem cell resource.
Numerous ethical considerations surrounding use of embryonic stem cells (ESCs) have triggered the scientist's interest in adult stem cells 5. Their most conventional source has been bone marrow, cell obtained from the iliac crest. Trans‐differentiation ability of bone marrow‐derived stem cells (BMSCs) into adipocytes consistent with differentiation into other lineages such as osteoblasts and chondrocytes that has been established in a variety of studies 6, 7. However, problems such as poor availability, invasive methods for sample collection and lower proliferation capacity compared to ESCs, limit clinical use of BMSCs.
Recently, menstrual blood (MB) has been suggested as a unique source of stem cells with some characteristics such as easy retrieval, no restriction in sample collection, and fewer ethical problems, compared to ESCs 8, 9, 10. It has been proposed that MB, in addition to cells shed from the endometrium, contains circulating bone marrow mesenchymal stem cells, which contribute to endometrial regeneration 11. Whether menstrual blood‐derived stem cells (MenSCs) have features in common with BMSCs, or have source‐specific peculiarities, is still under investigation. These cells appear to express markers of stromal/mesenchymal stem cells such as CD10, CD29, CD44, CD73, CD146, CD105, CD90, CD9, CD41a and CD59 which are also expressed by BMSCs. Likewise, MenSCs do not express haematopoietic stem cell markers such as CD14, CD45, CD34, CD38, CD133 similar to BMSCs. On the other hand, expression of OCT‐4 as pluripotency marker and lack of some mesenchymal markers (such as STRO‐1) distinguishes MenSCs from BMSCs 8, 9, 10, 12, 13, 14, 15.
Previous studies have demonstrated that growth ability of MenSCs is much greater than that of BMSCs 8, 16. The MenSCs population has different trans‐differentiation ability into osteogenic, chondrogenic and hepatic lineages compared to BMSCs 9, 10, 16, 17. At present, little information is available on differentiation potential of MenSCs into adipocytes, specially compared to BMSCs 9, 18, 19. Considering these differences, comparative evaluation of MenSCs and BMSCs in terms of differentiation into further lineages, including that of adipocytes would help us to understand any potential distinctive characterization that they may have. Meanwhile, study of adipogenic differentiation potential of MenSCs compared to BMSCs, as the well‐established stem cell source with adipogenic differentiation ability, will help researchers realize whether these cells could find a place for future stem cell therapy of soft tissue injuries.
To date, a variety of hormones and cytokines such as isobutylmethylxanthine (IBMX) as AMP cyclic activator, dexamethasone, retinoic acid (RA), rosiglitazone as PPAR‐γ agonist, indomethacin, recombinant human insulin, insulin‐like growth factor‐1, growth hormone, thiazolidinedione, fatty acids and cyclic adenosine monophosphate, are known to promote adipogenic differentiation of stem cells 15, 20, 21, 22, 23. However, efficiency of these factors in adipogenic development of MenSCs is unsolved. In this study, differentiation potential of MenSCs into adipocytes has been compared to BMSCs using cytochemical and molecular experiment. To find an appropriate stimulus to trigger adipogenic differentiation of MenSCs, we have developed protocols using combinations of known factors involved in adipogenic development of stem cells.
Material and methods
Isolation and culture of MenSCs and BMSCs
Menstrual blood donors were selected as healthy volunteers aged between 22 and 30 years. All donors signed informed consent forms approved by the medical ethics committee of Avicenna Research Institute, Tehran, Iran. Five millilitre MB was collected using sterile Diva cups (Diva International, Lunette, Finland) on the second day of the menstrual cycle. Bone marrow aspirates (5–10 ml) were also obtained from iliac crests of human donors at the Bone Marrow Transplantation Center, Shariati Hospital, Tehran, Iran. Samples were harvested after obtaining signed informed consent documentation, as above. The work was approved by the Medical Ethics Committee, Ministry of Health, Iran. Collected specimens were decanted into tubes containing 2.5 μg/ml fungizone (Gibco, Scotland, UK), 100 μg/ml streptomycin, 100 U/ml penicillin and 0.5 mm EDTA (Sigma‐Aldrich, St Louis, MO, USA) in phosphate buffered saline (PBS). Isolation of stem cells from menstrual blood and bone marrow was performed as described previously 9, 11.
Flow cytometric analysis and immunofluorescence staining of OCT‐4
For immunophenotyping of MenSCs, expression of mesenchymal stem cell markers such as CD73, CD44, CD29, CD10, CD90 and CD105 was evaluated by flow cytometric analysis as described in our team's recent publications 10, 12, 16, 17. Also, for evaluation of OCT‐4 expression level by flow cytometry, cultured cells at passage numbers 1 and 12, were detached from tissue culture flasks with trypsin‐EDTA and washed twice in PBS at room temperature (RT). Cell density was adjusted to 2 × 105 in 100 μl PBS containing 2% FBS (PBS–FBS) and fixed in 4% paraformaldehyde for 10 min at RT. Subsequently, cells were washed twice in PBS–FBS by centrifugation at 400 g for 10 min each, then resuspended in permeabilization buffer (0.1% saponin) and centrifuged at 200 g for 7 min. Then, cells were treated with primary rabbit anti‐human OCT‐4 polyclonal antibody (1:100) (Abcam, Cambridge, MA, USA) optimally diluted in permeabilization buffer and incubated for 40 min at 4 °C. Afterwards, cells were washed twice in permeabilization buffer and incubated in FITC‐conjugated sheep anti‐rabbit IgG (1:100) (Avicenna Research Institute, Tehran, Iran) for 30 min at 4 °C in dark. Finally, cell suspensions were washed twice in permeabilization buffer and analysed by flow cytometry (Partec GmbH, Munster, Germany) using appropriate isotype controls.
In addition, cells were fixed in acetone at −20 °C for 5 min and then subjected to OCT‐4 immunofluorescent staining. In brief, fixed cells were permeabilized with 0.4% triton X‐100 for 20 min. Washed cells were incubated for 1 h at RT in primary rabbit anti‐human OCT‐4 polyclonal antibody. Subsequently, cells were washed three times in PBS and incubated in FITC‐labelled sheep anti‐rabbit IgG at RT for 45 min in the dark. Thereafter, cells were incubated in 4, 6 diamidino‐2‐phenylindole (DAPI; 1 μg/ml) (Sigma‐Aldrich) for nuclear staining. Cells were visualized and photomicrographed using an epifluorescence microscope (Olympus BX51 microscope, Tokyo, Japan) connected to a digital camera (Olympus DP71).
Multiplex ligation‐dependent probe amplification (MLPA)
To investigate chromosomal imbalances in samples, MLPA analysis was performed on genomic DNA of cells at passages 1 and 12, using the SALSA MLPA kit P036‐E1 Human Telomer3 (MRC‐Holland, Amsterdam, the Netherlands) according to the manufacturer's protocol. The kit contains a probe for each human subtelomere. Briefly, total of 100 ng genomic DNA in a final volume of 5 μl was denatured and hybridized to SALSA probe mix, followed by incubation at 60 °C for 18 h. Subsequently, annealed probes were ligated using provided Ligase‐65 mix at 54 °C for 15 min. Next, 10 μl ligated products, as template, were used for DNA amplification. PCR amplicons were run on a Genetic Analyzer 3130 (Applied Biosystems, Foster City, CA), and results were analysed using GeneMarker software version 2.4 (SoftGenetics LLC, State College, PA). The normal pattern was expected to produce a normalized signal value ratio of 1:1; any value out of the ranges <0.75 or >1.35 was considered as abnormal and corresponded to a deletion or duplication respectively. In each MLPA reaction, four control samples were simultaneously used. In each MLPA reaction, four control samples (whole blood of adults with no evidence of genetic anomalies) were simultaneously used. In addition, all control samples were screened for chromosomal abnormality through karyotyping and normal 550 GTG banding protocol 24.
Adipogenic differentiation
For adipogenic differentiation, cells at passage 2–4 were plated at 4 × 104 cells/cm2 and induced under sequential exposures to three different combinations of cytokines, growth factors and hormones (as shown in Fig. 1). In protocol 1 (P1), cells were cultured in 0.5 ml DMEM‐F12 supplemented with 10% FBS, 1 μm dexamethasone (DEX), 10 μg/ml recombinant human insulin, 0.5 mm 3‐isobutyl‐1‐methyl‐xanthine (IBMX) and 200 μm indomethacin (Indo) (Sigma‐Aldrich) (called a conventional medium). After 3 days, conventional medium was replaced with normal medium consisting of DMEM‐F12 supplemented with 10% FBS (called maintenance medium). Replacement of conventional/maintenance medium was repeated up to day 21 9.
Figure 1.
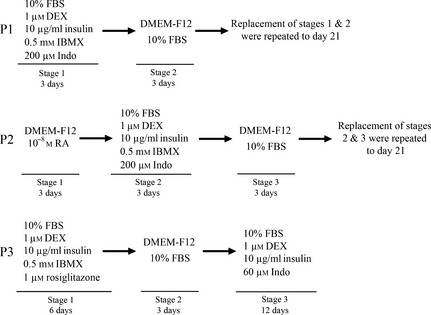
Schematic diagram of three differentiation protocols for adipogenic.
Protocol 2 (P2) was prepared in a similar manner with P1 with a amendments. Cells were treated with DMEM‐F12 supplemented with 10−8 m all‐trans RA (Sigma‐Aldrich) for 3 days, then conventional medium and maintenance medium were replaced every 3 days up to day 21 25.
Protocol 3 (P3), cells were cultured in conventional medium containing 1 μm rosiglitazone (Osvah Pharmaceutical Company, Tehran, Iran) instead of Indo for 6 days. Then, culture medium was replaced with maintenance medium and cells were cultured for the next 3 days. Then they were treated with DMEM supplemented with 10% FBS, 1 μm DEX, 10 μg/ml recombinant human insulin and 60 μm Indo up to 12 days 20. Cultured cells in maintenance medium were considered negative controls.
Oil red O staining
Adipogenic‐induced cells were stained for fat vacuoles using oil red O staining. Medium was removed from each well, and cells were rinsed in PBS and fixed in 3% glutaraldehyde (Sigma‐Aldrich) for 1 h at RT. They were then washed three times in PBS and twice in tap water before being stained in 0.6% oil red O solution (Sigma‐Aldrich) (three parts 1% oil red O in 99% isopropanol, and two parts PBS) for 30 min at RT, followed by washing in 1 ml water three times to remove unbound dye. Then cell nuclei were stained with haematoxylin solution. Lipid droplets of differentiated cells were then visualized using an inverted microscope (Olympus CKX41) connected to a digital camera (Olympus DP71). Cultured cells in maintenance medium were stained in the same manner as the controls.
Evaluation of adipocyte‐specific gene expression with quantitative real‐time reverse transcription polymerase chain reaction (QRT‐PCR)
Quantitative RT‐PCR was performed to assess expression of a set of adipocyte‐related genes including lipoprotein lipase (LPL), Peroxisome proliferator‐activated receptor gamma (PPAR‐γ) and leptin receptor (LEPR) (Table 1). Briefly, total RNA was isolated from 3 × 105 undifferentiated and differentiated MenSCs and BMSCs by standard RNA extraction protocol using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed using Super Script™ II Reverse Transcriptase kit (Life Technologies, Carlsbad, CA, USA). Reverse transcription was performed with 2 μg DNAse (Fermentase Inc, Vilnius, Lithuania)‐treated RNA, 1 μl SuperScriptTM II Reverse Transcriptase (200 U) (Life Technologies), 20 pm N6 Random‐Hexamer, 20 pm dNTP Mix, 4 μl 5X First Strand buffer, 2 μl dithiothreitol (0.1 m) and 1 μl RiboLockTM RNase inhibitor (all from Fermentas Inc) in a thermocycler (Eppendorf, Germany) at 25 °C for 10 min, 42 °C for 50 min and 70 °C for 15 min. Real time PCR was performed using SYBR Premix Ex Taq™ (Perfect Real Time) (Takara Bio Inc, Otsu, Shiga, Japan). Next, 2 μl cDNA was mixed with 1X SYBR Premix EX TaqTM (Takara Bio Inc), 0.2 μm of each primer and 0.4 μl ROXTM Reference Dye 50X, and amplified using a 7500 Real‐Time PCR System (Applied Biosystems) as follows: initial denaturation at 95 °C for 10 s, PCR amplification at 95 °C for 5 s and 60 °C for 30 s for 40 cycles. Ultimately, dissociation stage was performed at 95 °C for 15 s, 60 °C for 1 min and 95 °C for 15 s. Mean efficiencies and crossing point values for each gene were determined with LinRegPCR (version 11.0) 26 and normalized to values for GAPDH in differentiated cells in reference to undifferentiated cells using REST 2009 software (available at http://www.genequantification.de/rest-2009.htm).
Table 1.
Sequences of primers used for analysis of cell differentiation into the adipogenic lineage
| Gene names | Primer sequence | Product size (bp) | NCBI accession number |
|---|---|---|---|
| LEPR |
F5′‐CAATCTGAATGAAACCAAACCTC‐3′ R5′‐GGCTGCTCCTATGATACCTC‐3′ |
230 | NM_001198689.1 |
| LPL |
F5′‐CCCTACAAAGTCTTCCATTAC‐3′ R5′‐AGTTCTCCAATATCTACCTCTG‐3′ |
197 | NM_000237.2 |
| PPAR‐γ |
F5′‐CATTCTGGCCCACCAACTT‐3′ R5′‐CCTTGCATCCTTCACAAGCA‐3′ |
372 | NM_138711.3 |
| GAPDH |
F5′‐CTCTCTGCTCCTCCTGTTCG‐3′ R5′‐ACGACCAAATCCGTTGACTC‐3′ |
114 | NM_002046.4 |
LPL, lipoprotein lipase; PPAR‐γ, peroxisome proliferator‐activated receptor gamma; LEPR, leptin receptor; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Statistical analysis
All experiments were performed using samples from 3 to 6 donors. All measurements were performed in triplicate. Results of flow cytometry were presented as median and range. Statistical analysis of relative gene expression results in real time PCR was performed using REST© freeware according to formula presented by Pfaffl et al. 27.
Results
Characterization of isolated MenSCs
Immunophenotype analysis of cells revealed that MenSCs typically express surface antigens associated with mesenchymal stem cells such as CD73, CD44, CD29, CD 10, CD90 and CD105 (Fig. 2a). MenSCs were of spindle‐shape and fibroblast‐like morphology which was preserved even at high passage number up to passage 12 (Fig. 2b‐i). However, expression of OCT‐4 reduced over following passages. Based on flow cytometry data, while expression of OCT‐4 was 97.5% in cells at passage 1, only 19.43% of this marker expressed by cells at passage 12 (Fig. 2b‐ii). These data were confirmed by immunofluorescent staining results. As shown in Fig. 2b‐iii, typical expression of OCT‐4 reduced over passages.
Figure 2.
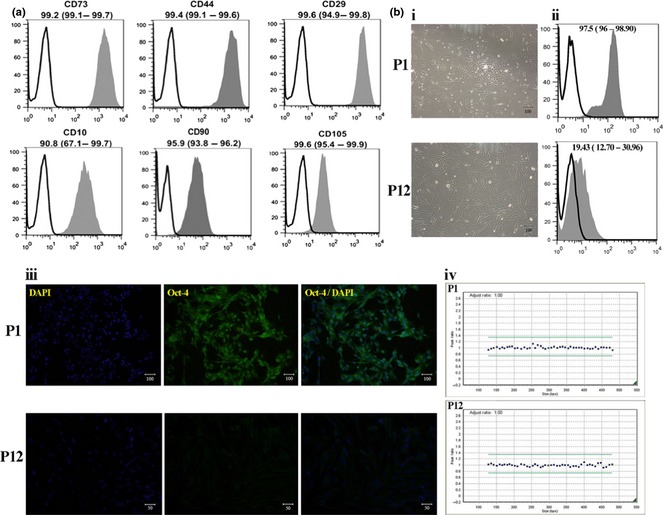
Characterization of isolated menstrual blood‐derived stem cells (MenSCs). (a) Immunophenotyping of MenSCs by flow cytometry. Representative histograms for CD markers are demonstrated (grey). Respective isotype control shown as black line. Results are presented as median (range) of three to five independent experiments. (b) Phase contrast morphology (i) and OCT‐4 expression of MenSCs judged by flow cytometry (ii) and immunofluorescence staining (iii) at passages 1 and 12. Chromatograms illustrating no chromosomal aberrations in MenSCs at passage 12 compared to cells at passage 1 (iv). GeneMarker plots showing results of MLPA analysis. Green lines illustrated the upper and lower limits of acceptable ranges of variations in MLPA analysis. Blue dots show the chromosomal locations which are balanced.
Based on MLPA analysis, MenSCs maintained diploid configuration without chromosomal aberration during passages. Passage 12 MenSCs had no chromosomal abnormalities (Fig. 2b‐iv).
Evaluation of adipogenesis of MenSCs and BMSCs
In the case of BMSCs, adipogenic differentiation usually takes place in the presence of conventional medium comprising DEX, recombinant human insulin, IBMX and Indo referred to P1. First, we investigated whether this medium would be potent enough to induce adipogenesis of MenSCs. As shown in Fig. 3a, MenSCs did not reach adipocyte characteristics this way, whereas BMSCs developed typical accumulation of oil vacuoles as shown by oil red O staining. Analysis of adipogenic genes using QRT‐PCR showed that although LPL mRNA was significantly up‐regulated in differentiated MenSCs under conventional medium with reference to undifferentiated MenSCs (P = 0.001), unexpectedly mRNA expression level of PPAR‐γ (P = 0.001) and LEPR (P = 0.17) was down‐regulated or was with no significant change in these cells. Contrary to MenSCs, differentiated BMSCs in the presence of conventional medium expressed typical levels of these genes with reference to undifferentiated cells (Fig. 3b).
Figure 3.
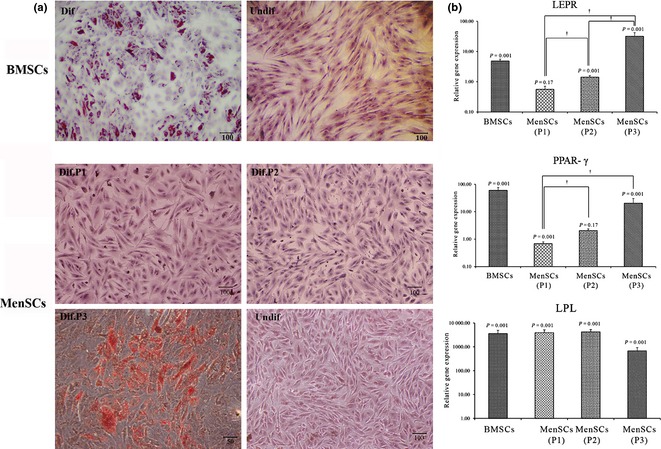
Histological and molecular evaluation of differentiated menstrual blood‐derived stem cells (MenSCs) and bone marrow‐derived stem cells (BMSCs). (a) assessment of oil vacuoles accumulation by Oil Red O staining in differentiated MenSCs under different protocols and BMSCs. (b) qRT‐PCR results; data of differentiated cells were normalized to corresponding GAPDH results and calculated with reference to undifferentiated cells. †Indicates significant difference (P < 0.05) between different protocols in MenSCs.
Influence of fortification of conventional medium with RA (referred as P2) for adipogenic differentiation of MenSCs was investigated thereafter. As shown in Fig. 3b, medium fortification with RA caused up‐regulation of LEPR mRNA in MenSCs differentiated under P2 compared to undifferentiated cells (1.42‐fold, P = 0.001), however, it had no considerable effect on expression of PPAR‐γ mRNA (2.04‐fold, P = 0.17). In addition, results of oil red O staining were negative in these cells.
Interestingly, protocol 3 produced an odd influence on mRNA expression level of all genes (LEPR, LPL and PPAR‐γ). The promoting efficacy of protocol 3 was more pronounced when treated MenSCs exhibited typical expression of LEPR (32.09‐fold, P = 0.001) compared to undifferentiated cells; a gene which had been down regulated under P1. Moreover, up‐regulation level of PPAR‐γ mRNA was significantly (20.43‐fold, P = 0.001) higher in differentiated MenSCs under P3 compared to undifferentiated cells. Oil red O staining confirmed lipid content of those cells indicating fat‐producing ability of induced MenSCs under P3.
Discussion
In recent years, advantages of menstrual blood stem cells, such as easy accessibility and high availability, have impelled scientists to search for the characteristics of these cells. However, more studies are required to achieve comprehensive information on their nature specially in comparison to other adult stem cells. They preserved their morphological appearance and karyotypic stability during passages. This preservation of normal karyotype at high passage number has also been shown by others 8, 28. In addition, Allickson et al. have reported that MenSCs are able to be subcultured more times before senescence compared to stem cells from other sources, such as bone marrow and dental pulp 23.
Consistent with our recent studies 10, 12, 16, 17, we have shown that MenSCs express markers of stromal/mesenchymal stem cells such as CD10, CD29, CD44, CD73, CD90 and CD105, while failing to express embryonic stem cell markers (ESC) such as SSEA‐4, Nanog and c‐kit. Regardless of these findings, in the present study, we demonstrated that MenSCs could not be classified simply as mesenchymal stem cells, due to their expression of ESC marker, OCT‐4, which is not typical of mesenchymal stem cells. However, expression of OCT‐4, critical for self‐renewal potency of MenSCs, was down‐regulated during subculturing. Therefore, although conservation of gross morphological properties and karyotypic normality as well as proliferation ability over long‐term cell culture, all hold promise for MenSCs expansion in quantities relevant to clinical application, these data imply gradual reduction in self‐renewal ability of MenSCs.
Considering the aforesaid differences, comparative evaluation of MenSCs and BMSCs in terms of differentiation, including the adipogenic lineage, would help us to understand other potential distinctive characteristics that they may present. To the best of our knowledge, such data have not been reported previously. In the present study, to determine adipogenic differentiation potential of MenSCs, specially compared to BMSCs, we developed three protocols (P1–P3) using cocktails consisting of dexamethasone, recombinant human insulin, IBMX, indomethacin and rosiglitazone.
There are inconsistent previous reports concerning differentiation potential of MenSCs into adipogenic lineages. While the experiments of Musina et al. to induce adipogenic differentiation of cultured MenSCs in well‐known adipogenic medium, were not successful 18 others have presented primary results implying positive adipo‐differentiation of MenSCs 9. Although this discrepancy can be attributed to donor differences, sample source, sampling day or stem cell isolation method, one limitation of the studies is to rely solely on the oil red O staining technique without use of multiple experiments to prove the obtained data. Here, we used both cytological and molecular examinations for comparative study between MenSCs and BMSCs required to obtain robust decisions on adipogenic differentiation capacity of MenSCs. The data of oil red O staining showed that BMSCs only, differentiated in well‐known adipogenic medium, formed lipid vacuoles; differentiated MenSCs failed to accumulate lipid droplets. Moreover, based on quantitative real‐time PCR results, while all three evaluated adipogenic markers including LPL, LEPR and PPAR‐γ up‐regulated in differentiated BMSCs, in the case of MenSCs, except LPL, two other assessed markers LEPR and PPAR‐γ were down‐regulated during differentiation into the adipogenic lineage.
The important role of RA for early adipogenic differentiation of ESCs has been shown previously 25. However, effectiveness of RA on adipogenic development of adult stem cells, especially MenSCs, had not previously been determined. Thus, influence of fortification of conventional medium with RA (referred as P2) on adipogenic differentiation of MenSCs was investigated thereafter. Although the result of oil red O staining was negative once more, the commitment of MenSCs to the adipogenic lineage was more pronounced under this protocol compared to P1, as LEPR was significantly up‐regulated as was LPL in differentiated MenSCs, with reference to undifferentiated cells.
Roziglitazon is known to be a promoting factor during adipogenic development of BMSCs 20, 29, 30. It has been demonstrated that treatment of rodents with rosiglitazone increased bone marrow adipocytes and concommitently reduced new bone formation 31, 32. Based on these reports, we evaluated the fortification effect of differentiation medium with rosiglitazone (referred as P3), which resulted in typical impact on intensity of lipid accumulation. Moreover, up‐regulation level of adipogenic markers LEPR, PPAR‐γ and LPL mRNA in differentiated MenSCs under P3 was significantly much higher than in differentiated cells under P1 or P2. Although the molecular mechanisms governing promoting effect of rosiglitazone on adipogenic differentiation of MenSCs is not clear, it can be attributed to PPAR‐γ agonist property of this agent 30. PPAR‐γ is a member of the nuclear hormone receptor gene superfamily of ligand‐activated transcription factors that acts as a pivotal transcription factor in the regulation of adipocyte gene expression and differentiation 33.
In conclusion, based on accumulative data, MenSCs could develop into adipocytes, but with a different differentiation potential and expression pattern compared to BMSCs. We have developed an efficient culture method for adipogenic differentiation of human MenSCs by combination of factors involved in adipogenic development of BMSCs. The differences between MenSCs and BMSCs may be attributed to different immunophenotypic features and signalling machinery. It is notable that considering the refreshing nature, accessibility and lack of ethical issues, MenSCs are superior to BMSCs and therefore this study may be valuable in future in vivo investigations concerning healing of soft tissue injuries.
Conflicts of interest
The authors indicate no potential conflicts of interest.
Acknowledgements
We thank the Iranian Council of Stem Cell Technology for providing research grant for this study. We also kindly appreciate the cooperation of Dr. Saeed Talebi and Dr. Ali Ahani in providing facilitations and equipment in QRT‐PCR and MLPA analysis.
M. Khanmohammadi and S. Khanjani contributed equally to the manuscript.
References
- 1. Langstein HN, Robb GL (1999) Reconstructive approaches in soft tissue sarcoma. Semin. Surg. Oncol. 17, 52–65. [DOI] [PubMed] [Google Scholar]
- 2. Klein AW, Elson ML (2000) The history of substances for soft tissue augmentation. Dermatol. Surg. 26, 1096–1105. [PubMed] [Google Scholar]
- 3. Gomillion CT, Burg KJ (2006) Stem cells and adipose tissue engineering. Biomaterials 27, 6052–6063. [DOI] [PubMed] [Google Scholar]
- 4. Leeper NJ, Hunter AL, Cooke JP (2010) Stem cell therapy for vascular regeneration: adult, embryonic, and induced pluripotent stem. Circulation 122, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henningson CT Jr, Stanislaus MA, Gewirtz AM (2003) Embryonic and adult stem cell therapy. J. Allergy Clin. Immunol. 111, S745–S753. [DOI] [PubMed] [Google Scholar]
- 6. Zheng YH, Xiong W, Su K, Kuang SJ, Zhang ZG (2013) Multilineage differentiation of human bone marrow mesenchymal stem cells in vitro and in vivo. Exp. Ther. Med. 5, 1576–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hung SC, Chang CF, Ma HL, Chen TH, Low‐Tone Ho L (2004) Gene expression profiles of early adipogenesis in human mesenchymal stem cells. Gene 340, 141–150. [DOI] [PubMed] [Google Scholar]
- 8. Meng X, Ichim TE, Zhong J, Rogers A, Yin Z, Jackson J et al (2007) Endometrial regenerative cells: a novel stem cell population. J. Transl. Med. 5, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel AN, Park E, Kuzman M, Benetti F, Silva FJ, Allickson JG (2008) Multipotent menstrual blood stromal stem cells: isolation, characterization, and differentiation. Cell Transplant. 17, 303–311. [DOI] [PubMed] [Google Scholar]
- 10. Khanjani S, Khanmohammadi M, Zarnani AH, Talebi S, Edalatkhah H, Eghtesad S et al (2013) Efficient generation of functional hepatocyte‐like cells from menstrual blood derived stem cells. J. Tissue Eng. Regen. Med. 2013 Mar 18. doi: 10.1002/term.1715. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11. Taylor HS (2004) Endometrial cells derived from donor stem cells in bone marrow transplant recipients. JAMA 292, 81–85. [DOI] [PubMed] [Google Scholar]
- 12. Khanmohammadi M, Khanjani S, Bakhtyari MS, Zarnani AH, Edalatkhah H, Akhondi MM et al (2012) Proliferation and chondrogenic differentiation potential of menstrual blood and bone marrow‐derived stem cells in two dimensional culture. Int. J. Hematol. 95, 484–493. [DOI] [PubMed] [Google Scholar]
- 13. Dominici M, Le Blanc K, Mueller I, Slaper‐Cortenbach I, Marini F, Krause D, et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- 14. Gargett CE, Masuda H (2010) Adult stem cells in the endometrium. Mol. Hum. Reprod. 16, 818–834. [DOI] [PubMed] [Google Scholar]
- 15. Sousa BR, Parreira RC, Fonseca EA, Amaya MJ, Tonelli FM, Lacerda SM et al (2014) Human adult stem cells from diverse origins: an overview from multiparametric immunophenotyping to clinical applications. Cytometry A 85, 43–77. [DOI] [PubMed] [Google Scholar]
- 16. Darzi S, Zarnani AH, Jeddi‐Tehrani M, Entezami K, Mirzadegan E, Akhondi MM et al (2012) Osteogenic differentiation of stem cells derived from menstrual blood versus bone marrow in the presence of human platelet releasate. Tissue Eng. Part A 18, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kazemnejad S, Akhondi MA, Soleimani M, Zarnani AM, Khanmohammadi M, Darzi S et al (2012) Characterization and chondrogenic differentiation of menstrual blood‐derived stem cells on a nanofibrous scaffold. Int. J. Artif. Organs 35, 55–66. [DOI] [PubMed] [Google Scholar]
- 18. Musina RA, Belyavski AV, Tarusova OV, Solovyova EV, Sukhikh GT (2008) Endometrial mesenchymal stem cells isolated from the menstrual blood. Bull. Exp. Biol. Med. 145, 539–543. [DOI] [PubMed] [Google Scholar]
- 19. Dimitrov R, Timeva T, Kyurkchiev D, Stamenova M, Shterev A, Kostova P et al (2008) Characterization of clonogenic stromal cells isolated from human endometrium. Reproduction 135, 551–558. [DOI] [PubMed] [Google Scholar]
- 20. Ninomiya Y, Sugahara‐Yamashita Y, Nakachi Y, Tokuzawa Y, Okazaki Y, Nishiyama M (2010) Development of a rapid culture method to induce adipocyte differentiation of human bone marrow‐derived mesenchymal stem cells. Biochem. Biophys. Res. Commun. 394, 303–308. [DOI] [PubMed] [Google Scholar]
- 21. Gregoire FM, Smas CM, Sul HS (1998) Understanding adipocyte differentiation. Physiol. Rev. 78, 783–809. [DOI] [PubMed] [Google Scholar]
- 22. Gargett CE, Schwab KE, Zillwood RM, Nguyen HP, Wu D (2009) Isolation and culture of epithelial progenitors and mesenchymal stem cells from human endometrium. Biol. Reprod. 80, 1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allickson JG, Sanchez A, Yefimenko N, Borlongan CV, Sanberg PR (2011) Recent studies assessing the proliferative capability of a novel adult stem cell identified in menstrual blood. Open Stem Cell J. 3, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Res. 30, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dani C, Smith AG, Dessolin S, Leroy P, Staccini L, Villageois P et al (1997) Differentiation of embryonic stem cells into adipocytes in vitro. J. Cell Sci. 110, 1279–1285. [DOI] [PubMed] [Google Scholar]
- 26. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ et al (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group‐wise comparison and statistical analysis of relative expression results in real‐time PCR. Nucleic Acids Res. 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zemel'ko VI, Grinchuk TM, Domnina AP, Artsybasheva IV, Zenin VV, Kirsanov AA et al (2011) Multipotent mesenchymal stem cells of desquamated endometrium: isolation, characterization and use as feeder layer for maintenance of human embryonic stem cell lines. Tsitologiia 53, 919–929. [PubMed] [Google Scholar]
- 29. Benvenuti S, Cellai I, Luciani P, Deledda C, Baglioni S, Giuliani C et al (2007) Rosiglitazone stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells. J. Endocrinol. Invest. 30, RC26–RC30. [DOI] [PubMed] [Google Scholar]
- 30. Valyasevi RW, Harteneck DA, Dutton CM, Bahn RS (2002) Stimulation of adipogenesis, peroxisome proliferator‐activated receptor‐gamma (PPARgamma), and thyrotropin receptor by PPARgamma agonist in human orbital preadipocyte fibroblasts. J. Clin. Endocrinol. Metab. 87, 2352–2358. [DOI] [PubMed] [Google Scholar]
- 31. Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka‐Czernik B (2004) Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology 145, 401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL (2005) Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology 146, 1226–1235. [DOI] [PubMed] [Google Scholar]
- 33. Bruedigam C, Eijken M, Koedam M, van de Peppel J, Drabek K, Chiba H et al (2010) A new concept underlying stem cell lineage skewing that explains the detrimental effects of thiazolidinediones on bone. Stem Cells 28, 916–927. [DOI] [PubMed] [Google Scholar]