

Abstract
Prostate cancer is one of the leading causes of death among men in the USA.
Objective: In this study, we investigated the role of atypical protein kinase C‐iota (PKC‐ι) in androgen‐independent prostate DU‐145 carcinoma cells compared to transformed non‐malignant prostate RWPE‐1 cells.
Materials and methods: Western blotting and immunoprecipitations demonstrated that PKC‐ι is associated with cyclin‐dependent kinase activating kinase (CAK/Cdk7) in RWPE‐1 cells, but not in DU‐145 cells.
Results: Treatment of prostate RWPE‐1 cells with PKC‐ι silencing RNA (siRNA) decreased cell viability, cell‐cycle accumulation at G2/M phase, and phosphorylation of Cdk7 and Cdk2. In addition, PKC‐ι siRNA treatment caused less phosphorylation of Bad at ser‐155, ser‐136, and greater Bad/Bcl‐xL heterodimerization, leading to apoptosis. In DU‐145 cells, PKC‐ι was anti‐apoptotic and was required for cell survival. Treatment with PKC‐ι siRNA blocked increase in cell number, and inhibited G1/S transition by accumulation of cells in G0/G1 phase. In addition to cell‐cycle arrest, both RWPE‐1 and DU‐145 cells underwent apoptosis due to mitochondrial dysfunction and apoptosis cascades, such as release of cytochrome c, activation of caspase‐7, and poly (ADP‐ribose) polymerase (PARP) cleavage.
Conclusion: Our results suggest that PKC‐ι is required for cell survival in both transformed non‐malignant prostate RWPE‐1 cells and androgen‐independent malignant prostate DU‐145 cells, whereas suppressing PKC‐ι lead to apoptosis in DU‐145 prostate cells.
Introduction
Protein kinase C (PKC) isozymes are serine‐theronine kinases that are involved in cell proliferation, survival, differentiation and apoptosis. Fourteen known members of the PKC family are classified as classical, novel and atypical. Classical PKCs (α, βΙ, βΙΙ, γ) are activated by calcium (Ca2+) and diacylglycerol or phorbol 12‐myristate 13‐acetate (PMA), also referred to as 12‐O‐tetradecanoyl phorbol 13‐acetate (TPA). Novel PKCs (δ, ɛ, η, θ) are activated by diacylglycerol and TPA (PMA). Both classical and novel PKCs have two zinc finger domains in the regulatory domain. However, atypical PKCs (ζΙ, ζΙΙ, ι/λ, µ) are not activated by calcium and phorbol ester and have only one zinc finger in the regulatory domain (1, 2, 3, 4). Although high homology between PKC isozymes in cell lines obscures their specific function, understanding roles of PKCs in the cell cycle is crucial to understanding cancer cell survival and to developing therapeutic agents.
Atypical PKC‐ι and classical PKC‐βΙΙ have been linked to phosphorylation of cyclin‐dependent kinase activating kinase (CAK/Cdk7) during cell proliferation in human glioma cells (5, 6). Atypical PKCs are likely to be anti‐apoptotic proteins in K562 leukaemia cells because overexpression of PKC‐ι prevented okadaic acid and taxol‐induced apoptosis (1). In non‐small cell lung cancer (NSCLC), PKC‐ι phosphorylated Bad, a pro‐apoptotic BH3‐only protein, and disrupted the Bad/Bcl‐xL dimer, leading to enhanced survival and chemoresistance (7, 8). PKC‐ι also regulates the Rac1/Pak/Mek/Erk signalling pathway involved in cell proliferation of NSCLC (8, 9, 10). Hence, evidence is accumulating to indicate a significant role of PKC in many types of cancer cells.
An increasing number of studies implicate PKC isozymes in prostate cancer cell regulation (11, 12). The PKC activator, phorbol ester (PMA), induces apoptosis in LNCaP, an androgen‐dependent, prostate cancer cells (13, 14, 15). However, DU‐145 cells are insensitive to PMA and taxol‐induced apoptosis (12). Thus, it is likely that atypical PKCs, which are insensitive to PMA, play a role in DU‐145 cell survival. In addition, DU‐145 cells have a mutated Rb gene, and do not express Bax (16, 17).
Apoptosis manifests extrinsic and intrinsic pathways (18, 19). The intrinsic pathway depends on depolarization of mitochondria and the extrinsic pathway involves activation of death receptors. Members of the Bcl‐2 family include anti‐apoptotic proteins (Bcl‐2, Bcl‐xL, and Bcl‐W) and pro‐apoptotic proteins (Bax, and Bak, and the ‘BH‐3 only protein‐Bad’ Bid, and Blk) (19). Many studies have shown that inactivation of Bad at single or multisite phosphorylation dictates whether the cell lives or dies. In addition, phosphorylation of Bad and Bad/Bcl‐xL interactions abrogates the anti‐apoptotic function of Bcl‐xL (7, 20, 21, 22, 23). It is not clear why anti‐apoptotic Bcl‐2 and Bcl‐xL are co‐expressed in some prostate cancer cells. Perhaps overexpression of Bcl‐xL and Bcl‐2 proteins might enable prostate cancer cells to survive in an androgen‐deprived environment (24, 25, 26, 27).
Here, we demonstrate that PKC‐ι is required for cell survival in both RWPE‐1 and DU‐145 cells. Treatment of RWPE‐1 cells with PKC‐ι silencing RNA (PKC‐ι siRNA) results in decreased cell viability, cell‐cycle accumulation at G2/M phase, and a decrease in phosphorylation of p‐Cdk‐7 (T170) and p‐Cdk2 (T160). In DU‐145 cells, PKC‐ι is required for cell survival and is anti‐apoptotic. Three‐fold greater concentration of PKC‐ι siRNA (compared to RWPE‐1) blocked an increase in DU‐145 cell number, inhibited G1/S transition, and induced apoptosis.
Materials and methods
Reagents and antibodies
Primary antibodies were purchased from the following companies: PKC‐α, βΙ, δ, ɛ, γ, θ (Santa Cruz Biotechnology, Santa Cruz, CA, USA); PKC‐ι (Transduction Laboratory, Lexington, KY, USA); PKC‐ζ (Upstate Biotechnology, Lake Placid, NY, USA); p‐Bad (ser‐112), p‐Bad (ser‐155), p‐Bad (ser‐136), Bad, PARP, cytochrome c, caspase‐7, survivin, Cdk2, Cdk7, β‐actin (Santa Cruz Biotechnology); cleaved PARP (Asp 214), Bcl‐xL, p‐Cdk2 (Ther160) (Cell Signalling Technology, Danvers, MA, USA); and phospho‐Cdk7‐T170 (Abgent, San Diego, CA, USA). Secondary antibodies were purchased from the following companies: horseradish‐peroxidase (HRP) goat × mouse immunoglobulin G (IgG), HRP goat × rabbit IgG (Accurate, Westbury, NY, USA); HRP bovine anti‐goat IgG (Santa Cruz Biotechnology); anti‐rabbit IgG HRP‐linked antibody (Cell Signalling Technology). Standards for PKCs are from various sources that came with the antibody: HeLa, Jurkat, K‐562, and whole cell lysates from Santa Cruz Biotechnology. For immunoprecipitation anti‐rabbit IgG (whole molecule)‐agarose beads (1 : 1 v/v) (Sigma, St. Louis, MO, USA); Bad mouse monoclonal 25% agarose conjugate, and normal mouse IgG 25% agarose conjugate, were from Santa Cruz Biotechnology. All other chemicals and reagents, unless otherwise stated, were obtained from Sigma Aldrich, Fisher Scientific (Norcross, GA, USA), Pierce (Rockford, IL, USA) and Bio‐Rad (Richmond, CA, USA).
Cell culture
RWPE‐1 and DU‐145 cell lines were obtained from American Type Tissue Culture Collection (Rockville, MD, USA). DU‐145 cells were seeded (1 × 106) and grown as a monolayer in 75 cm2 flasks containing 90% Minimum Essential Medium Eagle's (MEME) Earle's balanced salt solution, non‐essential amino acids, 1 mm sodium pyruvate, 2 mm l‐glutamine, 1500 mg sodium bicarbonate/L, 10% foetal bovine serum (FBS), and antibiotics 5 ml (penicillin 10 U/ml and streptomycin 10 µg/ml) in a 5% CO2 incubator at 37 °C. For RWPE‐1, cells were seeded (1 × 106) and grown as a monolayer in 75 cm2 flasks containing keratinocyte serum‐free media, epidermal growth factor (2.5 µg) and bovine pituitary extract (25 mg) (Gibco) and antibiotics 5 ml (penicillin 10 U/ml and streptomycin 10 µg/ml) in a 5% CO2 incubator at 37 °C.
Proliferating cells vs. contact inhibition
RWPE‐1 cells and DU‐145 cells were grown (1.25 × 105) in monolayer with their respective complete media. To reach 50% confluence, the cells were allowed to proliferate until they covered 50% of the area of T‐75 flask with complete media. The cell lysate was prepared as described in the ‘Immunoprecipitation and Western blot analysis’ section below. To achieve contact inhibition, the cells were grown to 100% confluence (that is, the monolayer of cells covered the whole surface area of T‐75 flask). Subsequently, they were serum starved by washing them twice with Dulbecco's phosphate‐buffered saline (DPBS). The starvation medium, which does not have FBS (for DU‐145) and bovine pituitary extract (for RWPE‐1), was added to the flasks and incubated for 48 h. Cell lysate was prepared as described in the ‘Immunoprecipitation and Western blot analysis’ section below.
Cell‐cycle time point
Both RWPE‐1 cells and DU‐145 cells were grown (5 × 105) in monolayers with their respective complete media for 24 h. The following day, cells were washed with DPBS (2×) to remove the complete media. Starvation medium, which does not have FBS (for DU‐145) and bovine pituitary extract (for RWPE‐1), was added to the flasks and incubated for 48 h. After being serum starved for 48 h (T = 0), starvation medium was removed and complete medium with serum was added to each flask. The cells in each flask received complete media from 3 to 36 h, this allowed the cells them complete the cell cycle.
Trypan blue dye exclusion assay
For siRNA experiments, 40–60% confluent cells were seeded. After siRNA treatment (detail in siRNA tansfections), cells were trypsinized, pelleted, washed with 1 ml of PBS, resuspended in 0.4% trypan blue solution (Sigma), and live (dye excluding) cells were counted using a haemacytometer. Results from three separate independent experiments were used to determine mean viability and standard deviation for each time point.
Cell‐cycle analysis by flow cytometry
Cells were washed in DPBS, trypsinized, and fixed by dropwise addition (while vortexing) of cold ethanol until a concentration of 60% ethanol was reached. The day before analysis, the 60% ethanol was decanted and PBTB (PBS, 0.2% Triton and 1% bovine serum albumin) was added. Cells were counted and diluted to 1 × 106 cells/ml with PBTB; they were then filtered and 50 µl of RNase was added. Nuclei were analysed for DNA content using a propidium iodide 10 µl staining protocol and flow cytometry (28). Distributions of 1 × 106 nuclei were quantified using FAC STARPlus flow cytometry (Becton Dickinson, San Jose, CA, USA) and the ModFitLT Cell Cycle Analysis program, version 2.0 (Verity Software House, Topsham, ME, USA). Results from three independent experiments were used to determine mean viability and standard deviation for each time point. Statistical significance was determined by Student's t‐test (Minitab Inc., State College, PA, USA).
Immunoprecipitation and Western blot analysis
Cells were placed on ice to terminate the incubation and cell extracts were prepared by washing twice with ice‐cold DPBS. Monolayers were scraped at 4 °C, resuspended and sonicated in 2 ml homogenization buffer (50 mm HEPES, pH 7.5), 150 mm NaCl, 0.5% Triton‐X100, 1 mm EDTA and 2 mm ethylene glycol bis(β‐aminoethyl ether)‐N,N,N′,N′, ‐tetraacetic acid (EGTA), 0.1 mm orthovanadate, 1 mm NaF, 2 mm PMSF, 2.5 µg/ml leupeptin, 1 mm DTT, and 0.15 U/ml aprotinin. Cell suspensions were centrifuged at 40 000 g for 30 min to obtain cell extracts and protein content was measured according to Bradford (29). Immunoprecipitation was carried out as follows: cell lysates (1 mg) were precleared for 30 min at 4 °C with anti‐rabbit IgG‐agarose beads (1 : 1 v/v, 10 µl) and incubated with 5 µg of rabbit polyclonal antibody or mouse agarose conjugate (according to experiment) overnight at 4 °C and then a further 1 h with anti‐rabbit IgG‐agarose beads (50 µl). Protein samples were separated by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) and were electroblotted onto supported nitrocellulose (30, 31). Each blot was blocked for 1 h with 5% fat‐free milk Tween Tris‐buffered Saline (TTBS) solution at room temperature. Protein bands were probed with primary antibody at 4 °C overnight, followed by horseradish peroxidase‐conjugated anti‐mouse, anti‐rabbit or anti‐goat secondary antibody. Immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrate according to the manufacturers’ instructions (Pierce).
Inhibition of gene expression with siRNA
For short interference RNAs: control siRNA‐A, PKC‐ι siRNA (h2) siRNA transfection reagent, and transfection media were purchased from Santa Cruz Biotechnology. RNA interference functions by a regulator mechanism for sequence‐specific gene silencing through double‐stranded RNA (dsRNA). Sequence‐specific RNAs that were 19–25 nucleotides in length were synthesized by Santa Cruz Biotechnology against PKC‐ι. PKC‐i siRNA consists of three combined RNA sequences. The gene accession number for PKC‐ι is NM_002740. The oligonucleotides sequence for PKC‐i siRNA are as follows: 663 5′‐CAAGCCAAGCGUUUCAACA‐3′; 5′‐UGUUGAAACGCUUGGCUUG‐3′; 739 5′‐GGAACGAUUGGGUUGUCAU‐3′, 5′‐AUGACAACCCAAUCGUUUCC‐3′; 2137 5′‐CCCAAUAUCUUCUCUUGUA‐3′, and 5′‐UACAAGAGAAGAUAUUGGG‐3′. Negative controls containing a scramble sequence that do not lead to specific degradation of any known cellular mRNA were synthesized. The control siRNA‐A sequence is proprietary and Santa Cruz Biotechnology does not reveal it. Effects of PKC‐ι and PKC‐ζ siRNA were determined in exponentially growing RWPE‐1 and DU‐145 prostate cells in complete media over 72 h. Cells were plated in 75 cm2 at a density of 1.5–2.5 × 105 cells/flask with complete media, respectively. The cells were not serum starved. Twenty‐four hours post‐plating, cells were incubated with either control siRNA‐A or PKC‐ι siRNA (50 nm for RWPE‐1 and 150 nm for DU‐145, respectively), according to the manufacturer's instructions (Santa Cruz Biotechnology). Following treatment, cells were washed with PBS, trypsinized and resuspended in 2–3 ml of PBS. Cell viability was quantified using tryphan blue exclusion assay and numbers of unstained cells were counted as mentioned above. Three independent experiments were carried out for each treatment with appropriate controls. Cell viability was determined relative to vehicle control using mean and standard deviation for each time point. Treatment means were separated by Student's t‐test using (Minitab Inc.)
Densitometry
Intensity of each band was measured using Gel Base/Gel Blot‐Pro software (Synoptics Ltd). Briefly, background intensity was subtracted from intensity of each band, to derive corrected intensity. Mean absorbance of three independent experiments were compared using Student's t‐test.
Results
PKC‐ι expression in dividing cells and arresting cells
First, we determined levels of PKC‐ι in both RWPE‐1 and DU‐145 cells that were 50% or 100% confluent (that is, contact inhibited). Cell‐cycle analysis showed accumulation of cells at G0/G1 cell cycle phase in 100% confluent cells. However, 50% confluent cells underwent regular cell‐cycle progression (Table 1). RWPE‐1 cells were slightly arrested whereas DU‐145 cells were highly contact inhibited. In RWPE‐1 cells G0/G1 increased by 14% while S decreased by 30% in 100% confluent cells (Table 1), likely due to perturbation of E6 and E7 gene products, which destabilize p53 and abrogates the function of Rb, respectively (32). In consequence, analysis of the effects of any manipulation of cell‐cycle kinetics in these cells is complicated, and we were unable to achieve higher G0/G1 arrest for RWPE‐1 cells. Nevertheless, RWPE‐1 cells were inhibited and PKC‐ι expression decreased compared to no inhibition (50% confluent), indicating its role for cell survival (Fig. 1). In DU‐145 cells, G0/G1 accumulation increased by 86%, S decreased by more than 95% and the cell cycle was significantly arrested (Table 1). PKC‐ι expression was also reduced in 100% confluent DU‐145 cells, suggesting that PKC‐ι is required for cell survival (Fig. 1a). Other PKC isoforms such as PKC‐α, δ, ζ, βΙ, ɛ were expressed constitutively with no differences between 50% and 100% contact‐inhibited (arrested) cells, demonstrating specificity of PKC‐ι expression at 50% and 100% confluence. Level of PKC‐ι in total cell lysates was higher in 50% confluent cells compared to 100% confluent cell‐cycle‐arrested cells (Fig. 1a). There was no caspase‐7 activation in serum starved (S) and non‐serum‐starved cells, indicating no cell death, when cells were temporally serum starved. Absorbance densitometry revealed a 2‐fold increase in PKC‐ι expression in rapidly dividing cells compared to arrested cells (Fig. 1b). Hence, our data demonstrate that PKC‐ι expression is lower in contact‐inhibited and arrested cells compared to rapidly dividing cells. Therefore, it is likely that PKC‐ι plays a role in cell survival.
Table 1.
Summary of cell‐cycle phases RWPE‐1 and DU‐145 a
| Cell type | G0/G1 | S | G2/M |
|---|---|---|---|
| RWPE‐1 (100% confluent) | 72 ± 1.21 | 23 ± 0.33 | 5 ± 1.67 |
| RWPE‐1 (50% confluent) | 63 ± 3.18 | 33 ± 3.33 | 4 ± 2.67 |
| DU‐145 (100% confluent) | 91 ± 3.38 | 2 ± 0.33 | 6 ± 3.71 |
| DU‐145 (50% confluent) | 49 ± 2.03 | 41 ± 1.15 | 9 ± 2.51 |
N = 3 experiments per cell line.
Figure 1.
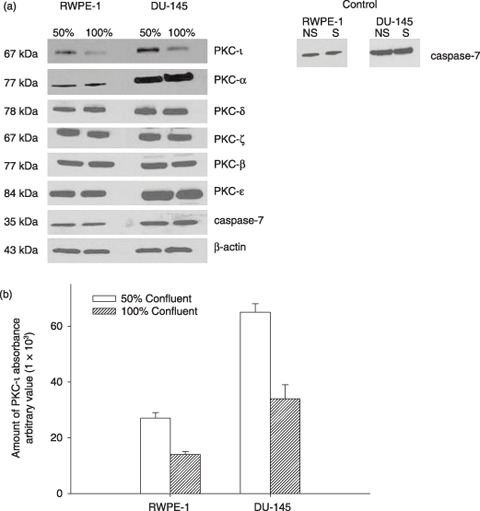
(a) Effect of cell density and cell‐cycle progression on PKC‐ι expression in RWPE‐1 cells and DU‐145 cells. FACS analysis of average DNA content in cell‐cycle distribution is shown in Table 1 . Serum starvation caused cell‐cycle arrest in both rapidly dividing cells (50% confluent) and contact‐inhibited (100% confluent) cells. (a) Western blotting of PKC isoforms was performed as described in the Materials and methods section. Immunoblots of PKC‐ι show fluctuations between 50% and 100% confluent cells. Randomly chosen PKC isoforms (α, δ, ζ, β, ɛ) depict no significant changes between 50% and 100% confluent cells. Inactivated caspase‐7 indicates survival in both cells. Non‐starved (NS) and serum starved (S) at 100% confluent cells. Western blotting of β‐actin indicates equal loading of protein in each lane. (b) Densitometry for PKC‐ι absorbance was calculated from three independent Western blots obtained from three independent experiments. Absorbance, mean and standard deviation were plotted. Average cell‐cycle distribution is shown in Table 1.
Protein kinase C isoforms in RWPE‐1 and DU‐145 cells
Progression through the cell‐cycle involves activation of cyclin‐dependent kinases (Cdks). They fluctuate throughout the cell cycle and are expressed in different cell‐cycle phases. Like Cdks, PKC isoforms may also be expressed transiently or constitutively in different phases of the cell cycle. Therefore, we first determined the doubling time for both RWPE‐1 and DU‐145 cells, which enabled us to determine whether PKC‐ι is associated with Cdks. After counting cells over a 5‐day period, extrapolation as judged by trypan blue exclusion assay revealed that the doubling time for each cell line was 36 h (data not shown) (33). In further experiments, we assumed that this doubling time would represent the length of a cell cycle. Next, we examined PKC isozyme profiles in both RWPE‐1 and DU‐145. The expression of PKCs was not always constitutive and fluctuated throughout the cell cycle (Fig. 2). Therefore, from the 36th hour (doubling time), we monitored expressions of PKC isoforms. In this experiment the cells were semisynchronized by serum starving for 48 h (T = 0), maintaining cell density at 50% confluence in each flask. Subsequent serum incubation from 3 to 36 h allowed the cells to divide for time point analysis without being contact inhibited (shown in Fig. 1). There were nine PKC isoforms (α, β, γ, δ, ɛ, θ, µ, ι and ζ) present in RWPE‐1 cells. PKC‐θ and PKC‐µ had minimal detection sizes even at high protein levels (150–200 µg) (Fig. 2f,g). There was constitutive expression of PKC‐ι in RWPE‐1 cells (32); all nine PKC isoforms (α, β, γ, δ, ɛ, θ, µ, ι and ζ) were detected in DU‐145 cells. PKC‐θ expression appeared to be regulated throughout the cell cycle (Fig. 2f, third column) was also constitutive. Hence, PKC‐ι is required for cell survival and its expression is constitutive.
Figure 2.
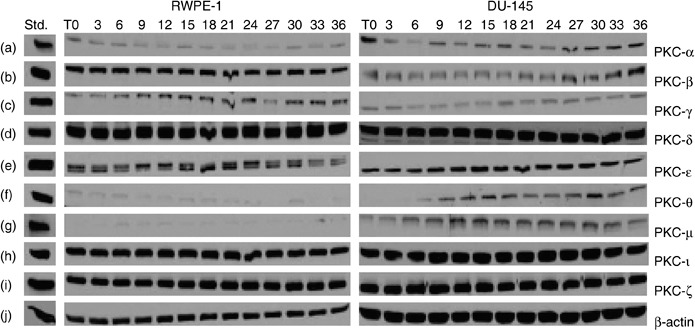
Protein kinase C (PKC) isoforms in RWPE‐1 and DU‐145 cells. Cells were grown to 50–60% confluence. At this confluence, cells have space for cell‐cycle progession and cell division, without contact inhibition. Growth medium was removed and cells were semisynchronized by serum starvation for 48 h (T = 0). Growth media were then added to flasks and cells were allowed to grow for the indicated time (3–36 h). Flasks were collected every 3 h and cells were lysed as described in the Material and methods section. Equal amounts of protein (50–100 µg) were loaded on each lane and ran on 10% SDS‐PAGE. Column 1 contains standards for PKCs, column 2 the PKCs isozymes from normal RWPE‐1 cells, and column 3 PKCs isozymes in androgen‐independent prostate carcinoma DU‐145 cells at indicated times. Only nine PKC isozymes were present: PKC‐α, PKC‐βΙ, PKC‐γ, PKC‐δ, PKC‐ɛ, PKC‐θ, PKC‐µ, PKC‐ι and PKC‐ζ in both RWPE‐1 and DU‐145 cell lines (a–i). Higher protein concentration was required to detect PKC‐θ and PKC‐µ (150 µg each) and only traces were observed in normal RWPE‐1 cells (f–g). Regulation of PKC‐θ expression with the cell cycle was observed in DU‐145 cells (f). Western blots for PKC‐βΙΙ and PKC‐η were performed at higher concentration of protein (200 µg) for each cell line but neither enzyme was expressed. β‐actin (j) verified that equal amounts of protein were loaded in each lane.
Association of PKC‐ι and Cdk7 in RWPE‐1 cells
To test whether PKC‐ι is associated with Cdk7, we immunoprecipitated PKC‐ι from RWPE‐1 cells and prepared Westerns blots probing for Cdk7. There was transient association of PKC‐ι and Cdk7 at the 30 h time point (Fig. 3a). We also performed reverse IP (that is, immunoprecipitating PKC‐iota and Western blotting for Cdk7), but no association was observed. Additionally, association of Cdk2 was observed with Cdk7 (Fig. 3c). However, there was no direct association between PKC‐ι and Cdk2 (data not shown). Phosphorylation of Cdk7 and Cdk2 were also observed throughout the cell cycle (Fig. 3d,e). These results suggest that PKC‐ι was transiently associated with Cdk7. Subsequently, Cdk7 phosphorylated Cdk2 in RWPE‐1 cells. However in DU‐145 cells, there was no association of PKC‐ι with Cdk7 (data not shown) nor were PKC isozymes α, βΙ, δ, θ, ɛ, γ, ζ associated with Cdk7.
Figure 3.
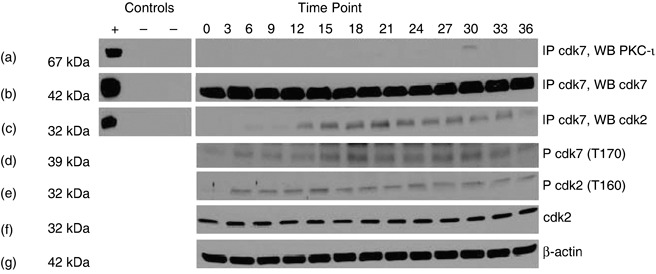
Association of PKC‐ι, with Cdk7 in RWPE‐1 cells. Whole cell extracts (1 mg) from each time point were immunoprecipitated with rabbit polyclonal anti‐Cdk7 (5 µg) as described in the Materials and methods section. Column 1 contains both positive (+) and two negative controls (–). Positive control (+) is the whole cell lysates (100 µg); the first negative control (–) contains whole cell lysates (1 mg) plus rabbit IgG whole molecule (50 µl of 1 : 1 v/v) and the second negative control contains whole cell lysate (1 mg) plus rabbit IgG whole molecule (50 µl) and normal rabbit IgG serum (5 µg). Column 2 is cell lysates taken at indicated time points. Immunoprecipitates were separated by SDS‐PAGE and were Western blotted with anti‐PKC‐ι mouse monoclonal antibody. Physical association of PKC‐ι and Cdk7 were observed at 30 h time points (a). Immunoprecipitation (IP) with rabbit polyclonal Cdk7 (b) showed that Cdk2 is also coimmunoprecipitated (c). Western blotting of phospho‐Cdk7 (p‐Cdk7; T170) (d) and phospho‐Cdk2 (p‐Cdk2; T160) were also observed (e). Presence of Cdk2 was observed throughout the cell cycle (f) and β‐actin (g) shows equal loading of samples in each lane.
Effects of PKC‐ι silencing RNA in RWPE‐1cells and DU‐145 cells
To further prove that PKC‐ι is required for survival of RWPE‐1 cells, PKC‐ι was temporarily inhibited using PKC‐ι siRNA. PKC‐ι siRNA (50 nm) decreased RWPE‐1 cells’ viability by 87–96% after 24–72 h compared to control cells (Fig. 4a). In DU‐145 cells, treatment with 3‐fold more PKC‐ι siRNA (150 nm) blocked any increase in cell number by 80% compared to control siRNA (Fig. 4a). Higher concentration of siRNA was applied due to overexpression of PKC‐ι in DU‐145 cells compared to RWPE‐1 cells (Fig. 3). Immunoblotting for PKC‐ι in PKC‐ι siRNA treated RWPE‐1 cells showed that expression decreased by 90–92% while in DU‐145 cells, a 60–70% decrease in PKC‐ι expression was observed between 24 and 72 h (Fig. 4b). Therefore, incomplete knockdown of PKC‐ι may have contributed to some viable DU‐145 cells. The PKC‐ι siRNA effect was isospecific to the PKC‐ι isozyme and did not affect atypical PKC‐ζ isoforms, which is highly homologus to PKC‐ι. Thus, PKC‐ι siRNA was isospecific to PKC‐ι and did not affect other PKC isoforms. Suppression of atypical PKC‐ι led to cell death and blocked cell population growth in both cell lines.
Figure 4.
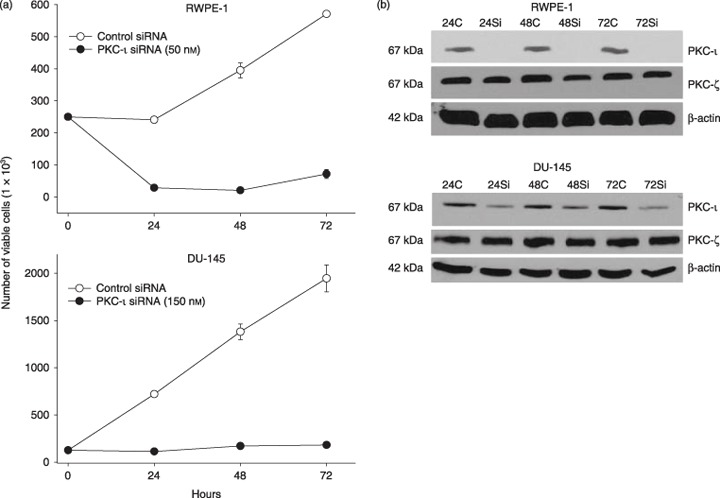
Effect of PKC‐ι siRNA on RWPE‐1 cells and DU‐145 cells. (a) Subconfluent cells (2.5 × 105) were treated with PKC‐ι siRNA complex (50 nm for RWPE‐1 and 150 nm for DU‐145) from 24 to 72 h as described in the Materials and methods section. At indicated times, viable cells were counted using trypan blue exclusion assay and haematocyometry. Three independent experiments were performed and mean number of viable cells and SD were plotted. There is a lower cell viability in PKC‐ι siRNA‐treated cells compared to control siRNA. (b) The same population of treated cells was immunobloted for PKC‐ι, as described in the Materials and methods section. Protein (15 µg) was loaded on each lane for each time point, separated by SDS‐PAGE and Western blotted with anti‐PKC‐ι mouse monoclonal or anti‐PKC‐ζ rabbit polyclonal antibody. There was no or very little PKC‐ι present in PKC‐ι siRNA‐treated cells compared to control siRNA. PKC‐ι siRNA has very little ot no effect on PKC‐ζ indicating specificity. Immunobloting with goat polyclonal anti‐β‐actin shows that the loading of protein is equal in all lanes.
Effects of PKC‐ι siRNA on the cell cycle
We have shown that PKC‐ι siRNA reduced population growth of RWPE‐1 cells while for DU‐145, cells remained cytostatic. Next we established by flow cytometry, which cell cycle phases were disrupted by treatment with PKC‐ι siRNA. RWPE‐1 cell cycle analysis showed a 44–63% decrease in S‐phase and a more than 2‐fold increase in G2/M phase compared to control siRNA throughout the time points (Table 2). However, we did not see any sub‐G0 population (which would represent apoptotic events). Thus, PKC‐ι siRNA inhibited the G2/M cell cycle phase in RWPE‐1. However in DU‐145 cells, PKC‐ι siRNA increased G0/G1 phase progression by 34–36%, and decreased DNA synthesis S‐phase by 41–44% throughout the time course (Table 3). Thus, PKC‐ι siRNA inhibited the G1/S transition in DU‐145 cells. Similar to RWPE‐1, no sub‐G0 population was observed in DU‐145 cells.
Table 2.
Summary of transformed non‐malignant prostate RWPE‐1 cell‐cycle phases after treatment with control siRNA and PKC‐ι siRNA at indicated time a
| Time (h) | G0/G1 | S | G2/M | |||
|---|---|---|---|---|---|---|
| Control | siRNA | Control | siRNA | Control | siRNA | |
| 24 | 59 ± 1 | 63 ± 1 | 32 ± 1 | 18 ± 1 | 8 ± 1 | 20 ± 1 |
| 48 | 56 ± 1 | 70 ± 1 | 36 ± 1 | 13 ± 1 | 15 ± 4 | 18 ± 2 |
| 72 | 59 ± 2 | 67 ± 1 | 32 ± 2 | 12 ± 1 | 9 ± 1 | 22 ± 2 |
N = 3 independent experiments.
Table 3.
Summary of androgen‐independent prostate carcinoma, DU‐145, cell‐cycle phases after treatment with control siRNA and PKC‐ι siRNA at indicated time a
| Time (h) | G0/G1 | S | G2/M | |||
|---|---|---|---|---|---|---|
| Control | siRNA | Control | siRNA | Control | siRNA | |
| 24 | 45 ± 1 | 61 ± 1 | 41 ± 2 | 23 ± 4 | 14 ± 3 | 16 ± 3 |
| 48 | 47 ± 2 | 63 ± 5 | 39 ± 5 | 23 ± 7 | 14 ± 4 | 15 ± 2 |
| 72 | 54 ± 3 | 57 ± 5 | 33 ± 5 | 25 ± 6 | 13 ± 3 | 17 ± 2 |
N = 3 independent experiments.
PKC‐ι siRNA leads to apoptosis
Although we did not see any sub‐G0 population in either cell line, it has been shown that disruption of the cell cycle leads to apoptosis in prostate cancer cells (34, 35). In addition, PKC‐ι siRNA reduced the number RWPE‐1 cells, as judged by phase contrast microscopy. Both RWPE‐1 and DU‐145 cells treated with PKC‐ι siRNA displayed cell rounding, cell membrane blebbing, and lifting of cells from the basebof the flask, indicating cell death, compared to control cells (data not shown). Therefore, RWPE‐1 whole cell extracts were analysed for apoptosis pathways involving activation of PARP, cleaved PARP (Asp214), activation of caspase‐7, survivin and cytochrome c (Fig. 5a, first column). In addition to apoptosis, a similar experiment was performed with RWPE‐1 cells to analyse cell survival pathway involvement of cyclin‐dependent kinases (Cdks). After 24 h treatment with PKC‐ι siRNA complex, cell lysates showed decrease in phosphorylation of Cdk7 (p‐Cdk7; T170) and p‐Cdk2 (T160) in PKC‐ι siRNA‐treated cells compared to control siRNA (P = 0.001 and 0.035, respectively) (Fig. 5b,c). There were no changes in expression of Cdk7 or Cdk2 protein. This corresponded to our earlier finding that PKC‐ι siRNA‐inhibited G2/M phase and decreased phosphorylation of Cdk7 and Cdk2.
Figure 5.
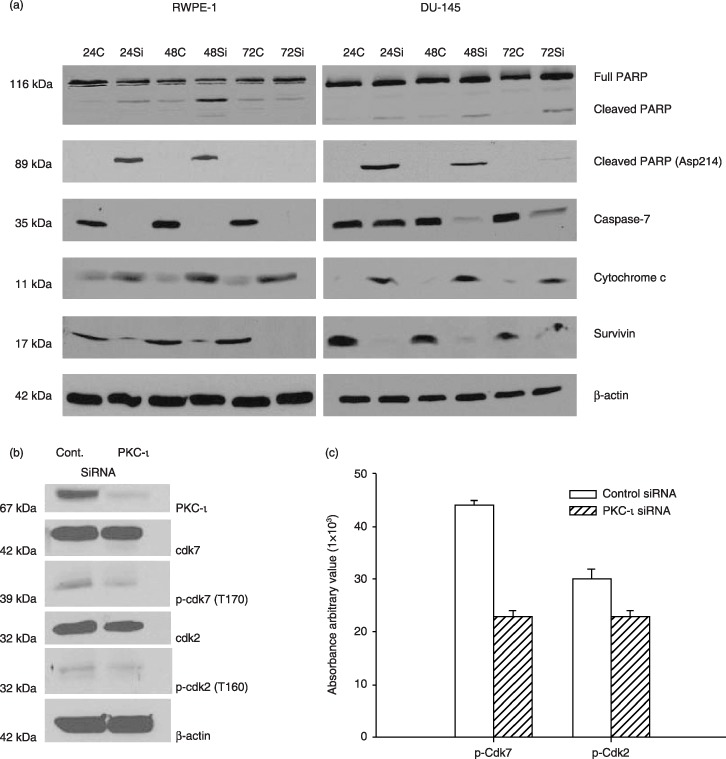
PKC‐ι siRNA leads to apoptosis. Whole cell extracts of RWPE‐1 treated with control siRNA and PKC‐ι siRNA were prepared as described in the Materials and methods section and immunoblot analysis of PARP and cleaved PARP (Asp214), indicated cells are undergoing apoptosis via activation of caspase‐7. Immunoblotting for survivin and cytochrome c further demonstrated apoptosis in PKC‐ι siRNA treated cells. Western blot analysis of β‐actin shows that an equal amount of protein was loaded in each lane. Similar immunoblots were performed for DU‐145 cells. Activation of PARP and caspase‐7, combined with an increase in cytochrome c and decrease in survivin indicated apoptosis in DU‐145 cells. (b) A separate experiment was repeated for RWPE‐1 cells with control siRNA and PKC‐ι siRNA and whole cells extracts (150 µg) were analysed for Cdk7, p‐Cdk7 (T170), Cdk2, p‐Cdk2 (T160), and β‐actin to verify equal loading of protein. (c) Densitometry for p‐Cdk7 and p‐Cdk2 showed a significant decrease in PKC‐ι siRNA‐treated cells compared to control siRNA (P = 0.001 and 0.035, respectively).
In addition, we have observed that PKC‐ι siRNA arrested DU‐145 cells in G0/G1. In addition, immunoblotting for PARP and cleaved PARP (Asp214) indicated that PKC‐ι siRNA‐treated cells experienced apoptosis. Activation of caspase‐7, combined with increased cytochrome c protein and decrease in survivin were also observed in PKC‐ι siRNA‐treated cells compared to cells treated with control siRNA (Fig. 5a, second column). Collectively, these results imply that PKC‐ι siRNA inhibited the cell cycle in the RWPE‐1 and DU‐145 cells leading to apoptosis. Hence, PKC‐ι is an anti‐apoptotic protein required for cell survival.
PKC‐ι siRNA effects on phosphorylation status of Bad and Bad/Bcl‐xL heterodimerization
PKC‐ι has been reported to be overexpressed in NSCLC and to phosphorylate Bad at multiple sites for cell survival to occur (8). To test whether PKC‐ι can directly phosphorylate endogenous Bad, Bad protein was immunoprecipitated from control siRNA and treated with PKC‐ι siRNA (incubation time 24 h) in RWPE‐1 and DU‐145 cells. Results demonstrated no association between PKC‐ι and Bad. We also increased protein level and performed reverse immunoprecipitation with PKC‐ι antibody, but no association of Bad with PKC‐ι was observed. Although immunoblotting with phospho‐specific Bad (ser‐112, ser‐155, ser‐136) showed no significant changes in phospho Bad ser‐112, phosphorylation of Bad ser‐155 and ser‐136 decreased in RWPE‐1 cells (Fig. 6a, second column) and slight increase in Bad/Bcl‐xL association was also observed. These results suggest that pro‐apoptotic Bad is activated by dephosphorylation and subsequently it heterodimerizes with anti‐apoptotic Bcl‐xL, thereby quenching the anti‐apoptotic function and exerting the pro‐apoptotic function of Bad (21). Because no direct association of PKC‐ι/Bad was observed, the results show only that PKC‐ι may indirectly influence phosphorylation of Bad, leading to cell death.
Figure 6.
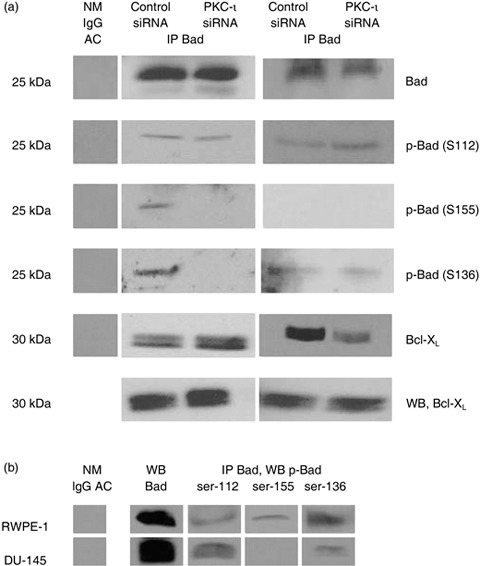
Effects of PKC‐ι siRNA on Bad phosphorylation and Bad/Bcl‐xL heterodimerization. (a) Both RWPE‐1 cells and DU‐145 cells were treated with PKC‐ι siRNA for 24 h and cell lysates (300 µg) were immuoprecipitated with Bad mouse monoclonal agarose‐conjugate (5 µg). The resulting supernatant and immunocomplex beads were subjected to SDS‐PAGE. The first column is the negative control containing normal mouse IgG agarose conjugate with cell lysate. Phosphorylation of Bad was analysed using phospho‐specific antibodies: Bad Ser‐112 (S112), Ser‐155 (S155) and Ser‐136(S136). Bound Bcl‐xL with Bad and unbound Bcl‐xL was also analysed. (b) Cell lysates of endogenous protein (1 mg) were immunoprecipitated with Bad and different level of phosphorylation status of Bad in normal RWPE‐1 cells and DU‐145 cells was also examined.
A similar experiment was performed with DU‐145 cells (Fig. 6a, third column), and we found no association of PKC‐ι and Bad (data not shown). However, we observed increase in phosphorylation of Bad at serine‐112 but no significant changes in phospho‐Bad serine‐136 and 155. An increase in phosphorylation of Bad in PKC‐ι siRNA‐treated cells could be due to Akt/PKB, which mediates phospho Bad at serine 136, while RSK2/PKA/PAK are involved with phosphorylation of Bad at ser‐112 and ser‐155 (36). Our results also showed disruption of Bad/Bcl‐xL heterodimerization in PKC‐ι siRNA‐treated cells compared to control siRNA but the anti‐apoptotic level of Bcl‐xL protein remained the same. Disruption of Bad/Bcl‐xL heterodimerization may play a critical role in mitochondrial dysfunction and apoptosis in PKC‐ι siRNA‐treated cells.
Higher concentration of protein (1 mg) was immunoprecipitated with Bad to demonstrate phosphorylation of Bad in both cell lines. We also found that there were different levels of phosphorylation of Bad between RWPE‐1 and DU‐145 cells (Fig. 6b). All three phospho serine sites (ser‐112, ser‐155, ser‐136) on Bad protein were expressed endogenously in RWPE‐1 cells while DU‐145 cells had only two phospho‐Bads (ser‐112 and ser‐136) (Fig. 6b). Collectively, these results suggest that in DU‐145 cells, phosphorylation of Bad (deactivation/survival) was not able to rescue the cells from PKC‐ι siRNA‐induced apoptosis.
Discussion
Although expression of PKC is regulated in early prostate cancer, the mechanisms are still unclear due to high PKC homology and specificity (2, 11). Here, we have demonstrated that PKC‐ι is required for cell survival in both RWPE‐1 and DU‐145 cells. Analysis of PKC isozyme profiles revealed nine PKC isoforms (α, βΙ, γ, δ, ɛ, θ, µ, ι, and ζ) in both cell lines throughout their cell cycle but protein kinases C‐βΙΙ and PKC‐η were not detected, even at higher concentrations (data not shown). PKC‐ι was constitutively expressed throughout cell cycle.
RWPE‐1 cells were only slightly arrested at high confluenc in G0/G1 (a 10% increase in the number of cells arrested in G0/G1 with a 30% decrease in the number of cells in S phase). This is consistent with apparent disruption of both Rb and p53 here (32). The RWPE‐1 cell line was derived from a prostatic epithelial cells and was immortalized by using HPV‐18 (32). This disrupts regulation of the cell cycle and cell death through interaction of the E6 and E7 gene products, which destabilize p53 and abrogate the function of Rb, respectively. In consequence, analysis of the effects of any manipulation of cell cycle kinetics in these cells is complicated. This is further highlighted by the fact that levels of PKC‐ι do not appear to change during the cell cycle. However, PKC‐ι decreases when RWPE‐1 cells are contact inhibited, indicating its role in cell survival.
DU‐145 cells in contrast, were highly contact inhibited and showed highly significant arrest in G0/G1. We found higher expression of PKC‐ι in dividing cells compared to contact‐inhibited arrested cells. The change in PKC expression is specific to PKC‐ι; other isoforms were constitutively present in both dividing cells and arrested cells. Hence, fluctuation of PKC‐ι expression may play a role in cell cycle progrression. Other studies have shown that PKC‐ι and PKC‐βΙΙ co‐localize with CAK/Cdk7 in human glioma cells and phosphorylated CAK/Cdk7 during cell‐cycle progression (5, 6). Similarly in RWPE‐1 cells, we found that PKC‐ι was transiently associated with Cdk7, which phosphorylates Cdk2 and drives cell‐cycle progression, whereas we found no association of PKC‐ι and Cdk7 in DU‐145 cells, nor did we find other PKC isoforms to be associated with Cdk7 in DU‐145 cells.
PKC‐ι is required for survival of RWPE‐1 cells, as PKC‐ι siRNA induced cell cycle arrest at G0/G1 and G2/M. The complex results seen in the RWPE‐1 cells may be related to the multiple Cdks that can be phosphorylated by Cdk7 and/or disruption of the cell cycle by HPV‐18 E6 and E7 oncoproteins (32). In addition, there was decrease in phosphorylation of Cdk7 and Cdk2 when the cells were arrested at G2/M phase. Therefore, it is possible that the association of PKC‐ι with Cdk7 during the G2/M phase is required for cell cycle progression and cell survival.
However, in androgen‐independent DU‐145 cells, PKC‐ι siRNA treatment leads to G0/G1 arrest, resulting in inhibition of G1/S transition. Consistent with G0/G1 arrest, and no association of PKC‐ι and Cdk7 in DU‐145 cells, it is possible that PKC‐ι may associate with other Cdks such as Cdk4 and Cdk6 (34, 35, 37). Besides atypical PKC‐ι, novel PKC‐δ is also involved in cell survival and phorbol ester‐induced apoptosis of prostate LNCaP carcinoma cells. However, PKC‐δ‐mediated apoptosis was independent of caspase activation, suggesting that allosteric activation of PKC‐δ is sufficient to trigger apoptosis in LNCaP cells (38). Suppression of PKC‐ι in RWPE‐1 cells resulted in G2/M arrest and cell death; in DU‐145 cells, PKC‐ι siRNA arrested cells at the G0/G1 phase, blocking increase in cell number.
Apoptotic cell death is characterized by morphological changes such as cell shrinkage, chromatin condensation and DNA fragmentation (39, 40). Transfecting RWPE‐1 cells and DU‐145 cells with PKC‐ι siRNA induced cells to demonstrate stress signals such as membrane blebbing, cell rounding, and lifting of cells from culture flask surfaces. This indicates apoptotic changes, which have both of intrinsic and extrinsic pathways (19, 41). The intrinsic pathway depends on depolarization of mitochondria and the extrinsic pathway involves activation of death receptors. Release of cytochrome c, activation of caspase‐3, caspase‐6 and caspase‐7, and cleaved PARP are relevant biomarkers in apoptotic induction of the intrinsic pathway, all of which occurred when PKC‐ι was suppressed with PKC‐ι siRNA in RWPE‐1 cells and DU‐145 cells (42, 43, 44). Survivin protein, which regulates cell division and apoptosis, is also linked to ‘intrinsic’ apoptosis (45, 46) and we showed that survivin was suppressed in PKC‐ι siRNA‐treated cells compared to control siRNA in RWPE‐1 and DU‐145 cells. Our data correspond to other reports where disruption of cell‐cycle leads to apoptosis in prostate cells (34, 35). Collectively, the data show that PKC‐ι is required for cell survival in RWPE‐1 cells and that it is an anti‐apoptotic protein required for DU‐145 cell survival.
There are a number of substrates for PKCs (7, 10, 47). In particular, PKC‐ι has been shown to phosphorylate Bad at three different serine sites (ser‐112, ser‐155 and ser‐136) for survival of NSCLC (7). Bad is a pro‐apoptotic protein that can translocate from cytosol to nucleus and disrupts Bad/Bcl‐2/Bcl‐xL heterodimerization, leading to apoptosis. Phosphorylated Bad (inactivated Bad) is then sequestered by 14‐3‐3 proteins for cell survival (48, 49). We also found that immunoprecipitation with either Bad antibody or PKC‐ι antibody showed no association of Bad with PKC‐ι in both non‐maglignant prostate and carcinoma prostate cell lines.
Transient suppression of PKC‐ι also induced no significant changes in phospho Bad ser‐112, but there was decrease in phospho Bad ser‐155 and ser‐136 in prostate, RWPE‐1, cells. A slight increase in association of Bad/Bcl‐xL was also observed in PKC‐ι siRNA‐treated cells, meaning pro‐apoptotic Bad had quenched anti‐apoptotic protein, Bcl‐xL, leading to cell death of RWPE‐1 cells. However, in DU‐145, phosphorylation of Bad increased at ser‐112, while ser‐136 showed no significant change in phosphorylation. Phosphorylation of Bad at ser‐155 was not observed in endogenous protein levels. Increased phosphorylation of Bad could be due to Akt/PKB, which mediates phosphorylation of Bad at serine‐136, while RSK2/PKA/PAK is involved with phosphorylation of Bad at ser‐112 and ser‐155 (36). We also observed reduced disassociation of Bad/Bcl‐xL. Taken together, PKC‐ι may not be directly involved in phosphorylation of Bad in prostate cells, however, transient suppression of PKC‐ι may indirectly influence mitochondrial dysfunction, independent of phosphorylation of Bad, and lead to apoptosis in both RWPE‐1 and DU‐145 cells. Our results indicate that RWPE‐1 and DU‐145 cells have different Bad phosphorylation patterns. In RWPE‐1 cells, there were endogenous levels of phospho Bad at ser‐112, ser‐136 and ser‐155. In DU‐145 cells, p‐Bad ser‐112 and p‐Bad ser‐136 were present. It is reasonable to assume that there could be kinetic effects between phosphorylation of Bad and PKC‐ι, an anti‐apoptotic protein. Increase in phosphorylated pro‐apoptotic Bad did not overcome suppression of anti‐apoptotic PKC‐ι in prostate cancer cells.
It has been hypothesized that a balance between prosurvial and prodeath Bcl‐2 family members determines whether a cell lives or dies (50), but the precise molecular mechanisms of how membrane‐bound Bcl‐2 proteins exert their control over caspase activation and apoptosis is still unclear. Anti‐apoptotic Bcl‐xL protein is overexpressed in prostate cancer cells and down‐regulation of Bcl‐xL leads to TGF‐βΙ‐induced apoptosis (18). Bcl‐xL also participates with Bcr‐Abl, a protein responsible for chemotherapeutic resistance and mediates apoptosis in transformed pro‐myelocytic HL‐60 cells (51, 52). Taxol and 2‐methoxyestradiol induced phosphorylation of Bcl‐xL at ser‐62–has induced apoptosis in prostate cancer cells (53). In addition, Bcl‐xL has been shown to be overexpressed in many tumour cells leading to cell survival (28). Our study found no decrease in Bcl‐xL protein level nor changes in phosphorylation status of Bcl‐xL. PKC‐ι siRNA increased association of Bad/Bcl‐xL in RWPE‐1 cells, but disassociated Bad and Bcl‐xL in DU‐145 cells, leading to mitochondrial dysfunction followed by apoptosis.
In summary, suppression of PKC‐ι leads to cell‐cycle arrest at G2/M, leading to apoptosis in RWPE‐1 cells. In DU‐145 cells, suppression of PKC‐ι leads to G0/G1 arrest and apoptosis independent of Bad phosphorylation.
Acknowledgements
We acknowledge the excellent contribution of the Flow Cytometry Core, H. Lee Moffitt Cancer Center. This project was supported in part by Inhibiton Therapeutics Inc., the Sapphire Foundation, and the Research Service of the Veterans’ Administration.
References
- 1. Murray NR, Fields AP (1997) Atypical protein kinase C ι protects human leukemia cells against drug‐induced apoptosis. J. Biol. Chem. 272, 27521–27524. [DOI] [PubMed] [Google Scholar]
- 2. Cornford P, Evans J, Dodson A, Parsons K, Woolfenden A, Neoptolemos J, Foster CS (1999) Protein kinase C isozyme patterns characteristically modulated in early prostate cancer. Am. J. Pathol. 154, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yuichi TM, Gavrielides V, Mitsuuchi Y, Fujii T, Kazanietz MG (2003) Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J. Biol. Chem. 278, 33753–33762. [DOI] [PubMed] [Google Scholar]
- 4. Jamieson L, Carpenters L, Biden TJ, Fields AP (1999) Protein kinase Cι activity is necessary for Bcr‐Abl‐mediated resistance to drug‐induced apoptosis. J. Biol. Chem. 274, 3927–3930. [DOI] [PubMed] [Google Scholar]
- 5. Acevedo‐Duncan M, Patel R, Whelan S, Bicaku E (2002) Human glioma PKC‐ι and PKC‐βII phosphorylate cyclin‐dependent kinase activating kinase during the cell cycle. Cell Prolif. 35, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bicaku E, Acevedo‐Duncan M (2001) Ultrastructural study of protein kinase C‐βII localization during the cell cycle of human glioma cells. Tissue Cell 33, 55–62. [DOI] [PubMed] [Google Scholar]
- 7. Zhaohui J, Meiguo X, Xingming D (2005) Survival function of protein kinase Cι as a novel nitrosamine 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone‐activated Bad kinase. J. Biol. Chem. 290, 16045–16052. [DOI] [PubMed] [Google Scholar]
- 8. Stallings‐Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP (2006) A novel small‐molecule inhibitor of protein kinase Cι blocks transformed growth of non‐small‐cell lung cancer cells. Cancer Res. 66, 1761–1774. [DOI] [PubMed] [Google Scholar]
- 9. Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP (2005) Atypical protein kinase Cι plays a critical role in human lung cancer cell growth and tumorigenicity. J. Biol. Chem. 280, 31109–31115. [DOI] [PubMed] [Google Scholar]
- 10. Gustafson WC, Ray S, Jamieson L, Thomson EA, Brasier AR, Fields AP (2004) Bcr‐Abl regulates protein kinase Cι (PKCι) transcription via an Elk1 site in the PKC‐ι promoter. J. Biol. Chem. 279, 9400–9408. [DOI] [PubMed] [Google Scholar]
- 11. Choi WC, Ahn CH (1993) Protein kinase C (PKC) in cellular signaling system: translocation of six protein kinase C isozymes in human prostate adenocarcinoma PC‐3 cell line. Korean J. Zool. 36, 439–451. [Google Scholar]
- 12. Henttu P, Vihko P (1998) The protein kinase C activator, phorbol ester, elicits separate functional responses in androgen‐sensitive and androgen‐independent human prostatic cancer cells. Biochem. Biophys. Res. Comm. 244, 167–171. [DOI] [PubMed] [Google Scholar]
- 13. Gonzalez‐Guerrico AM, Meshki J, Xiao L, Benavides F, Conti CJ, Kazanietz MG (2005) Molecular mechanisms of protein kinase C‐induced apoptosis in prostate cancer cells. J. Biochem. Mol. Biol. 38, 639–645. [DOI] [PubMed] [Google Scholar]
- 14. Powell CT, Brittis NJ, Stec D, Hug H, Heston WD, Fair WR (1996) Persistent membrane translocation of protein kinase C α during 12‐0‐tetradecanoylphorbol‐13‐acetate‐induced apoptosis of LNCaP human prostate cancer cells. Cell Growth Differ. 7, 419–428. [PubMed] [Google Scholar]
- 15. Fuji T, Garcia‐Bermejo ML, Bernabo JL, Caamaon J, Ohba M, Kuroki T, Li L, Yuspa SH, Kazanietz MG (2000) Involvement of protein kinase C δ (PKCδ) in phorbol ester‐induced apoptosis in LNCaP prostate cancer cells. J. Biol. Chem. 275, 7574–7582. [DOI] [PubMed] [Google Scholar]
- 16. Haldar S, Chintapalli J, Croce CM (1996) Taxol induces bcl‐2 phosphorylation and death of prostate cancer cells. Cancer Res. 56, 1253–1255. [PubMed] [Google Scholar]
- 17. Bookstein R, Shew JY, Chen PL, Scully P, Lee WH (1990) Suppression of tumorigenicity of human prostate carcinoma cells by replacing a mutated Rb gene. Science 247, 712–715. [DOI] [PubMed] [Google Scholar]
- 18. Chipuk JE, Bhat M, Hsing AY, Ma J, Danielpour D (2001) Bcl‐xL blocks transforming growth factor‐β1‐induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing apaf‐1‐dependent caspase activation in prostate epithelia cells. J. Biol. Chem. 276, 26614–26621. [DOI] [PubMed] [Google Scholar]
- 19. Gross A, McDonnell JM, Korsmeyer SJ (1999) Bcl‐2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911. [DOI] [PubMed] [Google Scholar]
- 20. Virdee K, Parone PA, Tolkovsky AM (2000) Phosphorylation of the pro‐apoptotic protein BAD on serine 155, a novel site, contribute to cell survival. Curr. Biol. 10, 1151–1154. [DOI] [PubMed] [Google Scholar]
- 21. Tan Y, Demeter MR, Ruan H, Comb MJ (2000) Bad ser‐155 phosphorylation regulates BAD/Bcl–XL interaction and cell survival. J. Biol. Chem. 275, 25865–25869. [DOI] [PubMed] [Google Scholar]
- 22. Yang CC, Lin HP, Chen CS, Yang YT, Tseng PH, Rangnekar VM, Chen CS (2003) Bcl‐xL mediates a survival mechanism independent of the phosphoinostide 3‐kinase/Akt pathway in prostate cancer. J. Biol. Chem. 278, 25872–25878. [DOI] [PubMed] [Google Scholar]
- 23. Zhou XM, Liu Y, Payne G, Lutz RJ, Chittenden T (2000) Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155 . J. Biol. Chem. 275, 25046–25051. [DOI] [PubMed] [Google Scholar]
- 24. Irina L, Robert R, Joshua O, Paul C, Stein CA (2000) Bcl‐xL in prostate cancer cells: effects of overexpression and down regulation of chemosensitivity. Cancer Res. 60, 6052–6090. [PubMed] [Google Scholar]
- 25. Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R (1995) Overexpression of bcl‐2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo . Cancer Res. 55, 4438–4445. [PubMed] [Google Scholar]
- 26. Dean GT, Li L, Dharam PC, Arthur TP (1998) Extended survivability of prostate cancer cells in the absence of trophic factors: increase proliferation, evasion of apoptosis, and the role of apoptosis proteins. Cancer Res. 58, 3466–3479. [PubMed] [Google Scholar]
- 27. Kazuki Y, Palma R, Hideaki M, Ladan F, Alan S, Uwe ZW, Martin EG (2006) Induction of apoptosis and enhancement of chemosensitivity in human prostate cancer LNCaP cells using bispecific antisense oligonucleotide targeting Bcl‐2 and Bcl‐xL genes. BJU Int. 97, 13000–11308. [DOI] [PubMed] [Google Scholar]
- 28. Carlton JC, Terry NHA, White A (1991) Measuring potential doubling times of murine tumors using flow cytometry. Cytometry 12, 645–6450. [DOI] [PubMed] [Google Scholar]
- 29. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 30. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 237, 680–685. [DOI] [PubMed] [Google Scholar]
- 31. Towbin H, Staehelin T, Gordon PE (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl Acad. Sci. USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS (1997) Androgen responsive adult human prostate epithelial cell lines immortalized by papillomavirus 18. Carcinogenesis 18, 1215–1223. [DOI] [PubMed] [Google Scholar]
- 33. Webber MM, Bello D, Quader S (1997) Immortalized and tumorigenic adult human prostatic epithelial cell lines: characteristics and applications. Part 2. Tumorigenic cell lines. Prostate 30, 58–64. [DOI] [PubMed] [Google Scholar]
- 34. Yim D, Sigh RP, Agarwal C, Lee S, Chi H, Agarwal R (2005) A novel anticancer agent, decursin, induces G1 arrest and apoptosis in human prostate carcinoma cells. Cancer Res. 65, 1035–1044. [PubMed] [Google Scholar]
- 35. Mantena SK, Sharma SD, Katiyar SK (2006) Berberine, a natural product, induces G1‐phase cell cycle arrest and caspase‐3‐dependent apoptosis in human prostate carcinoma cells. Mol. Cancer Ther. 5, 296–308. [DOI] [PubMed] [Google Scholar]
- 36. Downward J (1999) How Bad phosphorylation is good for survival. Nat. Cell Bio. 1, E33–E35. [DOI] [PubMed] [Google Scholar]
- 37. Gao N, Zhang Z, Jiang BH, Shi X (2003) Role of PI3/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophys. Res. Commun. 4, 1124–1132. [DOI] [PubMed] [Google Scholar]
- 38. Teruhiki F, Maria LGB, Juan LB, Jorge C, Motoi O, Toshio K, Luowei Li Stuart HY, Marcelo GK (2000) Involvement of protein kinase C δ (PKC δ) in phorbol ester‐induced apoptosis in LNCaP prostate cancer cells. J. Biol. Chem. 275, 7574–7582. [DOI] [PubMed] [Google Scholar]
- 39. Kerr JFK, Wyllie AH, Currie AH (1972) Apoptosis, a basic biological phenomenon with wider implication in tissue kinetics. Br. J. Cancer 26, 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wyllie AH, Keer JFK, Currie AR (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251–306. [DOI] [PubMed] [Google Scholar]
- 41. Chipuk JE, Green DR (2005) Do inducers of apoptosis trigger caspase‐independent cell death? Nature 6, 268–275. [DOI] [PubMed] [Google Scholar]
- 42. Kaufmann S, Desnoyers S, Ottaviano Y, Davidson N, Poirier G (1993) Specific proteolytic cleavage by poly (ADP‐ribose) polymerase: an early marker of chemotherapy‐induced apoptosis. Cancer Res. 53, 3976–3985. [PubMed] [Google Scholar]
- 43. Green GR, Reed JC (1998) Mitochondria and apoptosis. Science 283, 1309–1312. [DOI] [PubMed] [Google Scholar]
- 44. Marchetti P, Castedo M, Susin SA et al (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 184, 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grossman D, Kim PJ, Blanc‐Brude OP, Brash DE, Tognin S, Marchisio PC, Altieri DC (2001) Transgenic expression of survivin in keratinocytes counteracts UVB‐induced apoptosis and cooperates with loss of p53. J. Clin. Invest. 108, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mesri M, Wall NR, Li J, Kim RW, Altieri DC (2001) Cancer gene therapy using surviving mutant adenovirus. J. Clin. Invest. 108, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ying L, Lee J, Allan RB, Alan PF (2001) NF‐κB/RelA transactivation is required for atypical protein kinase Cι‐mediated cell survival. Oncogene 20, 4777–4792. [DOI] [PubMed] [Google Scholar]
- 48. Sandeep RD, Alex K, Linda H, Andrew P, Stephen WF, Michael BY, Micahel EG (2000) 14‐3‐3 proteins and survival kinases cooperate to inactivate Bad by BH3 domain phosphorylation. Mol. Cell 6, 41–51. [PubMed] [Google Scholar]
- 49. Jiping Z, Hisasha H, Elizabeth Y, Jennifer J, Stanley JK (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14‐3‐3 not BCL‐XL Cell 87, 619–628. [DOI] [PubMed] [Google Scholar]
- 50. Oltvai ZN, Korsmeyer SJ (1994) Checkpoints of dueling dimmers foil death wishes. Cell 79, 189–192. [DOI] [PubMed] [Google Scholar]
- 51. Amarante‐Mendes GP, McGahon AJ, Nishioka WK, Afar DEH, Witte ON, Green GR (1998) Bcl‐2‐independent Bcr‐Abl‐mediated resistance to apoptosis: protection is correlated with up regulation of Bcl‐xL. Oncogene 16, 1383–1390. [DOI] [PubMed] [Google Scholar]
- 52. Larisch‐Bloch S, Danielpour D, Roche NS, Lotan R, Hsing AY, Kerner H, Hajouj T, Lechleider RJ, Robers AB (2000) Selective loss of the transforming growth factor‐β apoptotic signaling pathway in mutant NRP‐154 rat prostatic epithelial cells. Cell Growth Differ. 11, 1–10. [PubMed] [Google Scholar]
- 53. Basu A, Haldar S (2003) Identification of a novel Bcl‐xL phosphorylation site regulating the sensitivity of taxol‐ or 2‐methoxyestradiol‐induced apoptosis. FEBS Lett. 538, 41–47. [DOI] [PubMed] [Google Scholar]