

Abstract
Abstract. Stem cells share many properties with malignant cells, such as the ability to self‐renew and proliferate. Cancer is believed to be a disease of stem cells. The gastrointestinal tract has high cancer prevalence partly because of rapid epithelial cell turnover and exposure to dietary toxins. The molecular pathways of carcinogenesis differ according to the tissue. Work on hereditary cancer syndromes including familial adenomatous polyposis (FAP) has led to advances in our understanding of the events that occur in tumour development from a gastrointestinal stem cell. The initial mutation involved in the adenoma–carcinoma sequence is in the ‘gatekeeper’ tumour‐suppressor gene adenomatous polyposis coli (APC). Somatic hits in this gene are non‐random in FAP, with the type of mutation selected for by the position of the germline mutation. In the stomach, a metaplasia–dysplasia sequence occurs and is often related to Helicobacter pylori infection. Clonal expansion of mutated cells occurs by niche succession. Further expansion of the aberrant clone then occurs by the longitudinal division of crypts into two daughter units – crypt fission. Two theories seek to explain the early development of adenomas – the ‘top down’ and ‘bottom up’ hypotheses. Initial studies suggested that colorectal tumours were monoclonal; however, later work on chimeric mice and a sex chromosome mixoploid patient with FAP suggested that up to 76% of early adenomas were polyclonal. Introduction of a homozygous resistance allele has reduced tumour multiplicity in the mouse and has been used to rule out random collision of polyps as the cause of these observations. It is likely that short‐range interaction between adjacent initiated crypts is responsible for polyclonality.
INTRODUCTION
The gastrointestinal tract has a rapid epithelial cell turnover that continues throughout life. Intestinal cells are also exposed to a hostile environment as they come into close contact with numerous toxins and carcinogens contained in digested food. Thus it is of little surprise that cancer of the digestive system is common, with 255 640 new cases in the USA alone in 2004 (American Cancer Society 2004). Partly because of its rapid cell turnover and high cancer prevalence, the gastrointestinal epithelium has become an important tissue in the understanding of cancer biology. Colonic polyposis syndromes were first recognized 200 years ago, and it has been 100 years since inflammatory and adenomatous polyps were characterized (Franzin et al. 1995). More recently, work on the familial colonic cancer syndromes including familial adenomatous polyposis (FAP) has led to a number of advances in the understanding of intestinal tumour initiation including the recognition that many colonic adenomas progress to adenocarcinomas (Morson 1974). This was termed the adenoma–carcinoma sequence, and has become established as a stepwise pattern of mutational activation of oncogenes and inactivation of tumour suppressor genes that result in cancer (Vogelstein et al. 1988). The rapid turnover of cells in the gastrointestinal tract means that differentiating cells are shed into the lumen and replaced every few days and thus do not have a sufficient lifespan to accumulate the mutations necessary for malignant change, thus it is widely believed that it is the perpetual stem cell that is the target of mutational changes (Potten & Loeffler 1990; Wong & Wright 1999; Bach et al. 2000). This review will summarize the molecular and cellular events involving the stem cell that occur at the birth of the adenoma, the spread of dysplastic tissue around the bowel and the development of malignancy.
Intestinal stem cells and their niches
The stomach and the small and large intestines are histologically distinct, although the various regions share a basic structure. There is a mucosal lining beneath which is a thin muscular layer known as the muscularis mucosae. Beneath the muscularis mucosae is the submucosa and the next layer is the muscularis propria. The luminal surface of the intestine is composed of a columnar epithelial mucosa, with glandular invaginations called crypts – the basic functional unit of the gut. In the small bowel, several of these crypts contribute epithelium to finger‐like projections called villi. The cells of the intestinal epithelium are arranged hierarchically, becoming progressively more differentiated as they age and pass along the crypt–villus axis. In the stomach, the epithelial lining is formed into long tubular glands, each subdivided into foveolus, isthmus, neck and base regions. The gastric pits open on to the surface of the gastric mucosa. Between three and five of the tubular gastric glands themselves open into the bottom of one pit. The foveolae and mucosal surface is made up of gastric foveolar mucous cells. The acid‐secreting parietal (oxyntic) cells and the pepsinogen‐secreting peptic/chief (zymogenic) cells are found in the base of the gastric glands, in the body and the fundus/body of the stomach, respectively (Wright 1984). Within the gastric glands cell migration is bi‐directional, with the differentiating mucous cells migrating upwards and the developing parietal and chief cells moving down towards the gland base. The putative stem cell compartment is thus believed to lie in the neck/isthmus region of the gastric gland. The immature, relatively undifferentiated nature of gastrointestinal epithelial stem cells means that they are not directly identifiable, and researchers in this field in the past have had to rely on ingenious indirect methods to identify their position and track their progeny (reviewed in Preston et al. 2004). Recent work on molecules uniquely involved in the biochemical pathways of the stem cell may provide useful tools for cell identification. One such protein is Mushashi‐1, the mammalian equivalent of a Drosophila protein. It is responsible for the up‐regulation of expression of the transcriptional repressor Hes‐1, a protein involved in neural stem cell self‐renewal (Nakamura et al. 2000). Both these proteins are coexpressed in cells superior to the Paneth cells in the mouse intestine, but Hes‐1 alone is only seen in differentiating cells, thus it is hypothesized that the co‐localization of these two markers may denote the small bowel stem cell population (Kayahara et al. 2003). Other putative markers for gastrointestinal stem cells may include members of the integrin family, which are differentially expressed along the crypt–villus axis (Teller & Beaulieu 2001). The stem cell compartment is believed to be at the origin of the crypt–villus axis, the base of the colonic crypt and at cell position 4–5 in the small bowel (reviewed in Brittan & Wright 2002). The number of stem cells within this compartment is debated but is generally believed to be between four and six (Bjerknes & Cheng 1999; Marshman et al. 2002). Stem cells themselves divide infrequently and it is the first few generations of stem cell daughters, known as transit‐amplifying cells, that proliferate in the lower part of the crypt (Bach et al. 2000). Stem cells reside within a stem cell compartment or ‘niche’. This is a group of epithelial and mesenchymal cells and extra‐cellular substrates that provide an optimal microenvironment for stem cells to give rise to their differentiated progeny. In the intestinal crypts this is formed by a fenestrated sheath of surrounding mesenchymal cells that regulate stem cell behaviour through paracrine secretion of growth factors and cytokines (Powell et al. 1999). Functionally, a niche is characterized by its persistence on removal of stem cells and the cessation of stem cell potential when cells are removed from this niche (Spradling et al. 2001).
Stem cell mutations and the path to cancer
The different behaviour of the stem cells within the different tissue niches along the gastrointestinal tract is reflected in the marked difference in the prevalence of digestive system malignancies. Small intestinal crypts rarely undergo malignant transformation despite large numbers of stem cells undergoing rapid cell division (Marshman et al. 2002). Unfortunately, the stem cells of the oesophagus, stomach and colon frequently acquire the genetic defects associated with cancer induction; however, the route taken in carcinogenesis differs in each organ.
Stomach
The gastrointestinal tract is one of the most frequent sites of carcinogenesis because of its continual self‐renewal and the resultant large numbers of daily mitotic events. In the stomach, resulting carcinomas are the second most common cause of cancer‐related mortality worldwide. Gastric adenocarcinomas can be subdivided histopathologically according to Lauren into two types termed the intestinal or well‐differentiated type and the diffuse or poorly differentiated type. The intestinal‐type adenocarcinomas show intestinal‐gland‐like structures and develop in a stepwise transition from normal mucosa through atrophic gastritis, intestinal metaplasia and dysplasia. The diffuse‐type cancer is only poorly differentiated, lacks any glandular structure and in its development no association with atrophy or metaplasia can be found. The two of them also differ substantially in epidemiology and pathogenesis. What both of them seem to have in common is their most important aetiological factor, Helicobacter pylori.
Helicobacter pylori infection
Infection with this bacterium, which has been classified by the WHO as a class 1 carcinogen in 1994, is usually acquired orally early in life and it is estimated that more than 50% of the world's population are affected by it (Marshall & Warren 1984; Danesh 1999). Helicobacter pylori is the only known bacterium that is able to set up a persistent monoculture in the normal stomach and leads to a 2.1–16.7‐fold increased risk of gastric cancer (GC) (Nomura et al. 1991; Parsonnet et al. 1991). This colonization of the gastric epithelium induces an inflammatory reaction, which might persist throughout the patient's lifetime (Graham & Go 1993). A link between inflammation and cancer has been established for many tissues, but the underlying mechanisms still remain unclear. The inflammatory reaction leads to the generation of reactive oxygen species and an increased level of nitric oxide synthase (Moss 1998; Li et al. 2001; Sepulveda 2001). This results in mutations by deamination of DNA (Correa 1995; Davies et al. 1994; Kuipers & Meuwissen 1996), and the activation of DNA methyltransferases leads to gene silencing by methylation of promoters containing CpG islands (Hmadcha et al. 1999; Tamura 2004). Chronic inflammation also promotes apoptosis of normal cells, which increases proliferation as a compensatory response of the remaining tissue. Indeed, oxidative DNA stress, increased cell proliferation and a decrease of the apoptotic index with a resulting increased proliferation of the epithelial cells have been shown in H. pylori infections (Farinati et al. 1998; Moss 1998). The development of gastric carcinoma is certainly triggered by H. pylori, but it is promoted by a variety of other factors such as host genetic susceptibility, immune response and environmental factors as well (Graham & Go 1993; Ming 1998). Recently, the influence of the host immune response to the infection on the risk of GC has been investigated. In these studies, it was shown that T cells play a crucial role in disease initiation and progression and possibly influence the risk for further progression to GC (Roth et al. 1999; Eaton et al. 2001).
The metaplasia–dysplasia sequence
The stage of chronic inflammation is the first step in both the intestinal type and the diffuse type of GC, but the subsequent pathways are different. The diffuse type develops in the stomach without passing through intermediate steps. It tends to be more aggressive and might be primarily genetic in origin like the familial diffuse form of GC, which is linked to an E‐cadherin gene mutation (Guilford et al. 1998). The intestinal type of GC arises through a cascade of molecular and morphological changes. According to Correa, there is a sequence of chronic gastritis associated with multifocal atrophic gastritis, which frequently advances to intestinal metaplasia, then occasionally to dysplasia, and rarely to carcinoma (Correa 1995). The chronic inflammation results in gastric atrophy, which represents a loss of specialized glands and associated lower acidity of gastric juice. At this point it is possible for other bacteria to colonize the stomach and additionally trigger carcinogenesis by producing further carcinogenic substances (Leach et al. 1987; McColl et al. 1997). As the enzyme ornithine decarboxylase (ODC) is significantly increased in areas of gastric atrophy or intestinal metaplasia (IM) it might be considered as a marker of pre‐malignancy in the stomach (Patchett et al. 1995). The next step is IM, a non‐neoplastic change in cell phenotype. Gastric mucosa is replaced by an epithelium that histologically resembles the intestinal mucosa. The IM can be subdivided in the complete type I and the incomplete types II and III (Jass & Filipe 1980; Reis et al. 1999). They differ histologically by the composition pattern of specialized cells and histochemically by the different expression patterns of mucin core (MUC) protein (Ho et al. 1995; Reis et al. 1999). The risk of GC is related to the type of IM and seems to be increased in type III IM (Filipe et al. 1994). A wide range of molecular alterations, affecting transcription factors like CDX1 and CDX2, telomerases, microsatellite instability, mutations of p53 protein, overexpression of COX‐2, cyclin D2, and decreased expression of p27 are shown to be involved in the progress from IM to GC (reviewed in Nardone et al. 2004). Gastric dysplasia is the next step in this sequence. Usually, this develops in the metaplastic tissue (Correa 1995). Mutations in genes include p53, loss of heterozygosity (LOH) of the adenomatous polyposis coli (APC) gene, overexpression of the antiapoptotic gene bcl‐2 and a mixture of polyploidy and aneuploidy (reviewed in Nardone et al. 2004). The dysplastic cells resemble malignant cells found in the final carcinoma and might in fact be already malignant. During the progression of normal tissue through the metaplasia–dysplasia sequence in the development of gastric cancer various gene mutations occur and accumulate. However, gastric cancer does not follow a pattern similar to colorectal carcinoma progression and it is not clear if and which mutation plays an initiating role in malignant transformation (Kim et al. 1991; Stemmermann et al. 1994; Maesawa et al. 1995).
The colorectum
The colorectum is the third most common site of cancer development, and is responsible for 10% of cancer deaths (American Cancer Society 2004). Numerous steps are involved in the progression of normal tissue through dysplasia to malignancy and it is estimated that a typical colorectal tumour contains at least 11 000 genomic alterations (Stoler et al. 1999).
‘Gatekeeper’ and ‘caretaker’ gene mutations
The observation that the accumulation of mutations in tumour suppressor and proto‐oncogenes seemed to parallel the clinical progression of bowel tumours led Vogelstein et al. (1988) to propose a stepwise progression of colorectal tumorigenesis. The molecular pathogenesis of FAP has shed much light on the initial mutations required in this step‐like progression. FAP results in the formation of multiple bowel adenomas in the second and third decades of life. Colonic cancer is inevitable in these patients who therefore require prophylactic colectomy. The heritable nature of FAP was first recognized at the end of the 19th century; however, it was not until 1986 that an interstitial deletion of chromosome 5q was observed in an FAP patient (Herrera et al. 1986). This prompted linkage analysis studies which co‐demonstrated tight linkage of the condition to markers on chromosome 5q21 (Bodmer et al. 1987; Leppert et al. 1987). The gene responsible was APC (Groden et al. 1991; Kinzler et al. 1991) that encodes a large (approximately 2800 amino acids) multifunctional cytoplasmic protein (Joslyn et al. 1993). This important protein is an essential component of the Wnt signalling ‘destruction complex’ involved in the binding and down‐regulation of β‐catenin. A functioning APC protein is therefore vital in maintaining low levels of cytosolic β‐catenin in the absence of a Wnt signal, preventing excessive cell proliferation. Additionally, APC has important roles in control of intracellular adhesion as an integral part of adherens junctions, regulation of apoptosis, cell‐cycle progression and chromosomal stability (reviewed in Goss & Groden 2000; Sieber et al. 2000; Kaplan et al. 2001; van Es et al. 2001). Subsequent work revealed that mutations in APC are also found in 63% of sporadic adenomas (Powell et al. 1992) and up to 80% of sporadic colorectal cancers (Miyoshi et al. 1992). This led to the proposal that APC acts as a ‘gatekeeper’ gene – a gene involved in the control of normal epithelial cell proliferation required for cellular homeostasis. Mutation of a gatekeeper gene results in an imbalance of cell division over death, thus FAP is a disease of accelerated tumour initiation (Kinzler & Vogelstein 1996). Other hereditary bowel cancer syndromes have been used in the identification of alternative pathogenetic mechanisms. Hereditary non‐polyposis coli (HNPCC) is a condition that predisposes to cancers of the colon, endometrium and several other extra‐colonic sites, notably without prior polyp formation (reviewed in Lynch et al. 1996). The use of microsatellite markers to try to identify allelic losses in this syndrome led to the discovery of multiple new di‐ and trinucleotide repeats throughout the genome. This was termed microsatellite instability and is explained by defects in DNA mismatch repair (MMR) genes, which normally recognize and repair single base and larger strand slippage mismatches in DNA replication. The mutated MMR genes in the majority of HNPCC are hMSH2, hMLH1 and hPMS2 (reviewed in Kinzler & Vogelstein 1996). This led to the proposal of MMR genes as ‘caretaker’ genes and that HNPCC is a disease of tumour progression, with impaired DNA repair accelerating the process. The observation that the median age of cancer development in both HNPCC and FAP is 42 demonstrates the importance of both ‘gatekeeper’ and ‘caretaker’ gene functions (Kinzler & Vogelstein 1996). Gatekeeper gene mutations, such as APC, are present in very early adenomas (Powell et al. 1992) and are sufficient to promote small adenoma growth in the absence of microsatellite instability, K‐ras or β‐catenin mutation or allelic loss of 1p (Lamlum et al. 2000). Thus, APC inactivation provides a stem cell with a selective growth advantage by allowing unregulated activation of Wnt signalling. Mutations in β‐catenin, preventing its breakdown, can also promote adenoma initiation; however, small adenomas with β‐catenin mutations alone do not progress to larger adenomas or carcinomas as frequently as adenomas with APC mutations (Samowitz et al. 1999). Therefore, although APC's role in the regulation of Wnt signalling is the most important in the prevention of tumour initiation, its involvement in apoptosis and chromosomal stability also have an effect on the progression of the adenoma growth (reviewed in Fodde et al. 2001).
APC mutations in FAP
Knudson's two‐hit hypothesis postulates that inactivation of a tumour suppressor gene occurs only after two independent mutation events, and certainly bi‐allelic mutation of APC can be detected in early intestinal tumours (Powell et al. 1992). In FAP, initial germline mutations are frequently small‐scale truncating mutations. Second hit, somatic mutations may be point mutations, which commonly cluster in a region between codon 1286 and 1513 termed the mutation cluster region (MCR) (Miyoshi et al. 1992), or large‐scale genetic changes that are collectively termed allelic loss or loss of heterozygosity (Tomlinson et al. 2001). Careful mutation analysis of multiple polyps from FAP patients showed that the position and type of the somatic hit in the APC gene depended on the position of the germline mutation. Lamlum et al. (1999) studied 210 early polyps from 35 different patients from 26 FAP families. They found loss of heterozygosity as the cause of the somatic mutation in 20% of adenomas and this was strongly associated with cases whose germline mutations were around codon 1300. Patients with germline mutations away from this region tended to acquire their second hits by point mutation within the MCR (Lamlum et al. 1999). A strong genotype–phenotype correlation was noted with more severe polyposis associated with patients whose germline mutations were between codons 450 and 1600, especially those with mutations around codon 1300, who had thousands of colonic polyps. Patients with mutations at the 3′ or 5′ end of the gene had less severe diseases with tens of polyps and later age of onset (Nagase et al. 1992; Fodde & Khan 1995). It appears that different APC mutations provide a cell with different selective advantages, and the somatic mutations are non‐random events, that are selected for the growth advantage they confer to the tumour cell. The greatest growth advantage is conferred by germline mutation around codon 1300, which then obtains a second hit by allelic loss, an event that occurs at a high spontaneous frequency. Patients with germline mutations that provide less growth advantage to the cell need to acquire second or even third hits, often in the MCR to compensate for the weaker selective advantage provided by the first mutation (Lamlum et al. 1999). Based on this work, Albuquerque et al. (2002) and Crabtree et al. (2003) were able to define the ‘just right’ and ‘loose fit’ models to explain these findings. These models suggest that for a cell to acquire a selective advantage the function of APC must be impaired to a specific degree to allow sufficient nuclear accumulation of β‐catenin promoting excess proliferation, but not so much as to cause cell apoptosis. The regulation of β‐catenin by APC is achieved by three 15‐amino acid and seven 20‐amino acid repeats that, respectively, bind and down‐regulate β‐catenin. The number of amino acid repeats left in an abnormal APC protein depends upon the position of the gene mutation, with an early mutation leaving fewer repeats. As an optimal ‘just‐right’ level of β‐catenin activity is required for tumour cell growth advantage, the somatic mutation is selected for by the number of amino acid repeats it will code for, usually resulting in one or two residual β‐catenin binding amino acid repeats (see Fig. 1) (Albuquerque et al. 2002). The ‘loose fit’ model adapts this slightly to show that some variation in APC is tolerable (Crabtree et al. 2003).
Figure 1.
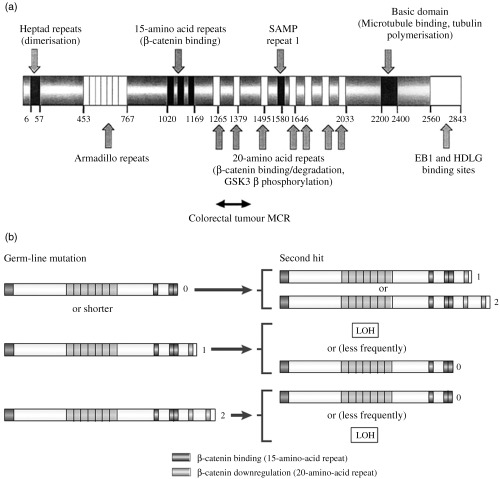
(a) The adenomatous polyposis protein showing major functional domains by amino acid position. Reprinted by permission from Oncogene, Reference (Crabtree et al. 2003), (b) Interdependence of germline and somatic APC mutations according to the ‘just right’ hypothesis. The germline and somatic mutations are represented by the truncated protein they produce. Numbers to the right of the protein are the remaining number of 20 amino acid repeats after protein truncation. Germline mutations that result in a protein lacking all amino acid repeats tend to acquire somatic mutations that result in a protein that retains one or two repeats. When the germline mutation results in a protein containing one amino acid repeat, the second hit tends to be allelic loss, or more rarely point mutations resulting in short proteins. When the germline hit leaves a protein with two residual repeats then somatic mutations causing allelic loss or short proteins are selected for. In all cases the combination of germline and somatic mutations results in a truncated protein that retains one or two amino acid repeats. LOH, loss of heterozygosity. Reprinted with permission from Nature Reviews Cancer (Fodde et al. 2001).
Crypts as clonal populations
In the colon, the importance of the APC protein in a number of different cell regulatory functions means that mutations in the APC gene alone are sufficient to provide a cell with a selective growth advantage (Lamlum et al. 2000). To determine the next steps in the pathway from stem cell genetic mutations to the smallest recognizable adenomas, it is important to understand the organization and cell dynamics of the basic functional unit of the gut – the crypt. The clonality of the gastrointestinal crypt has been extensively studied in both the mouse and the human.
Mouse
In the mouse, clonality experiments frequently involve the formation of aggregation chimeras and/or the induction of somatic mutations in crypt stem cells. Studying the contribution to the crypt structure of the different cell populations in the chimera, or the cells bearing a new induced somatic mutation helps determine the cell lineages of the crypt occupants. The chimeric B6/SWR mouse is an aggregation chimera wherein the two populations of cells can be simply distinguished. The lectin Dolichos biflorus agglutinin (DBA) binds to sites on the B6‐derived but not the SWR‐derived cells in B6/SWR chimeras. Staining for DBA binding thus segregates cells from the different strains. In neonatal B6/SWR chimeras, the intestinal crypts exhibit mixed staining patterns for the first 2 weeks of life (Schmidt et al. 1988). After this time and in the adult mouse, the crypts are either positive or negative for DBA staining, with no mixed crypts in the many thousands studied (Ponder et al. 1985). This suggests that during development, multiple stem cells are present in the crypt, but as neonatal development progresses the crypts undergo a process of monoclonal conversion, resulting in a clonal population of cells where all cell lineages are derived from a single parental strain. Chemical mutagenesis has been used in other chimeric mice to demonstrate the dynamics of adult crypt populations. C57BL/6 J/SWR mice show heterozygous expression at the DBA binding site so that in the non‐mutated state crypt cells stain positive for bound agglutinin. Spontaneous or ethyl nitrosurea (ENU) induced mutation at the Dlb‐1 locus on chromosome 11 abolishes the DBA binding site, and mutated cells are then distinguished by the loss of ability to bind DBA. After ENU treatment, crypts emerge that are partially and then completely negative for DBA staining, suggesting mutation of a stem cell, which forms a clone that expands stochastically. Eventually, the mutated progeny populate the entire crypt (Winton et al. 1988). Bjerknes et al. (1997) used a knock‐in model to study cell dynamics and fate. SWR mice, normally negative for DBA binding, can be induced to bind the agglutinin by ENU treatment. By observing the morphology, location and longevity of the mutant clones in the mouse small intestine, they suggested that stem cell division gives rise to committed epithelial progenitor cells – the columnar cell progenitor, the mucus cell progenitor and to a lesser extent, the mixed progenitor. These cells undergo further transit‐amplifying divisions but are committed to differentiation down their cell line (Bjerknes & Cheng 1999). In the stomach, studies on the clonal architecture of mouse gastric glands using chimeric and transgenic mice have revealed that in the first weeks of life, all gastric glands show a mixed pattern but over the next weeks there occurred a process of monoclonal conversion in which 90% of gastric glands became monoclonal. The time to conversion is approximately 6 weeks in mouse stomach (Thompson et al. 1990; Tatematsu et al. 1994; Nomura et al. 1998).
Human
Studies in the human rely on natural mutations and polymorphisms. One such mutation is in the gene coding for the enzyme O‐acetyl transferase (OAT). This enzyme is responsible for the O‐acetylation of sialic acid in goblet cell mucus. Non‐O‐acetylated mucus will stain positively with mild periodic acid–Schiff reagent (mPAS). Approximately 9% of the Caucasian population have a homozygous mutation (OAT‐/–), resulting in mPAS staining of goblet cells carrying the mutation, allowing their identification. Forty‐two percent of the population are heterozygotes (OAT ±) and will not stain with mPAS unless further mutation causes loss of the remaining allele. Second hits in the heterozygote increase with age, resulting in randomly located positive mPAS‐stained crypts with all intracryptal goblet cells affected from base to lumen (Fuller et al. 1990). The frequency of positive crypts was increased after irradiation and initially crypts showed a partial crypt staining pattern, which then became uniform. The time taken for partially transformed crypts to become clonal after radiation damage was about a year and was called the ‘clonal stabilization time’ (Campbell et al. 1996). The best evidence for the clonality of human colonic crypts came from the fortuitous discovery of an XO/XY chimeric patient who required a colectomy for FAP. Non‐isotopic in situ hybridization for the Y chromosome revealed that all, except 4 of 12 614 colonic crypts studied, were composed of Y chromosome‐positive or ‐negative cells (see Fig. 2a,b). The four mixed clonality crypts were explained by non‐disjunction with loss of the Y chromosome in a stem cell.
Figure 2.
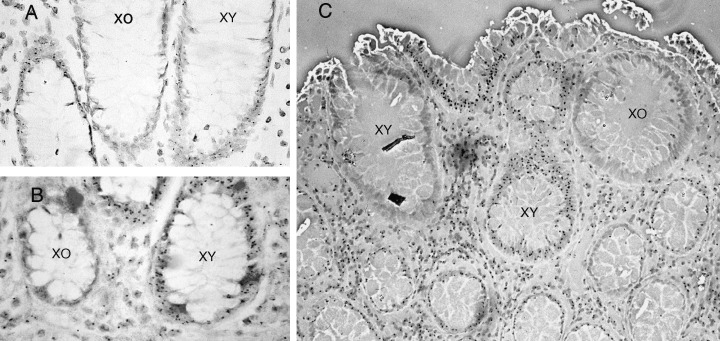
Monoclonal origins of human colonic crypts. Normal colonic mucosa from a rare XO/XY patient with FAP (a, b). Crypts stained by in situ hybridization with a Y chromosome‐specific probe, either stain positively or negatively for the Y chromosome (small black dot within XY cells). (c) Polyclonal adenoma in the same chimeric patient, with a mixture of XO and XY crypts. Reprinted with permission from Preston et al (2003).
More recently, Taylor et al. (2003) have shown that the colonic crypt cells accumulate sufficient mitochondrial DNA (mtDNA) mutations with age to cause a biochemical defect in the mtDNA coded subunits of cytochrome c oxidase (COX). This defect can also be stained for with enzyme histochemistry. Normal colonic tissue shows numerous completely COX‐deficient crypts, but also a few partially stained crypts. Serial sections of these partial crypts allowed them to reconstruct three‐dimensional images of the crypt revealing a ribbon of COX‐negative cells extending from the base of the crypt to the top. The ribbon of mutated, COX‐negative cells appear to be the progeny of one of the small number of stem cells in the niche, and that the partially negative crypts are likely to be intermediate steps in the expansion of the mutated clone with eventual formation of a completely clonal COX‐deficient crypt (Taylor et al. 2003). In the human gastric gland, the situation is complex. The antral gastric glands are clonal in derivation and in the body, the foveolae appear homotypic. The region that contains the putative gastric stem cell population in the neck of the gland is heterotypic on human androgen receptor analysis (HUMARA) and thus might be of polyclonal structure. At this time, clonal purification has not been shown in human gastric glands (Nomura et al. 1996).
Clonal expansion of mutated cells – niche succession and crypt fission
There are three possibilities that can result from a stem cell division (Loeffler et al. 1993): (1) the production of one stem cell and one daughter cell – asymmetric division; (2) symmetric division with self‐replication, where two stem cells are produced; (3) symmetric division with stem cell loss, where both daughter cells go on to differentiate. The majority of divisions are thought to be asymmetric and there is some evidence supporting the retention of the template DNA strand within the stem cell located in the niche – the so‐called immortal strand hypothesis (Cairns 1975). This allows any DNA replication errors to pass into the differentiating, short‐lived daughter cell affording a mechanism of stem cell genome protection (Potten et al. 2002). Park et al. used ethyl nitrosurea (ENU) to induce mutations in the X‐linked gene for glucose‐6‐phosphate dehydrogenase (G6PD) to demonstrate the expansion of a mutated clone within the crypt. G6PD gene mutation resulted in loss of staining in affected cells. After ENU treatment, they initially observed crypts that were only partially stained for G6PD, which eventually disappeared with the contemporaneous emergence of fully mutated crypts (monoclonal conversion or crypt purification). These eventually gave rise to patches of crypts that failed to stain with G6PD (Park et al. 1995). More recently, Yatabe et al. (2001) used CpG methylation patterns in three non‐expressed genes to study the dynamics of the stem cells within the niche, and proposed the niche succession model. They showed that the differences in methylation tag sequence between cells in adjacent crypts (inter‐crypt variation) were more pronounced than the tag variation seen between cells in the same crypt (intracrypt variation). They proposed that intracrypt variation was a consequence of multiple, yet related stem cells within each crypt. Stochastic extinction or amplification of one stem cell line by occasional symmetrical division results in a ‘bottleneck’ effect, wherein all cells in the crypt are descended from an original stem cell. Mathematical modelling suggested that this bottleneck occurs once every 8.2 years in the normal human colon (Yatabe et al. 2001). In normal‐appearing crypts from FAP patients, however, there was a greater intracrypt variation in methylation tags suggesting slower niche succession, probably as a consequence of enhanced stem cell survival. The longevity of APC + ‐stem cells in FAP patients increases the chances of receiving or selecting for a second hit in the gene (Kim et al. 2004a). Once APC protein function is impaired, a growth advantage is bestowed on the cell and clonal expansion would then occur much more quickly (see Fig. 3). Niche succession is a way by which a single stem cell line can ‘hitchhike’ its way to clonal dominance of a single crypt (Fig. 3) (Kim & Shibata 2002), but then how does a stem cell line expand into adjacent tissue? Clonality experiments in both mice and humans have shown clustering of mutated, phenotypically similar crypts together in patches (Park et al. 1995; Taylor et al. 2003). It is thought that a process called crypt fission, where crypts undergo basal bifurcation followed by longitudinal division, with the ultimate formation of two daughter crypts, is responsible for the clustering of apparently related crypts. This process is central in the massive increase in crypt number (in both the small and large intestine) in the post‐natal period (Maskens & Dujardin‐Loits 1981) and in the regenerative phase following radiation (Cairnie & Millen 1975). The crypt cycle – crypts born by crypt fission gradually increasing in size until they, themselves, divide by crypt fission – takes approximately 108 days in the mouse jejunum and 9–18 years in the human large intestine (Bjerknes 1986; Totafurno et al. 1987). Studies on the methylation patterns of adjacent crypts showed significant inter‐crypt variation, both in adjacent crypts and in those up to 15 cm apart. It was proposed that this was a consequence of the time taken for crypts to divide, allowing neighbouring crypts to develop different methylation patterns during the process (Kim & Shibata 2004b). It was originally suggested that a crypt would be prompted to go into fission once it had reached a threshold size; however, attention has now focused on the stem cell number being the important factor. The rate of crypt fission is increased in pathological conditions such as ulcerative colitis and Crohn's disease (Cheng et al. 1986), as well as in the flat mucosa of FAP (Bjerknes et al. 1997; Wasan et al. 1998) and sporadic and hyperplastic adenomas (Wong et al. 2002). In adenoma growth, fission is asymmetrical with atypical branching and budding (Wasan et al. 1998). Clusters of dysplastic crypts are termed microadenomas or dysplastic aberrant crypt foci (ACF), and are thought to be morphologically and genetically distinct lesions that are the precursors of adenomas and cancers (1998, 2001). Studies have shown that dysplastic ACFs are clonal populations (Siu et al. 1999), and expand by crypt fission (Fujimitsu et al. 1996); however, the expansion of a mutated clone from a single cell to form small adenomas is contentious, with two main theories – the top‐down and bottom‐up models.
Figure 3.
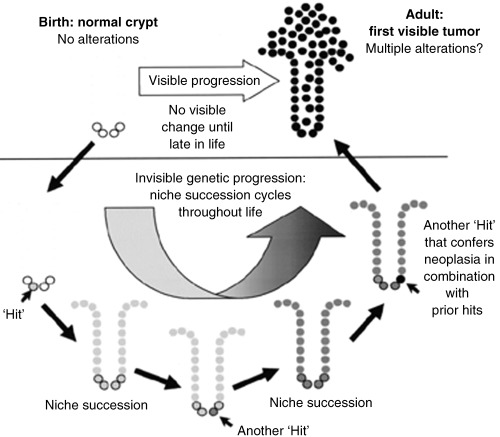
Niche succession sequence. Although most mutations will be lost by stochastic extinction of stem cell lines, random genetic mutations in a stem cell may be passively fixed as a consequence of niche succession. Rarely, combinations of mutations in the same cell can hitchhike their way to clonal dominance, through successive niche succession cycles (small arrows) and can then confer a tumour phenotype on the crypt. Reprinted by permission from Oncogene (Kim & Shibata 2002).
Top down or bottom up?
The top‐down model is based on the frequent observation of dysplastic cells solely at the luminal surface of the crypts (Maskens 1979; Lightdale et al. 1982; Shih et al. 2001) (Fig. 4), along with apparent retrograde migration of adenomatous cells from the surface to the base of the crypt (Lightdale et al. 1982). Shih et al. (2001) examined the morphology and molecular characteristics of small (1–3 mm), well‐orientated specimens from sporadic adenomas. Using digital SNP analysis of four single‐nucleotide polymorphisms (SNP) within the APC gene, they assessed for LOH of APC in cells in the upper portion of the crypts, most of which had truncating APC mutations on nucleotide sequence analysis. These were not seen in the histologically normal crypt bases. Only these upper crypt cells showed prominent proliferative activity and nucleur localization of β‐catenin. These observations were not easily reconciled with the conventional view of the stem cell origin of cancer, and the authors proposed two possible explanations to explain their findings. First, they considered a relocation of the stem cell area to the intracryptal zone, and secondly, they suggested that a mutated stem cell migrates from the base of the crypt to the luminal surface before expanding laterally and downwards. Lamprecht & Lipkin (2002) adjusted the latter model slightly to suggest that APC mutations occur within a transit‐amplifying cell, preventing it from terminally differentiating and altering the cell's migration dynamics, allowing it to remain in the mucosa as an incipient aberrant clone (Lamprecht & Lipkin 2002). The bottom‐up model involves the recognition of the earliest lesion in tumour development, the monocryptal adenoma, where the dysplastic cells occupy an entire single crypt. These lesions are common in FAP (Nakamura & Kino 1984), and although rare in non‐FAP cases, have been described (Woda et al. 1977). Clonality studies in the XO/XY FAP patient have shown that monocryptal adenomas are clonal populations (Novelli et al. 1996). Analysis of tiny (< 3 mm) adenomas in FAP patients showed increased proliferative activity and nucleur β‐catenin translocation in morphologically dysplastic cells from the crypt base to the luminal surface. Additionally there was a sharp cut‐off between the dysplastic surface epithelium with nucleur β‐catenin activity, and the normal mucosa in a neighbouring unaffected crypt. The observation of an increased, asymmetrical crypt fission index in adenomatous tissue led the researchers to propose the bottom up model – an abnormal stem cell clone with a growth advantage expands from the stem cell niche at the crypt base, to fill an entire crypt. Thereafter, initial spread is by crypt fission to form aberrant crypt foci, with top‐down spread undoubtably occurring in slightly larger lesions (see Fig. 4) (Preston et al. 2003).
Figure 4.
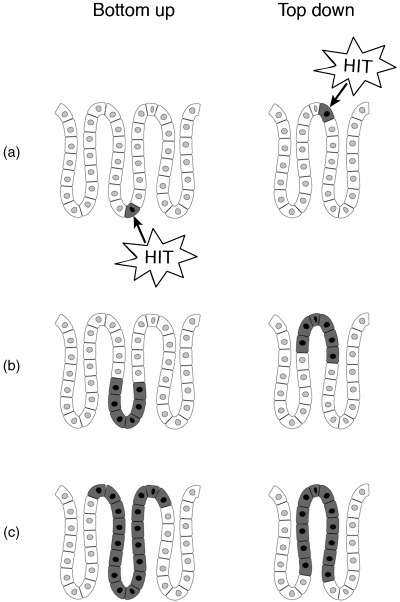
Top‐down, bottom‐up growth of colorectal adenomas. Bottom up: the stem cell, located in the crypt base undergoes APC mutation (a). The mutated cell proliferates (b) and spreads to the top of a crypt to form a monocryptal adenoma (c). Initial further expansion is by crypt fission. Based on (Preston et al. 2003). Top down: the initial transformation event occurs in a cell in the intracryptal zone (a) and then spreads laterally and downwards (b) eventually filling the whole crypt (c). Adapted from (Shih et al. 2001).
The clonality of tumours
It is widely accepted that tumours arise from mutations in a single cell, possibly a stem cell, which then clonally expands, with resultant common ancestry of all neoplastic cells. Evidence suggests that in the very early stages of tumour progression, from monocryptal adenoma to aberrant crypt foci, the dysplastic cells do comprise a clonal population (Novelli et al. 1996; Siu et al. 1999). However, the clonality of larger lesions and tumours is more widely debated. If indeed stem cells are the original targets for the mutation(s) required to initiate a neoplasm, then whether such a cell acts alone or in cooperation with other mutated stem cells becomes important. In this respect, Fearon et al. (1987), studying restriction fragment length polymorphisms of inactivation of the X chromosome‐linked phosphoglycerate kinase (PGK) gene in colorectal adenomas and carcinomas from 50 female patients, found that in adjacent normal epithelia, X chromosome inactivation was random and therefore polyclonal, but in the adenomas and carcinomas, a monoclonal pattern of X chromosome inactivation was seen (Fearon et al. 1987). Conversely, in female patients with Gardner's syndrome, a pre‐cancerous bowel condition in which patients develop multiple adenomas of the gastrointestinal mucosa, the adenomas have a multiclonal origin, expressing both forms of G6PD (Hsu et al. 1983). A CAG trinucleotide repeat polymorphism adjacent to methylation sites in the X chromosome‐linked human androgen receptor gene (HUMARA) is present in approximately 90% of female subjects, and provides a method of looking at tumour clonality by PCR amplification of a 600‐bp DNA fragment encompassing the polymorphic BstXI site and methylation‐sensitive HpaII sites. In 15 female patients with gastrointestinal tumours, the majority revealed an unequivocal monoclonal origin with random pattern of X‐inactivation in the normal surrounding mucosa, consistent with Southern blot analysis for phosphoglycerate kinase (PGK) and the M27β probe (DXS255) which detects X chromosome tandem repeat polymorphisms in about 90% of female subjects (Kopp et al. 1997). However, it is important to note that patch size was overlooked in these studies, and it is possible that tumours were covertly polyclonal but appeared monotypic as they arose within a large X‐linked patch. In this respect, Novelli et al. studied Sardinian females heterozygous for the glucose‐6‐phosphatase (G6PD) Mediterranean mutation (563 C→T) and heat‐deactivated the defective gene product by preheating at 53 °C for 5 min followed by enzyme histochemistry to demonstrate glucose‐6‐phosphate activity. Crypts were arranged in hexagonal arrays in large patches, with irregular patch borders, containing up to 450 individual crypts (Novelli et al. 2003). These direct observations suggest that, because of the large patch size in the colon, X‐inactivation studies are heavily biased toward showing that tumours are monoclonal. To exclude the possibility that all adenomas are polyclonal in origin, every crypt in at least 43 adenomas would need to be shown to be monophenotypic (95% confidence interval). To exclude the possibility that 10% of adenomas are polyclonal, 430 adenomas would need to be examined. Thus, patch size confounds measurements of tumour clonality in the colon, and in many other tissues (Novelli et al. 2003).
However, direct observation, rather than the indirect methods described above tell a different story. Novelli and colleagues (Novelli et al. 1996) studied a highly unusual individual who not only had familial polyposis coli (FAP) but also but was also a sex chromosome mixoploid chimera, presumably as a consequence of a dicentric Y chromosome. Thus, his tissues were a mosaic with the majority, some 80% of cells, being XY and the remaining 20%, XO. Therefore, the detection of the Y chromosome in the excised colonic tissues provides an excellent binary marker for lineage analysis. Of the 263 adenomas analysed, 246 were wholly XY, 4 were entirely XO, whereas 13 were of mixed XO/XY genotype. Simplistically, therefore, and considering only the tumours containing the XO lineage, 13/(13 + 4) or 76% of all adenomas would appear to be polyclonal. Subsequent studies in mice heterozygous for the Min germline mutation and chimeric for the lineage reporter gene ROSA26 used the same ratio, now called the Novelli ratio (Newton 2005), rather startlingly produced the same value – 76% were polyclonal. However, there are several problems with the Novelli ratio, not the least of which is that the proportion of XO/XY and XO tumours that are polyclonal may not reflect the whole population of tumours leading to an overestimate (Newton 2005), and this appears confounded when the minority component, here the XO and XY tumours, is small, as in this individual. Making reasonable assumptions about the mechanism of polyclonality, the proportion of polyclonal adenomas becomes closer to 50–60% (Newton 2005). Three possible explanations were considered for tumour polyclonality in the Novelli paper. First, true polyclonality with adenomas inducing dysplastic growth in adjacent crypts. Second, the XO/XY adenomas may have been clonal XY lesions that had focally lost their Y chromosome. Third, because of the large numbers of polyps in FAP, mixed tumours may have arisen through random collision of two different lesions. Novelli et al. (1996) considered the first explanation the most plausible. Chromosomal loss after tumour initiation is usually a late phenomenon in colorectal adenoma formation, and no Y chromosome loss was seen in any larger adenomas from control FAP patients. Additionally, chromosomal loss did not explain the findings in the chimeric mouse analysis of Merritt et al. (1997), so this explanation for tumour polyclonality seems unlikely. However, there remains the problem of random collision of adjacent tumours that looms large in a patient with many hundreds of adenomas or the Min, mouse, where tumours are also very numerous. Although Novelli et al. (1996) discounted this possibility through statistical inference, an important advance has come from Thliveris et al. (2005), where tumour multiplicity in the chimeric mouse model described above was reduced by introducing homozygosity for the tumour resistance allele of the Mom1 locus (of genotype B6 Apc Min/+ Mom1 R/R ↔ B6 Ap Min/+ ROSA26/+Mom R/R). The percentage of mixed tumours ranged from 8 to 63%, with a mean of 22%. This is much higher than would be expected if heterotypic tumours are formed by random collision. Consequently, the mechanism of this polyclonality becomes pivotal: we have seen above that crypts are clonal populations, so how could tumours arise which require the interaction of at least two clones? Such considerations are of course anathema to some, reared on the concept of the monoclonality of tumours as a modern shibboleth. It could be of course that the colon is heterogeneous in respect of its tumour susceptibility, but in the study, as mixed tumours were more frequent in the proximal small bowel, where tumour multiplicity is lower than the distal small intestine. Thliveris et al. (2005) analysed the chimeric architecture of the intestine in relationship to the appearance of the mixed tumours, and considered the spatial extent over which clonal interactions could occur. They concluded that very short‐range interactions were important, between clones as close as one to two crypts apart. But is such interaction required for the formation of all adenomas? Their analysis was consistent with a proportion of polyclonal adenomas reaching as high as 100%, although higher resolution in terms of the binary marker is required for firm conclusions, and what might be the mechanism of such clonal interactions? Possibilities include microheterogeneity of tumour susceptibility where local stroma promotes loss of heterozygosity in adjacent crypts, or induction of elevated proliferative rates in the stem cells of adjacent crypts by a mutated clone, increasing the chance of further mutation in an adjacent stem cell (Thliveris et al. 2005). These considerations introduce the concept of stem cell interactions, as yet an unexplored territory in mucosal regeneration and carcinogenesis in the gut.
Conclusion and future work
Recently, cancer biology investigators mostly working in the haematological malignancy field have focused on the similarities between normal stem cells and malignant cells and have developed the term ‘cancer stem cell’ to describe the ability of certain human cancer cells to regenerate tumour clones in immunocompromised animals, an ability not possessed by the vast majority of the abnormal cells within a neoplasm (reviewed in Reya et al. 2001). Kirkland showed the multilineage differentiation potential of the human rectal adenocarcinoma cell line HRA19a, after injection of the progeny of a single cell into an immunocompromised mouse, caused neoplastic growth containing all three epithelial cell lineages (Kirkland 1988). This work has profound implications for the biology of metastasis and for chemotherapy of tumours, as direct targeting of cancer stem cells would prevent recurrence of the tumour rather than simple destruction of the non‐tumorigenic daughter cells. As yet the identification of a cancer stem cell in gastrointestinal malignancy has not been achieved, however, a recent experiment by Houghton et al. (2004) suggested that bone marrow could be a source of cancer stem cells. Mice with a chronic Helicobacter felis infection underwent a bone marrow transplant. Donor‐derived cells were found to engraft within the stomach glands and subsequently progressed through a metaplasia–dysplasia sequence to intra‐epithelial cancer (Houghton et al. 2004). Bone marrow‐derived cells have also been found contributing to chronic inflammation in the mouse colon (Brittan et al. 2005), and tumour stroma in the pancreas (Direkze et al. 2004). Further studies on the contribution of bone marrow‐derived cells to neoplasia in other tissues are essential.
The adenoma–carcinoma sequence is established as a road map of tumour progression and has altered clinical practice. Intestinal polyps are now routinely removed during endoscopic procedures and surveillance programs established to follow up on these patients in the long term. The molecular changes that occur in a single cell at the very beginning of this process have been elucidated over the last 20 years of careful experimentation and future further discovery of important tumour suppressor and oncogenes involved in carcinogenesis is likely. The events that occur during the growth of the intestinal adenoma remain unresolved but future work in this field may help to clarify the process, and would have profound implication for the understanding of tumour biology in this field.
REFERENCES
- Albuquerque C, Breukel C, van Der Luijt R, Fidalgo P, Lage P, Slors FJ, Leitao CN, Fodde R, Smits R (2002) The ‘just‐right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta‐catenin signaling cascade. Hum. Mol. Genet. 11, 1549. [DOI] [PubMed] [Google Scholar]
- American Cancer Society Statistics (2004) Cancer facts and figures 2004. http://www.cancer.org. (Last accessed: 12 July 2005).
- Bach SP, Renehan AG, Potten CS (2000) Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis 21, 469. [DOI] [PubMed] [Google Scholar]
- Bjerknes M (1986) A test of the stochastic theory of stem cell differentiation. Biophys. J. 49, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H (1999) Clonal analysis of mouse intestinal epithelial progenitors. Gastroenterology 116, 7. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H, Hay K, Gallinger S (1997) APC mutation and the crypt cycle in murine and human intestine. Am. J. Pathol. 150, 833. [PMC free article] [PubMed] [Google Scholar]
- Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P, Sheer D, Solomon E, Spurr NK. (1987) Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328, 614. [DOI] [PubMed] [Google Scholar]
- Brittan M, Braun KM, Reynolds LE, Con Ti FJ, Reynolds AR, Poulsom R, Alison MR, Wright NA, Hodivala‐Dilke KM (2005) Bone marrow cells engraft within the epidermis and proliferate in vivo with no evidence of cell fusion. J. Pathol. 205, 1. [DOI] [PubMed] [Google Scholar]
- Brittan M, Wright NA (2002) Gastrointestinal stem cells. J. Pathol. 197, 492. [DOI] [PubMed] [Google Scholar]
- Cairnie AB, Millen BH (1975) Fission of crypts in the small intestine of the irradiated mouse. Cell Tissue Kinet. 8, 189. [DOI] [PubMed] [Google Scholar]
- Cairns J (1975) Mutation selection and the natural history of cancer. Nature 255, 197. [DOI] [PubMed] [Google Scholar]
- Campbell F, Williams GT, Appleton MA, Dixon MF, Harris M, Williams ED (1996) Post‐irradiation somatic mutation and clonal stabilisation time in the human colon. Gut 39, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Bjerknes M, Amar J, Gardiner G (1986) Crypt production in normal and diseased human colonic epithelium. Anat. Rec. 216, 44. [DOI] [PubMed] [Google Scholar]
- Correa P (1995) Helicobacter pylori and gastric carcinogenesis. Am. J. Surg. Pathol. 19, S37. [PubMed] [Google Scholar]
- Crabtree M, Sieber OM, Lipton L, Hodgson SV, Lamlum H, Thomas HJ, Neale K, Phillips RK, Heinimann K, Tomlinson IP (2003) Refining the relation between ‘first hits’ and ‘second hits’ at the APC locus: the ‘loose fit’ model and evidence for differences in somatic mutation spectra among patients. Oncogene 22, 4257. [DOI] [PubMed] [Google Scholar]
- Danesh J (1999) Helicobacter pylori infection and gastric cancer: systematic review of the epidemiological studies. Aliment. Pharmacol. Ther. 13, 851. [DOI] [PubMed] [Google Scholar]
- Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, Blake DR, Rampton DS (1994) Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo . Gut 35, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Direkze NC, Hodivala‐Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA (2004) Bone marrow contribution to tumor‐associated myofibroblasts and fibroblasts. Cancer Res. 64, 8492. [DOI] [PubMed] [Google Scholar]
- Eaton KA, Mefford M, Thevenot T (2001) The role of T‐cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J. Immunol. 166, 7456. [DOI] [PubMed] [Google Scholar]
- van Es JH, Giles RH, Clevers HC (2001) The many faces of the tumor suppressor gene APC . Exp. Cell Res. 264, 126. [DOI] [PubMed] [Google Scholar]
- Farinati F, Cardin R, Degan P, Rugge M, Mario FD, Bonvicini P, Naccarato R (1998) Oxidative DNA damage accumulation in gastric carcinogenesis. Gut 42, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Hamilton SR, Vogelstein B (1987) Clonal analysis of human colorectal tumors. Science 238, 193. [DOI] [PubMed] [Google Scholar]
- Filipe MI, Munoz N, Matko I, Kato I, Pompe‐Kirn V, Jutersek A, Teuchmann S, Benz M, Prijon T (1994) Intestinal metaplasia types and the risk of gastric cancer: a cohort study in Slovenia. Int. J. Cancer 57, 324. [DOI] [PubMed] [Google Scholar]
- Fodde R, Khan PM (1995) Genotype–phenotype correlations at the adenomatous polyposis coli (APC) gene. Crit. Rev. Oncog. 6, 291. [DOI] [PubMed] [Google Scholar]
- Fodde R, Smits R, Clevers H (2001) APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 1, 55. [DOI] [PubMed] [Google Scholar]
- Franzin G, Zamboni G, Scarpa A (1995) Polyposis syndromes In: Whitehead R, ed. Gastrointestinal and Oesophageal Pathology, Vol. 1, p. 892 Edinburgh: Churchill Livingstone. [Google Scholar]
- Fujimitsu Y, Nakanishi H, Inada K, Yamachika T, Ichinose M, Fukami H, Tatematsu M (1996) Development of aberrant crypt foci involves a fission mechanism as revealed by isolation of aberrant crypts. Jpn. J. Cancer Res. 87, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller CE, Davies RP, Williams GT, Williams ED (1990) Crypt restricted heterogeneity of goblet cell mucus glycoprotein in histologically normal human colonic mucosa: a potential marker of somatic mutation. Br. J. Cancer 61, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KH, Groden J (2000) Biology of the adenomatous polyposis coli tumor suppressor. J. Clin. Oncol. 18, 1967. [DOI] [PubMed] [Google Scholar]
- Graham DY, Go MF (1993) Helicobacter pylori: current status. Gastroenterology 105, 279. [DOI] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M et al. (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589. [DOI] [PubMed] [Google Scholar]
- Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE (1998) E‐cadherin germline mutations in familial gastric cancer. Nature 392, 402. [DOI] [PubMed] [Google Scholar]
- Herrera L, Kakati S, Gibas L, Pietrzak E, Sandberg AA (1986) Gardner syndrome in a man with an interstitial deletion of 5q. Am. J. Med. Genet. 25, 473. [DOI] [PubMed] [Google Scholar]
- Hmadcha A, Bedoya FJ, Sobrino F, Pintado E (1999) Methylation‐dependent gene silencing induced by interleukin 1 beta via nitric oxide production. J. Exp. Med. 190, 1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SB, Shekels LL, Toribara NW, Kim YS, Lyftogt C, Cherwitz DL, Niehans GA (1995) Mucin gene expression in normal, preneoplastic, and neoplastic human gastric epithelium. Cancer Res. 55, 2681. [PubMed] [Google Scholar]
- Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC (2004) Gastric cancer originating from bone marrow‐derived cells. Science 306, 1568. [DOI] [PubMed] [Google Scholar]
- Hsu SH, Luk GD, Krush AJ, Hamilton SR, Hoover HH Jr (1983) Multiclonal origin of polyps in Gardner syndrome. Science 221, 951. [DOI] [PubMed] [Google Scholar]
- Jass JR, Filipe MI (1980) Sulphomucins and precancerous lesions of the human stomach. Histopathology 4, 271. [DOI] [PubMed] [Google Scholar]
- Joslyn G, Richardson DS, White R, Alber T (1993) Dimer formation by an N‐terminal coiled coil in the APC protein. Proc. Natl Acad. Sci. U S A 90, 11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS (2001) A role for the adenomatous polyposis coli protein in chromosome segregation. Nat. Cell Biol. 3, 429. [DOI] [PubMed] [Google Scholar]
- Kayahara T, Sawada M, Takaishi S, Fukui H, Seno H, Fukuzawa H, Suzuki K, Hiai H, Kageyama R, Okano H et al. (2003) Candidate markers for stem and early progenitor cells, Musashi‐1 and Hes1, are expressed in crypt base columnar cells of mouse small intestine. FEBS Lett. 535, 131. [DOI] [PubMed] [Google Scholar]
- Kim KM, Calabrese P, Tavare S, Shibata D (2004) Enhanced stem cell survival in familial adenomatous polyposis. Am. J. Pathol. 164, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Shibata D (2002) Methylation reveals a niche: stem cell succession in human colon crypts. Oncogene 21, 5441. [DOI] [PubMed] [Google Scholar]
- Kim KM, Shibata D (2004) Tracing ancestry with methylation patterns: most crypts appear distantly related in normal adult human colon. BMC Gastroenterol. 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Takahashi T, Chiba I, Park JG, Birrer MJ, Roh JK, De Lee H, Kim JP, Minna JD, Gazdar AF (1991) Occurrence of p53 gene abnormalities in gastric carcinoma tumors and cell lines. J. Natl. Cancer Inst. 83, 938. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D et al. (1991) Identification of FAP locus genes from chromosome 5q21. Science 253, 661. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87, 159. [DOI] [PubMed] [Google Scholar]
- Kirkland SC (1988) Clonal origin of columnar, mucous, and endocrine cell lineages in human colorectal epithelium. Cancer 61, 1359. [DOI] [PubMed] [Google Scholar]
- Kopp P, Jaggi R, Tobler A, Borisch B, Oestreicher M, Sabacan L, Jameson JL, Fey MF (1997) Clonal X‐inactivation analysis of human tumours using the human androgen receptor gene (HUMARA) polymorphism: a non‐radioactive and semiquantitative strategy applicable to fresh and archival tissue. Mol. Cell. Probes 11, 217. [DOI] [PubMed] [Google Scholar]
- Kuipers EJ, Meuwissen SG (1996) Helicobacter pylori and gastric carcinogenesis. Scand. J. Gastroenterol. Suppl. 218, 103. [PubMed] [Google Scholar]
- Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, Frayling I, Efstathiou J, Pack K, Payne S, Roylance R, Gorman P, Sheer D, Neale K, Phillips R, Talbot I, Bodmer W, Tomlinson I (1999) The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's ‘two‐hit’ hypothesis. Nat. Med. 5, 1071. [DOI] [PubMed] [Google Scholar]
- Lamlum H, Papadopoulou A, Ilyas M, Rowan A, Gillet C, Hanby A, Talbot I, Bodmer W, Tomlinson I (2000) APC mutations are sufficient for the growth of early colorectal adenomas. Proc. Natl Acad. Sci. U S A 97, 2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht SA, Lipkin M (2002) Migrating colonic crypt epithelial cells: primary targets for transformation. Carcinogenesis 23, 1777. [DOI] [PubMed] [Google Scholar]
- Leach SA, Thompson M, Hill M (1987) Bacterially catalysed N‐nitrosation reactions and their relative importance in the human stomach. Carcinogenesis 8, 1907. [DOI] [PubMed] [Google Scholar]
- Leppert M, Dobbs M, Scambler P, O'Connell P, Nakamura Y, Stauffer D, Woodward S, Burt R, Hughes J, Gardner E et al. (1987) The gene for familial polyposis coli maps to the long arm of chromosome 5. Science 238, 1411. [DOI] [PubMed] [Google Scholar]
- Li CQ, Pignatelli B, Ohshima H (2001) Increased oxidative and nitrative stress in human stomach associated with cagA + Helicobacter pylori infection and inflammation. Dig. Dis. Sci. 46, 836. [DOI] [PubMed] [Google Scholar]
- Lightdale C, Lipkin M, Deschner E (1982) In vivo measurements in familial polyposis: kinetics and location of proliferating cells in colonic adenomas. Cancer Res. 42, 4280. [PubMed] [Google Scholar]
- Loeffler M, Birke A, Winton D, Potten C (1993) Somatic mutation, monoclonality and stochastic models of stem cell organization in the intestinal crypt. J. Theor. Biol. 160, 471. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Smyrk T, Lynch JF (1996) Overview of natural history, pathology, molecular genetics and management of HNPCC (Lynch Syndrome). Int. J. Cancer 69, 38. [DOI] [PubMed] [Google Scholar]
- Maesawa C, Tamura G, Suzuki Y, Ogasawara S, Sakata K, Kashiwaba M, Satodate R (1995) The sequential accumulation of genetic alterations characteristic of the colorectal adenoma–carcinoma sequence does not occur between gastric adenoma and adenocarcinoma. J. Pathol. 176, 249. [DOI] [PubMed] [Google Scholar]
- Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311. [DOI] [PubMed] [Google Scholar]
- Marshman E, Booth C, Potten CS (2002) The intestinal epithelial stem cell. Bioessays 24, 91. [DOI] [PubMed] [Google Scholar]
- Maskens AP (1979) Histogenesis of adenomatous polyps in the human large intestine. Gastroenterology 77, 1245. [PubMed] [Google Scholar]
- Maskens AP, Dujardin‐Loits RM (1981) Kinetics of tissue proliferation in colorectal mucosa during post‐natal growth. Cell Tissue Kinet. 14, 467. [DOI] [PubMed] [Google Scholar]
- McColl KE, El‐Omar E, Gillen D, Banerjee S (1997) The role of Helicobacter pylori in the pathophysiology of duodenal ulcer disease and gastric cancer. Semin. Gastrointest. Dis. 8, 142. [PubMed] [Google Scholar]
- Merritt AJ, Gould KA, Dove WF (1997) Polyclonal structure of intestinal adenomas in ApcMin/+ mice with concomitant loss of Apc+ from all tumor lineages. Proc. Natl Acad. Sci. U S A 94, 13927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming SC (1998) Cellular and molecular pathology of gastric carcinoma and precursor lesions: a critical review. Gastric Cancer 1, 31. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y (1992) Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum. Mol. Genet. 1, 229. [DOI] [PubMed] [Google Scholar]
- Morson BC (1974) Evolution of cancer of the colon and rectum. Cancer 34, 845. [DOI] [PubMed] [Google Scholar]
- Moss SF (1998) Review article: cellular markers in the gastric precancerous process. Aliment. Pharmacol. Ther. 12, 91. [DOI] [PubMed] [Google Scholar]
- Nagase H, Miyoshi Y, Horii A, Aoki T, Ogawa M, Utsunomiya J, Baba S, Sasazuki T, Nakamura Y (1992) Correlation between the location of germ‐line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res. 52, 4055. [PubMed] [Google Scholar]
- Nakamura S, Kino I (1984) Morphogenesis of minute adenomas in familial polyposis coli. J. Natl. Cancer Inst. 73, 41. [PubMed] [Google Scholar]
- Nakamura Y, Sakakibara S, Miyata T, Ogawa M, Shimazaki T, Weiss S, Kageyama R, Okano H (2000) The bHLH gene hes1 as a repressor of the neuronal commitment of CNS stem cells. J. Neurosci. 20, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone G, Rocco A, Malfertheiner P (2004) Review article: Helicobacter pylori and molecular events in precancerous gastric lesions. Aliment. Pharmacol. Ther. 20, 261. [DOI] [PubMed] [Google Scholar]
- Newton M (2005) On estimating the polyclonal fraction in lineage‐marker studies of tumor origin. University of Wisconsin Department of Statistics Technical Report 1099 (University of Wisconsin‐Madison).
- Nomura S, Esumi H, Job C, Tan SS (1998) Lineage and clonal development of gastric glands. Dev. Biol. 204, 124. [DOI] [PubMed] [Google Scholar]
- Nomura S, Kaminishi M, Sugiyama K, Oohara T, Esumi H (1996) Clonal analysis of isolated single fundic and pyloric gland of stomach using X‐linked polymorphism. Biochem. Biophys. Res. Commun. 226, 385. [DOI] [PubMed] [Google Scholar]
- Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez‐Perez GI, Blaser MJ (1991) Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 325, 1132. [DOI] [PubMed] [Google Scholar]
- Novelli M, Cossu A, Oukrif D, Quaglia A, Lakhani S, Poulsom R, Sasieni P, Carta P, Con Tini M, Pasca A, Palmieri G, Bodmer W, Tanda F, Wright N (2003) X‐inactivation patch size in human female tissue confounds the assessment of tumor clonality. Proc. Natl Acad. Sci. U S A 100, 3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli MR, Williamson JA, Tomlinson IP, Elia G, Hodgson SV, Talbot IC, Bodmer WF, Wright NA (1996) Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 272, 1187. [DOI] [PubMed] [Google Scholar]
- Park HS, Goodlad RA, Wright NA (1995) Crypt fission in the small intestine and colon. A mechanism for the emergence of G6PD locus‐mutated crypts after treatment with mutagens. Am. J. Pathol. 147, 1416. [PMC free article] [PubMed] [Google Scholar]
- Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK (1991) Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127. [DOI] [PubMed] [Google Scholar]
- Patchett SE, Alstead EM, Butruk L, Przytulski K, Farthing MJ (1995) Ornithine decarboxylase as a marker for premalignancy in the stomach. Gut 37, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder BA, Schmidt GH, Wilkinson MM, Wood MJ, Monk M, Reid A (1985) Derivation of mouse intestinal crypts from single progenitor cells. Nature 313, 689. [DOI] [PubMed] [Google Scholar]
- Potten CS, Loeffler M (1990) Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110, 1001. [DOI] [PubMed] [Google Scholar]
- Potten CS, Owen G, Booth D (2002) Intestinal stem cells protect their genome by selective segregation of template DNA strands. J. Cell Sci. 115, 2381. [DOI] [PubMed] [Google Scholar]
- Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB (1999) Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am. J. Physiol. 277, C183. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer‐Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359, 235. [DOI] [PubMed] [Google Scholar]
- Preston SL, Direkze NC, Brittan M, Wright NA (2004) Stem cells in the gastrointestinal tract In: Lanza R, ed. Handbook of Stem Cells, Vol. 2, p. 521 Boston: Elsevier Scientific Press. [Google Scholar]
- Preston SL, Wong WM, Chan AO, Poulsom R, Jeffery R, Goodlad RA, Mandir N, Elia G, Novelli M, Bodmer WF, Tomlinson IP, Wright NA (2003) Bottom‐up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 63, 3819. [PubMed] [Google Scholar]
- Reis CA, David L, Correa P, Carneiro F, de Bolos C, Garcia E, Mandel U, Clausen H, Sobrinho‐Simoes M (1999) Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res. 59, 1003. [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414, 105. [DOI] [PubMed] [Google Scholar]
- Roth KA, Kapadia SB, Martin SM, Lorenz RG (1999) Cellular immune responses are essential for the development of Helicobacter felis‐associated gastric pathology. J. Immunol. 163, 1490. [PubMed] [Google Scholar]
- Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML (1999) Beta‐catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res. 59, 1442. [PubMed] [Google Scholar]
- Schmidt GH, Winton DJ, Ponder BA (1988) Development of the pattern of cell renewal in the crypt‐villus unit of chimaeric mouse small intestine. Development 103, 785. [DOI] [PubMed] [Google Scholar]
- Sepulveda AR (2001) Molecular testing of Helicobacter pylori‐associated chronic gastritis and premalignant gastric lesions: clinical implications. J. Clin. Gastroenterol. 32, 377. [DOI] [PubMed] [Google Scholar]
- Shih IM, Wang TL, Traverso G, Romans K, Hamilton SR, Ben‐Sasson S, Kinzler KW, Vogelstein B (2001) Top‐down morphogenesis of colorectal tumors. Proc. Natl Acad. Sci. U S A 98, 2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber OM, Tomlinson IP, Lamlum H (2000) The adenomatous polyposis coli (APC) tumour suppressor – genetics, function and disease. Mol. Med. Today 6, 462. [DOI] [PubMed] [Google Scholar]
- Siu IM, Robinson DR, Schwartz S, Kung HJ, Pretlow TG, Petersen RB, Pretlow TP (1999) The identification of monoclonality in human aberrant crypt foci. Cancer Res. 59, 63. [PubMed] [Google Scholar]
- Spradling A, Drummond‐Barbosa D, Kai T (2001) Stem cells find their niche. Nature 414, 98. [DOI] [PubMed] [Google Scholar]
- Stemmermann G, Heffelfinger SC, Noffsinger A, Hui YZ, Miller MA, Fenoglio‐Preiser CM (1994) The molecular biology of esophageal and gastric cancer and their precursors: oncogenes, tumor suppressor genes, and growth factors. Hum. Pathol. 25, 968. [DOI] [PubMed] [Google Scholar]
- Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez‐Bigas MA, Petrelli NJ, Anderson GR (1999) The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc. Natl. Acad. Sci. U S A 96, 15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, Kato J, Kogawa K, Miyake H, Niitsu Y (1998) Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 339, 1277. [DOI] [PubMed] [Google Scholar]
- Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S et al. (2001) Analysis of K‐ras, APC, and beta‐catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology 121, 599. [DOI] [PubMed] [Google Scholar]
- Tamura G (2004) Promoter methylation status of tumor suppressor and tumor‐related genes in neoplastic and non‐neoplastic gastric epithelia. Histol. Histopathol. 19, 221. [DOI] [PubMed] [Google Scholar]
- Tatematsu M, Fukami H, Yamamoto M, Nakanishi H, Masui T, Kusakabe N, Sakakura T (1994) Clonal analysis of glandular stomach carcinogenesis in C3H/HeN<==>BALB/c chimeric mice treated with N‐methyl‐N‐nitrosourea. Cancer Lett. 83, 37. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ, Greaves LC, Kirkwood TB, Turnbull DM (2003) Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 112, 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teller IC, Beaulieu JF (2001) Interactions between laminin and epithelial cells in intestinal health and disease. Expert Rev. Mol. Med. 2001, 1. [DOI] [PubMed] [Google Scholar]
- Thliveris AT, Halberg RB, Clipson L, Dove WF, Sullivan R, Washington MF, Stanhope S, Newton MA (2005) Polyclonality of familial murine adenomas: Analyses of mouse chimeras with low tumor multiplicity suggest short‐range interactions. Proc. Natl Acad. Sci. USA 102, 6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M, Fleming KA, Evans DJ, Fundele R, Surani MA, Wright NA (1990) Gastric endocrine cells share a clonal origin with other gut cell lineages. Development 110, 477. [DOI] [PubMed] [Google Scholar]
- Tomlinson IP, Roylance R, Houlston RS (2001) Two hits revisited again. J. Med. Genet. 38, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totafurno J, Bjerknes M, Cheng H (1987) The crypt cycle. Crypt and villus production in the adult intestinal epithelium. Biophys. J. 52, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL (1988) Genetic alterations during colorectal tumor development. N. Engl. J. Med. 319, 525. [DOI] [PubMed] [Google Scholar]
- Wasan HS, Park HS, Liu KC, Mandir NK, Winnett A, Sasieni P, Bodmer WF, Goodlad RA, Wright NA (1998) APC in the regulation of intestinal crypt fission. J. Pathol. 185, 246. [DOI] [PubMed] [Google Scholar]
- Winton DJ, Blount MA, Ponder BA (1988) A clonal marker induced by mutation in mouse intestinal epithelium. Nature 333, 463. [DOI] [PubMed] [Google Scholar]
- Woda BA, Forde K, Lane N (1977) A unicryptal colonic adenoma, the smallest colonic neoplasm yet observed in a non‐polyposis individual. Am. J. Clin. Pathol. 68, 631. [DOI] [PubMed] [Google Scholar]
- Wong WM, Mandir N, Goodlad RA, Wong BC, Garcia SB, Lam SK, Wright NA (2002) Histogenesis of human colorectal adenomas and hyperplastic polyps: the role of cell proliferation and crypt fission. Gut 50, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WM, Wright NA (1999) Cell proliferation in gastrointestinal mucosa. J. Clin. Pathol. 52, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright NA, , Alison M (1984) The Biology of Epithelial Cell Populations. Oxford: Oxford University Press. [Google Scholar]
- Yatabe Y, Tavare S, Shibata D (2001) Investigating stem cells in human colon by using methylation patterns. Proc. Natl Acad. Sci. U S A 98, 10839. [DOI] [PMC free article] [PubMed] [Google Scholar]