

Abstract
Objectives: This study aims to compare pharmacokinetics and pharmacodynamics of pegfilgrastim, a pharmaceutical recombinant human granulocyte colony‐stimulating factor (rhG‐CSF), with that of a newly developed reagent, Maxy‐G34. This comparison was performed using rat experiments and biomathematical modelling of granulopoiesis.
Methods: Healthy rats and those with cyclophosphamide‐induced neutropenia were treated with either pegfilgrastim or Maxy‐G34 under various schedules. Time courses of absolute neutrophil count (ANC) and G‐CSF serum level were measured and we constructed a combined pharmacokinetic/pharmacodynamic model of both drugs. Neutropenic episodes were assessed by experimental data and model simulations.
Results: Both Pegfilgrastim and Maxy‐G34 showed strong dose‐dependent efficacy in reducing neutropenic episodes. However, time courses of ANC and G‐CSF serum levels were markedly different. The biomathematical model showed good agreement with these data. We estimated that differences between the two drugs could be explained by lower bioavailability and reduced elimination of Maxy‐G34. Based on the data and model interpolations, we estimated that Maxy‐G34 is superior in reducing neutropenic episodes. Also, we predicted that G‐CSF administration 48 h after cyclophosphamide would be superior to its administration after 2 or 24 h, for both derivatives.
Conclusion: Maxy‐G34 is a highly potent drug for stimulation of neutrophil production in rats. By our modelling approach, we quantified differences between Maxy‐G34 and pegfilgrastim, related to pharmacokinetic parameters. Model simulations can be used to estimate optimal dosing and timing options in the present preclinical rat model.
Introduction
Chemotherapy is commonly used for treatment of malignant tumours but myelosuppression is a serious adverse event associated with the use of most chemotherapeutic agents. Severe neutropenia is a dose‐limiting factor in many chemotherapy regimens, since it is associated with increased incidence of infection, hospitalization, antibiotic treatment, reduction of treatment intensity, therapy dropouts and therapy‐associated deaths (1, 2, 3, 4, 5, 6). Thus, recombinant human granulocyte colony‐stimulating factor (rhG‐CSF) is routinely given to patients undergoing chemotherapy to ameliorate neutropenia. rhG‐CSF acts in a lineage specific manner by increasing mitotic activity of granulopoietic progenitors and precursors, by accelerating maturation of cells and improving bone marrow release of mature granulocytes (7, 8, 9, 10, 11, 12, 13).
The first generation of pharmaceutical rhG‐CSF was filgrastim, which has a short half‐life in vivo due to rapid renal clearance and specific degradation mediated by G‐CSF receptors or neutrophil elastase (14, 15, 16, 17, 18, 19, 20, 21). To be effective against neutropenia, it must be given daily during each cycle of chemotherapy. Subsequently, PEGylated filgrastim (pegfilgrastim) has been developed, which has a considerably longer half‐life in vivo with reduced rate of renal elimination (22, 23, 24). A single pegfilgrastim injection given 24 h after chemotherapy is at least as effective in reducing severe neutropenia and prevention of infection as multiple injections of filgrastim (2, 25, 26, 27, 28). Even though pegfilgrastim is effective in reducing incidence and duration of severe neutropenia, myelosuppression still remains a serious risk in many chemotherapy regimens. Hence, there is an opportunity to further improve properties of rhG‐CSF pharmaceuticals.
Maxygen Inc. (Redwood City, CA, USA) has developed a new PEGylated recombinant human G‐CSF derivative, Maxy‐G34. In contrast to other recombinant G‐CSF molecules, Maxy‐G34 has been modified to reduce both renal and G‐CSF receptor‐mediated clearance. In this study, we have explored the potency of Maxy‐G34 in comparison to pegfilgrastim in a preclinical rat model with cyclophosphamide (CP) induced neutropenia. Based on experimental data from the rat studies, we constructed a mathematical model that would allow optimization of dosing and timing schedules.
Administration of G‐CSF after cytotoxic chemotherapy results in complex dynamic behaviour of granulopoiesis, which cannot easily be predicted (29, 30). It has been shown that the effectiveness of G‐CSF treatment depends on many factors, such as the chemotherapeutic agent used, patient’s individual factors and specially, dosing and timing of G‐CSF administration itself (31). Because of this large set of variable therapy parameters, optimal G‐CSF treatment cannot be developed based on preclinical or clinical trial data alone.
In the past we have shown that a biomathematical model of granulopoiesis, including a pharmacokinetic (PK) model of filgrastim administration, can be a valuable tool to pre‐select optimal filgrastim schedules in various therapy situations (32). In this study, we aim to include the new drug Maxy‐G34 and pegfilgrastim in our existing granulopoiesis model, in order to optimize dosing and timing schedules. We first constructed a PK model of both drugs based on experimental data of rat studies, and then combined the PK model with a simple model of rat granulopoiesis. The combined model allows prediction of the time course of G‐CSF serum levels and circulating granulocytes in rats, treated with different dosing and timing modes of pegfilgrastim or Maxy‐G34 with or without CP administration. Since we used the same model structure for both pegfilgrastim and Maxy‐G34, differences between the drugs can be reduced to differences in model parameters, which allow comparisons of PK and pharmacodynamic (PD) properties between the two drugs.
Methods
Description of the new G‐CSF derivative Maxy‐G34
Maxy‐G34 was designed to be an improved next‐generation G‐CSF for treatment of neutropenia. Maxy‐G34 is a PEGylated recombinant human G‐CSF, modified to contain five amino acid substitutions: K16R, K34R, K40R, T105K, and S159K (denoted using the one‐letter code for amino acid residues with human G‐CSF numbering). Lysine (K) to arginine (R) substitutions were made to remove potential PEGylation sites within wild‐type human G‐CSF protein. Conversely, threonine (T) and serine (S) to lysine (K) substitutions at positions 105 and 159 selectively introduced two new PEGylation sites. Maxy‐G34 is produced from the variant protein backbone through chemical coupling of amine‐specific activated 5 kDa methoxypoly(ethylene glycol) (mPEG) moieties. Following PEGylation, a selective dePEGylation step is performed to remove mPEG groups attached to the protein through unstable ester linkages. Purified Maxy‐G34 drug product predominantly carries three mPEG groups.
Rat study design and experimental procedures
Several dosing and timing schedules using the study drugs (Maxy‐G34 and pegfilgrastim) were investigated in healthy rats or in rats after CP administration. G‐CSF compounds were administered either 2 h (same day) or 24 h (next day) after CP administration. Groups tested can be found in Table 1.
Table 1.
Experimental outline of the study. Groups differ in G‐CSF derivatives applied and corresponding dosing schedules in healthy rats or dosing and timing schedules after CP administration
| No. | G‐CSF derivative | Dose of G‐CSF (μg/kg) | Timing of G‐CSF | CP administration |
|---|---|---|---|---|
| 1 (control group) | Vehicle | – | Same day | Yes |
| 2 (control group) | Vehicle | – | Next day | Yes |
| 3 | Maxy‐G34 | 30 | Same day | Yes |
| 4 | Maxy‐G34 | 100 | Same day | Yes |
| 5 | Maxy‐G34 | 300 | Same day | Yes |
| 6 | Maxy‐G34 | 30 | Next day | Yes |
| 7 | Maxy‐G34 | 100 | Next day | Yes |
| 8 | Maxy‐G34 | 100 | – | No |
| 9 | Maxy‐G34 | 300 | – | No |
| 10 | Maxy‐G34 | 1000 | – | No |
| 11 | Pegfilgrastim | 100 | Same day | Yes |
| 12 | Pegfilgrastim | 300 | Same day | Yes |
| 13 | Pegfilgrastim | 1000 | Same day | Yes |
| 14 | Pegfilgrastim | 100 | Next day | Yes |
CP, cyclophosphamide; G‐CSF, granulocyte colony‐stimulating factor.
Male Sprague–Dawley Ntac:SD rats (M&B Taconic, Lille Skensved, Denmark), weighing about 190 g at arrival, were housed in a defined environmentally controlled animal facility and were kept for a 2‐week acclimatization period before we performed the procedures. All procedures were performed as agreed in international laws for the care and use of laboratory animals and were approved by the national animal ethical committee (Denmark).
To induce a neutropenic episode, rats were injected intraperitoneally with 90 mg/kg CP (Sendoxan, Baxter AS, Oslo, Norway) dissolved in an injection volume of 2.7 ml/kg. Two hours (same‐day administration) or 24 h (next‐day administration) after CP treatment, the rats were injected subcutaneously with G‐CSF derivate in an injection volume of 1 ml/kg [pegfilgrastim 100, 300 or 1000 μg/kg (Neulasta, Amgen, Thousand Oaks, CA, USA); Maxy‐G34 30, 100, 300 or 1000 μg/kg (Maxygen Inc.)]. The control group received 10 mm sodium acetate (vehicle) instead of G‐CSF in an injection volume of 1 ml/kg. Blood samples were taken from tail veins before and after administration of vehicle or the study drugs, at time points indicated in Fig. 2. For PD evaluation, four drops of blood (around 160 μl) were collected in 1 ml MiniCollect tubes containing EDTA (Greiner Bio‐One, Frickenhausen, Germany) and tubes were stored at 4 °C until assayed. For PK evaluation, blood was transferred to a thrombin tube (Microvette 300Z, Sarstedt AG & Co., Nuernbrecht, Germany) and serum was separated by centrifugation, before being transferred to an Eppendorf tube and stored at –80 °C until assayed.
Figure 2.
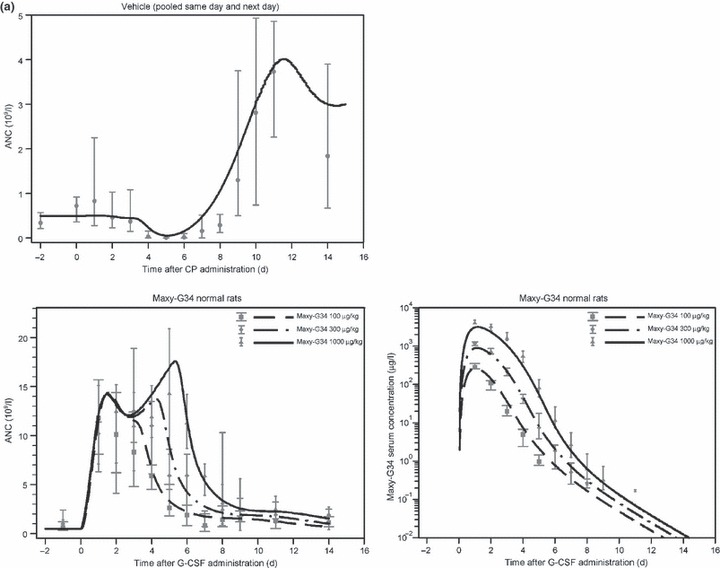
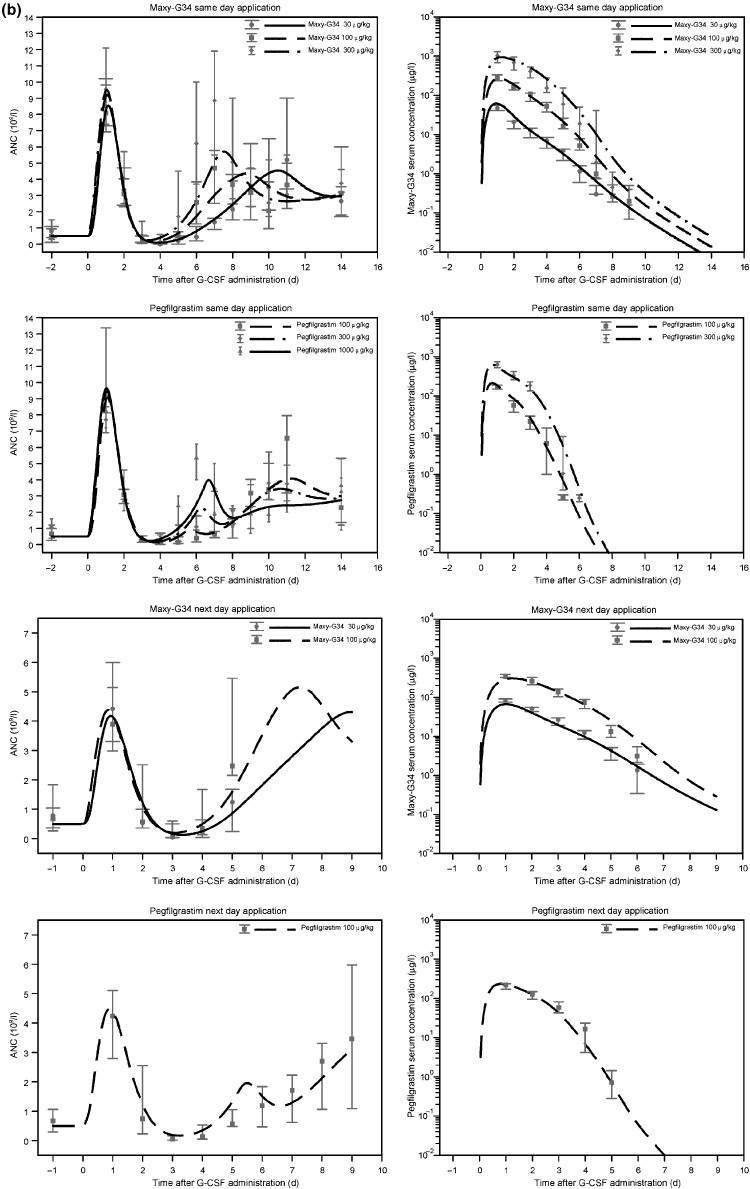
(a) Comparison of the estimates of the full PK‐PD model and the data for the control groups [cyclophosphamide (CP) administration with vehicle; single dose of Maxy‐G34 in normal rats]. We present median and range of data for both absolute neutrophil count (ANC) and cytokine concentrations. The lines are the model predictions for the corresponding scenarios. Maxy‐G34 administration in normal rats was used for model validation. (b) Comparison of the estimates of the full PK‐PD model and the data from the experimental groups (CP administration combined with a single dose of Maxy‐G34 or pegfilgrastim in different dosing schedules). We present median and range of data for both ANC and cytokine concentrations. Lines mark the model predictions for the corresponding scenarios. Note that the scales are different for same‐day and next‐day administrations.
The effect of Maxy‐G34 or pegfilgrastim on white blood cell counts was determined using an ABX Pentra 120 counter (Horiba ABX, F‐34184 Montpellier, France). Relative neutrophil count was manually performed from blood smears. Absolute neutrophil count (ANC) was calculated from relative levels of neutrophils to total white blood cells. Level of G‐CSF present in plasma was assessed using a commercially available ELISA kit (Human G‐CSF DuoSet ELISA, R&D Systems Inc., Minneapolis, MN, USA). Briefly, Maxy‐G34 or pegfilgrastim was captured via a mouse monoclonal antihuman G‐CSF antibody coated on 96‐well plates and detected by a biotinylated polyclonal goat antihuman G‐CSF antibody. Streptavidin‐conjugated horseradish peroxidase (HRP) was added and HRP activity was measured by addition of a chemiluminescent HRP substrate. The assay was performed according to the manufacturer’s instruction (R&D Systems Inc.). Measurements were supported by repeats in six or seven rats.
Statistical analysis
Number of neutropenic days with ANCs lower than the geometric mean in untreated rats were determined in the nadir phase (between days 1 and 9 after administration of CP) and were compared between the different groups of G‐CSF treatment, using the Jonckheere‐Terpstra test for trends. These calculations were performed using statistical software package R 2.7.0 (33).
Modelling
First we constructed a PK model of both pegfilgrastim and Maxy‐G34. For this purpose, assumptions about the absorption, distribution and elimination kinetics of G‐CSF derivatives have been transformed into ordinary differential equations. Hereby, we also exploited knowledge of the PK of filgrastim, which has already been investigated more intensively. We used the same model equations but different parameters for both drugs. Unknown parameters were determined by fitting the model to measured time courses of G‐CSF serum concentrations, minimizing the difference between model and logarithmized median of data. Experiments with Maxy‐G34 injections into healthy rats were not used for any model fitting but were used for later model validation. Since elimination of drugs is mainly determined by the numbers of circulating ANC (specific elimination; 14,16–18, 34–36, among others), measured ANC have been linearly interpolated and imprinted into the PK model. This approach leads to a model that predicts G‐CSF serum concentrations when the initial dose and ANC time course is known. Hence, the model is not fully independent of data at this stage.
In the next step, we combined the PK model with a simplified version of a PK‐PD model of granulopoiesis, which has been established recently for humans (29, 30). It is a compartment model describing time‐dependent dynamics of concatenated cell compartments in the bone marrow and mature circulating granulocytes. Bone marrow compartments represent different cell stages of granulopoiesis starting from early stem cells over different division and maturation stages to mature granulocytes in blood. The system is regulated by growth factor‐mediated feedback loops (Fig. 1).
Figure 1.
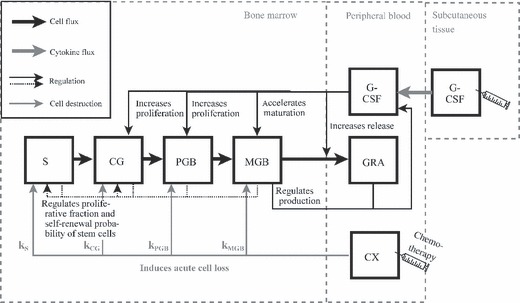
Schematic structure of the combined pharmacokinetic–pharmacodynamic (PK‐PD) model of murine granulopoiesis. The model is based on concatenated cell compartments. S represents early stem cells; CG, CFU‐G; PGB, proliferating granulocytic precursors; MGB, maturing granulocytic precursors; and GRA, circulating granulocytes (ANC). The identified PK model is attached to the PD model. Toxicity is modelled by an instantaneous cell depletion. The model is explained in detail in the literature (30).
Basic model assumptions are that G‐CSF increases the number of cell divisions for mitotic precursors (PGB), improves maturation of post‐mitotic precursors (MGB) and enhances release of mature granulocytes from bone marrow to the blood. The latter effect is also called ‘post‐mitotic amplification’ (9, 10, 12). These regulation processes are modelled by sigmoidal functions that regulate corresponding quantities (for example, number of cell divisions or maturation time) between a minimum and a maximum value in dependence on serum concentration of G‐CSF. The steepness of these regulation functions is variable by so‐called sensitivity parameters.
Parameters of the rat PD model, such as number of cell divisions, transition time between two cell stages and maturation time, were the same for both pegfilgrastim and Maxy‐G34, except for sensitivity parameters of corresponding regulation functions (eqn 1). Furthermore, there is a normalization constant, k shift, which translates serum concentration of both pegfilgrastim and Maxy‐G34 into effective bone marrow stimulation used for all regulation functions. This constant causes a shift in the regulation function without changing steepness of the function. We assumed that the shift parameter is also different between pegfilgrastim and Maxy‐G34. Hence, a total of five parameters were assumed to be different between Maxy‐G34 and pegfilgrastim, which are four sensitivity parameters (with respect to amplification in CG, amplification in PGB, post‐mitotic amplification in MGB and maturation time in MGB) and one shift parameter.
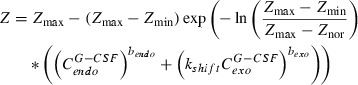 |
(1) |
Equation 1: Regulation function of amplification of CG and PGB, maturation and post‐mitotic amplification of MGB. Z min, Z nor and Z max are the conditions under minimal, normal and maximal stimulation, respectively. C G‐CSF endo and C G‐CSF exo are endogenous and exogenous G‐CSF, respectively. b endo and b exo are the sensitivity parameters with respect to endogenous and exogenous G‐CSF, respectively. The latter is assumed to be different between Maxy‐G34 and pegfilgrastim; shift parameter, k shift, is also assumed to be different but is constant for all regulation functions.
The effect of CP has been modelled by instantaneous and temporary cell depletion in all bone marrow cell stages. These toxicities can be represented by a drug‐specific set of cell stage‐specific toxicity parameters (see Scholz et al. for further details (30)). To model recovery from CP in the control groups, we included a model of endogenous G‐CSF production as in the human situation. All equations relating to the model can be found elsewhere (30).
Unknown parameters of the PD model were determined by fitting the model to the logarithmized median of granulocyte data of all experiments, except for those of Maxy‐G34 administration in healthy rats (which have been used to validate the model). Parameters of the PK model were not changed by the second fitting procedure. Because of the relatively low number of measurements per time point, we present the range of data as a measure of variance. We claim that the model curve should be between minimum and maximum of measurements.
Different G‐CSF derivatives were represented by different parameter settings but not by different model equations. This allows comparisons between the PK and PD properties of the drugs. Since the model is now fully independent of data, it can be used to simulate yet untested dosing and timing schedules of the G‐CSF derivatives in order to make predictions of corresponding granulotoxicity.
Differential equations and simulations have been implemented in the mathematical software package MATLAB 7.0.4.365 with SIMULINK toolbox (The MathWorks Inc. Natick, MA, USA).
Results
Experimental data
ANC values in normal rats. The normal value of ANC was determined in untreated rats; geometric mean was 6.6 × 108/l with a geometric standard deviation of 1.73.
ANC profile in rats with CP‐induced neutropenia. Administration of 90 mg/kg CP to healthy rats resulted in deep, but reversible, nadir of low ANC values (Fig. 2a), which occurred between days 4 and 6 after CP administration. ANC counts had recovered at day 9 followed by a period of overcompensation. Time courses of the two control groups that were given vehicle fluid (same‐day and next‐day administration) were similar and, therefore, were pooled for further analysis.
PD and PK profile in normal rats administered Maxy‐G34. Subcutaneous administration of Maxy‐G34 to normal rats resulted in a strong dose‐dependent increase of ANC values over time (Fig. 2a). In the 100 μg/kg dose group, a peak ANC of 11.8 × 109/l was observed on day 1, with high ANC values ranging from days 1 to 3. A peak ANC of 12.5 × 109/l was reached on day 3 in the 300 μg/kg dose group with high ANC values lasting from days 1 to 4. In the 1000 μg/kg dose group, ANC time course was clearly biphasic with peak ANC of 14.3 × 109/l on day 5 and high ANC values ranging from days 1 to 6. These data indicate that higher doses of Maxy‐G34 result in prolonged elevation of high ANC values and increased peak ANC values.
The pharmacokinetic profile of Maxy‐G34 showed clear dose dependence. Administration of higher doses of Maxy‐G34 resulted in its increased serum levels and prolonged drug elimination phase. Drug elimination was not constant but was highest during the corresponding phases of high ANC elevation, indicating a strong specific elimination process.
Efficacy of Maxy‐G34 or pegfilgrastim in rats with CP‐induced neutropenia. Neutropenic rats were administered Maxy‐G34 (30, 100, 300 μg/kg) or pegfilgrastim (100, 300, 1000 μg/kg) in same‐day administration (2 h after CP) or next‐day administration (24 h after CP) treatment groups. ANC and PK profiles for the different treatment groups are shown in Fig. 2b. PK profiles for Maxy‐G34 and pegfilgrastim showed clear dose dependence. Administration of Maxy‐G34 and pegfilgrastim resulted in high level of granulocytosis on day 1 followed by a neutropenic episode that was significantly shorter than in the control groups, injected with the vehicle. Rats that were given Maxy‐G34 in the same‐day administration group reached their nadir phase on day 4. Rats in the same‐day administration group for pegfilgrastim reached their nadir on day 3 in 4 cases (22%), on day 4 in 12 cases (67%) and later in 2 cases (11%). In the next‐day treatment group, all rats treated with pegfilgrastim reached the nadir on day 3. In the Maxy‐G34 next‐day treatment group, 1 animal reached nadir on day 2 (7%), 11 animals on day 3 (79%) and 2 animals on day 4 (14%).
For all dosing and timing schedules, we analysed number of neutropenic days, which are days in the nadir phase with ANC values below the normal values (Table 2). Both Maxy‐G34 and pegfilgrastim showed clear dose‐dependent reduction in number of neutropenic days in the nadir phase (pegfilgrastim same day: P = 0.00017; Maxy‐G34 same day: P = 0.00056; Maxy‐G34 next day: P = 0.036). Furthermore, comparing efficacy of the same doses of Maxy‐G34 and pegfilgrastim in the 100 and 300 μg/kg dose groups, Maxy‐G34 was superior to pegfilgrastim (100 μg/kg same day: P = 0.008; 100 μg/kg next day: P = 0.022). Observed difference in the 300 μg/kg same‐day regimen was not significant (P = 0.24).
Table 2.
Length of neutropenic episode defined as days with absolute neutrophil count (ANC) below normal. Number of animals with specified number of neutropenic days is presented for all dosing and timing schedules
| G‐CSF schedule | Pegfilgrastim | Maxy‐G34 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Days with neutropenia | Same day | Next day | Same day | Next day | |||||
| 100 μg/kg (N = 6) | 300 μg/kg (N = 6) | 1000 μg/kg (N = 6) | 100 μg/kg (N = 6) | 30 μg/kg (N = 6) | 100 μg/kg (N = 6) | 300 μg/kg (N = 6) | 30 μg/kg (N = 7) | 100 μg/kg (N = 7) | |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| 2 | 0 | 4 | 6 | 1 | 0 | 2 | 2 | 1 | 3 |
| 3 | 2 | 2 | 0 | 2 | 2 | 4 | 2 | 5 | 3 |
| 4 | 2 | 0 | 0 | 2 | 4 | 0 | 0 | 1 | 0 |
| 5 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
G‐CSF, granulocyte colony‐stimulating factor.
The model: PK model for pegfilgrastim and Maxy‐G34. The following compartments were considered in the model: subcutaneous tissue into which the drugs were injected, central compartment representing the blood system in which the drugs were haematologically active, and a peripheral compartment where the drugs were temporarily removed from the central compartment due to diffusion into other tissues or to protein binding (37).
Dynamics of G‐CSF serum concentrations can be modelled by the fluxes between these compartments and the degradation processes as well. The basic structure of the model can be found in Fig. 3.
Figure 3.
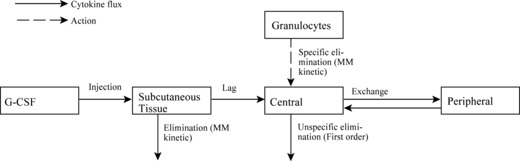
Schematic structure of the pharmacokinetic model for pegfilgrastim and Maxy‐G34. Compartments and model equations are explained in the text.
Major features of the model are: dose‐dependent bioavailability of the drugs after subcutaneous administration, modelled by Michaelis‐Menten elimination of drugs from subcutaneous tissue, delayed influx from subcutaneous tissue to central compartment, which is modelled by gamma distributed absorption kinetic represented by two delay compartments (see Scholz et al. for further details of this procedure (30)), non‐specific elimination from the central compartment (renal clearance) modelled by a first‐order elimination, specific elimination from the central compartment which is a nonlinear function of the ANC, and Michaelis‐Menten kinetic and exchange between central and peripheral compartments modelled by first order transition between the central and peripheral compartments. More specifically these properties have been transformed into an ordinary differential equation system describing change of compartment contents over time. The system has the following form:
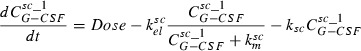 |
(2) |
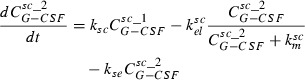 |
(3) |
The subcutaneous compartment is divided into two identical sub‐compartments to model lag of the influx of drugs into the central compartment. G‐CSF is injected into the first sub‐compartment, which can be modelled by an injection function ‘Dose’, which is positive and constant during the injection time (tinf) and zero elsewhere. The second summand on the right‐hand side of the two equations is a Michaelis‐Menten‐like elimination of drugs resulting in dose‐dependent bioavailability.
The third summand of the right‐hand side of the first equation is equal to the first summand in the second equation describing efflux and influx of drug from first to the second sub‐compartment. Finally, the third summand in the second equation is efflux of drug from the subcutaneous compartment to the central compartment. The equation for the central compartment has the following form:
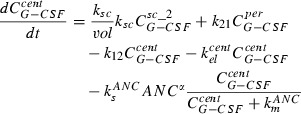 |
(4) |
where the first summand on the right‐hand side is influx from the subcutaneous compartment divided by distribution volume, vol. The second and the third summands describing exchange of drugs between central and peripheral compartments, modelled by a first order transition (C per G‐CSF influx from peripheral to central, C cent G‐CSF efflux from central to peripheral compartment). The fourth summand is the model for the first‐order non‐specific (renal) elimination of drugs and the fifth term is specific degradation mediated by the ANC. Again, this mechanism is modelled by a Michaelis‐Menten elimination kinetic.
The equation for the peripheral compartment has the simple form
| (5) |
which only describes a first order transition between the central and peripheral compartments.
An overview over all model parameters and quantities can be found in Table 3.
Table 3.
Quantities of the pharmacokinetic (PK) model for both pegfilgrastim and Maxy‐G34. We present type, unit and explanation of all quantities. Furthermore, PK parameter estimates are presented for both pegfilgrastim and Maxy‐G34 based on fitting the PK model to the data imprinting measured absolute neutrophil count (ANC) levels. Estimates were compared between the drugs. Parameter estimates were considered comparable if the model fit did not change by more than 10% by exchanging the estimates
| Symbol | Type | Unit | Explanation | Fitted value for pegfilgrastim | Fitted value for Maxy‐G34 | Relation between parameter estimates |
|---|---|---|---|---|---|---|
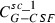 , , 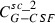
|
Function of time to be calculated | ng | Contains of the sub‐compartments of the subcutaneous tissue | – | – | – |
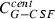
|
Function of time to be calculated | ng/ml | Contain of the central compartment (serum concentration) | – | – | – |
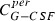
|
Function of time to be calculated | ng/ml | Contain of the peripheral compartment | – | – | – |
| Dose | Given function of time | ng/h | Applied dose per hour | – | – | – |
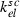
|
Parameter | ng/h | Elimination in subcutaneous tissue | 35 | 150 | Greater for Maxy‐G34 |
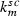
|
Parameter | ng | Michaelis‐Menten constant of subcutaneous elimination | 190 | 1900 | Greater for Maxy‐G34 |
| k sc | Parameter | 1/h | Transition constant from subcutaneous tissue to central compartment causing a time delay (lag) | 0.07 | 0.06 | Comparable |
| vol | Parameter | ml | Volume of distribution | 15.0 | 17.0 | Comparable |
| k 12 | Parameter | 1/h | Transition from central to peripheral compartment | 0.0003 | 0.0004 | Negligible for both derivatives |
| k 21 | Parameter | 1/h | Transition from peripheral to central compartment | 0.13 | 0.12 | Comparable |
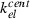
|
Parameter | 1/h | Unspecific (renal) elimination | 0.07 | 0.02 | Greater for pegfilgrastim |
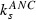
|
Parameter | ng/(ml*h) | Specific elimination via G‐CSF receptors or neutrophil elastase | 0.04 | 0.02 | Greater for pegfilgrastim |
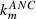
|
Parameter | ng/ml | Michaelis‐Menten constant of specific elimination | 0.1 | 0.5 | Greater for Maxy‐G34 |
| ANC | Given function of time | – | Circulating ANC in 109/l | – | – | – |
| α | Parameter | – | Exponent of ANC | 0.6 | 0.5 | Comparable |
G‐CSF, granulocyte colony‐stimulating factor.
Model fitting resulted in excellent fit of all scenarios with neutropenic rats and also the validation data set of healthy rats. Results are not shown separately but differ only slightly from the diagrams in Fig. 2a and 2b regarding G‐CSF serum concentrations, which will be presented for the full PK‐PD model later. Results for all fitted model parameters can be found in Table 3.
Analysis of estimated parameter values and comparison of these values between pegfilgrastim and Maxy‐G34 revealed that the exchange between peripheral and central compartments was small for both drugs. Furthermore, both drugs had low non‐specific degradation, indicating that they were mainly eliminated specifically. However, this parameter was even lower for Maxy‐G34. Finally, it has been estimated that pegfilgrastim has higher specific degradation, higher bioavailability (Fig. 4) and lower distribution volume than Maxy‐G34. All other parameters were within the same order of magnitude.
Figure 4.
Bioavailability of pegfilgrastim and Maxy‐G34. Curves are based on model simulations of the final pharmacokinetic model.
Combined PK‐PD model for pegfilgrastim and Maxy‐G34. Model fitting resulted in a unique parameter setting of the PD parameters valid for all experimental scenarios. Results of all model curves in comparison to data can be found in Fig. 2a and 2b. We obtained a good fit of all 13 data sets for both ANC and G‐CSF serum concentrations as well. This held true specially for data sets of healthy rats used only for model validation but not for model fitting.
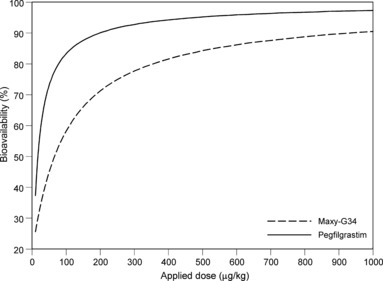
PD properties assumed to be different between pegfilgrastim and Maxy‐G34 were analysed as shown in Fig. 5. It revealed that there is a large scale of possible regulation functions that result in a good fit of data (less than 10% deviation from optimum). Confidence ranges of regulation functions of Maxy‐G34 and pegfilgrastim always intersect. Hence, within the range of observed G‐CSF serum concentration, no clear differences in PD could be identified between Maxy‐G34 and pegfilgrastim.
Figure 5.
Estimated regulation functions for Maxy‐G34 (black) and pegfilgrastim (grey) in a range of G‐CSF serum concentrations supported by measurements. Solid lines are based on parameter estimates resulting in optimal agreement between model and data. Dotted lines indicate a confidence range of the regulation function based on parameter settings resulting in less than 10% deviation from optimal fitness. G‐CSF, granulocyte colony‐stimulating factor.
Model predictions. To demonstrate how the model can be used in order to predict results of new dosing and timing schedules for both pegfilgrastim and Maxy‐G34 in our chemotherapy rat model, we performed a model simulation of G‐CSF administration starting 2 days after CP administration, for which we had no experimental data. We used the same dose levels for both drugs as tested in previous experiments. Results of these model simulations can be found in Fig. 6.
Figure 6.
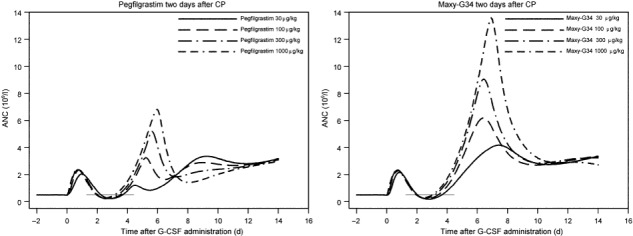
Model simulations of the timing of pegfilgrastim and Maxy‐G34 2 days after cyclophosphamide administration. We simulated four dose levels (30, 100, 300, 1000 μg/kg) and present model estimates of the absolute neutrophil counts for these scenarios. The grey baseline marks the nadir phase of simulated cell counts.
In order to compare toxicity of these schedules with toxicity of schedules already considered, we also simulated dose levels above for same‐day and next‐day timing after CP treatment. In order to evaluate and compare toxicity of the schedules, we calculated minimal ANC, duration of neutropenia (DN) and area over the curve (AOC) which is the area between simulated time course of ANC in the nadir phase and baseline (Table 4). There is clear dose–response relationship for Maxy‐G34, with estimated optimal timing 2 days after CP administration. For pegfilgrastim, higher doses were (in general, but not always) better than lower doses. As for Maxy‐G34, optimal timing was estimated to be 2 days after CP treatment. For fixed dosing and timing schedules, Maxy‐G34 was estimated to be superior in comparison to pegfilgrastim except for scheduling 30 μg/kg on day 2.
Table 4.
Model predictions and interpolation of toxicity of different dosing and timing schedules of pegfilgrastim and Maxy‐G34 after cyclophosphamide administration (CP). We simulated three different timing schedules and four doses in a full‐factorial design. Area over the curve (AOC) is the area between the baseline and the simulated time course of ANC in the nadir. DN and minimal ANC were calculated to evaluate toxicity. Both, high AOC, high DN and low minimal cell count indicate high toxicity as well
| Timing/dosing (μg/kg) | Pegfilgrastim | Maxy‐G34 | ||||
|---|---|---|---|---|---|---|
| DN (h) | Minimal ANC (109/l) | AOC (109h/l) | DN (h) | Minimal ANC (109/l) | AOC (109h/l) | |
| Same day | ||||||
| 30 | 76 | 0.061 | 23.0 | 62 | 0.088 | 17.8 |
| 100 | 49 | 0.13 | 12.4 | 45 | 0.17 | 10.0 |
| 300 | 45 | 0.17 | 10.3 | 32 | 0.26 | 5.2 |
| 1000 | 35 | 0.24 | 6.0 | 26 | 0.31 | 3.3 |
| Next day | ||||||
| 30 | 73 | 0.23 | 14.6 | 50 | 0.14 | 12.4 |
| 100 | 43 | 0.17 | 9.9 | 39 | 0.21 | 7.7 |
| 300 | 38 | 0.22 | 7.4 | 29 | 0.28 | 4.4 |
| 1000 | 30 | 0.29 | 4.3 | 24 | 0.33 | 2.8 |
| Two days after CP | ||||||
| 30 | 34 | 0.24 | 6.0 | 40 | 0.19 | 8.1 |
| 100 | 36 | 0.21 | 7.1 | 35 | 0.22 | 6.5 |
| 300 | 33 | 0.23 | 6.0 | 30 | 0.26 | 4.7 |
| 1000 | 29 | 0.28 | 4.3 | 26 | 0.30 | 3.4 |
Discussion
By a combined experimental and modelling approach, we constructed a comprehensive model of rat granulopoiesis under combined administration of CP and the G‐CSF derivatives pegfilgrastim or the new Maxy‐G34. To our knowledge the model presented here is the first one that describes different G‐CSF derivatives in the same model, allowing comparisons between the drugs to be made. The model allows for prediction of both G‐CSF serum concentration and ANC time course after CP treatment, for different dosing and timing schedules of pegfilgrastim or Maxy‐G34. Therefore, it can be used to optimize G‐CSF scheduling.
Recombinant G‐CSF is routinely given in many chemotherapy regimens to prevent and treat severe neutropenia (1, 2, 3, 4, 5, 38). Although a variety of G‐CSF derivatives is available now, high potency of the drug and its wide field of possible application warrant further research; there are several avenues for further improvement of their pharmaceutical properties. Most promising results can be expected from G‐CSF derivatives with prolonged half‐life in vivo. Beside the aspect of convenience for patients to receive single administration of G‐CSF with the same efficacy as multiple injections, it is believed that constant stimulation of granulopoiesis is favourable to markedly fluctuating serum concentrations of the drug (39). PEGylated G‐CSF (pegfilgrastim) has been developed for that purpose. Recently, an alternative product, Maxy‐G34, has been developed by Maxygen Inc.
It was the aim of this study to explore PK and PD properties of this new G‐CSF derivative in a preclinical, rat model of CP‐induced neutropenia. Second, comparisons with pegfilgrastim were performed. We showed that Maxy‐G34 has high potential in reducing the neutropenic period after administration of 90 mg/kg CP in rats. We found clear dose‐dependent reduction in number of neutropenic days in the nadir phase for both administration schedules tested (same‐day and next‐day administration). We also collected evidence that Maxy‐G34 is more effective in reduction of neutropenic days in comparison to the same protein mass of pegfilgrastim. We performed no functional studies of circulating cells. Hence, this study only considers number of ANC and not their effectiveness in preventing adverse events, such as febrile neutropenia. However, in clinical practice it has been shown that ANC values are correlated to adverse events (40).
In order to understand better the differences between the two G‐CSF derivatives, we combined our experiments with a systems‐biological approach of modelling physiological PK and PD properties of both drugs, in a comprehensive compartment model of rat granulopoiesis. Since there is a strong relation between G‐CSF‐induced granulocytosis and the level of G‐CSF elimination in dependence on circulating ANC itself (17, 20, 41, 42, 43, 44, 45), it is necessary to model both PK and PD in order to construct a model that is independent of experimental data. It has been shown in the past that such modelling can provide valuable insights into the complex interaction of the various stimulating effects of G‐CSF after chemotherapy‐induced neutropenia (24, 29, 32, 46, 47).
To reduce complexity of modelling, we constructed an isolated PK model first. Model assumptions were based on physiological knowledge of the drug and its elimination processes. We assumed that both drugs would have dose‐dependent bioavailability. Increasing bioavailability with dose has been found, at least for filgrastim (48). Influx from subcutaneous to the central compartment was delayed for both drugs, which can be observed from data showing delayed occurrence of maximum serum concentration after administration. The drugs are removed from the central compartment by non‐specific renal clearance. Parameter estimates revealed that this mechanism was very low for pegfilgrastim according to literature (24). It is even lower for Maxy‐G34, which can be explained by the additional PEGylation of the molecule. The major degradation mechanism is specifically via receptor binding or neutrophil elastase activity (14, 16, 18, 35, 36, 49). Both mechanisms depend on numbers of circulating ANC. However, it cannot be expected that this degradation mechanism is proportional to ANC since there is enrichment of younger granulocytes in the blood after G‐CSF administration. Furthermore, there is a high number of G‐CSF receptors in bone marrow that might contribute to the elimination process (20, 50). This bone marrow cellularity is correlated, but not necessarily proportional, to ANC. Therefore, we assumed that the degradation mechanism would be proportional to a (nonlinear) power function of the ANC. By model fitting, we have shown that its exponent is far below 1 for both derivatives. The Michaelis‐Menten factor for specific degradation was lower for Maxy‐G34 than for pegfilgrastim, which may be explained by the higher degree of PEGylation of the Maxy‐G34 molecule, that reduces receptor binding affinity. As a consequence, Maxy‐G34 has a longer half‐life. Finally, we assumed an exchange of G‐CSF between the central blood compartment and a peripheral compartment, which can be explained by diffusion processes, or protein binding without internalization (37). However, our parameter estimates suggest that this mechanism might only play a minor role for the dynamics of serum concentrations of the drugs considered.
In the next step of modelling, we combined the PK model with a PD model that is based on a simplified version of a human model of granulopoiesis established recently (29, 30). We kept the model as simple as possible to reduce the number of unknown parameters and to prevent over‐fitting of data. Assumptions of this model are discussed elsewhere (30). In our simplified model, we neglected spleen‐ and cytokine‐mediated regulation, which are not based on G‐CSF. This might be justified in our situation of G‐CSF administration in all experiments, except for the control group, since in these cases the system is predominantly regulated by G‐CSF dynamics.
Again we assumed the same model parameters for both pegfilgrastim and Maxy‐G34 except for those that were related to PD properties of the drugs. We assumed that sensitivity parameters, which are the steepness of regulation functions, are different for each drug. Furthermore, blood concentrations of the drugs were normalized by a shift parameter that is constant, but might be different for the drugs. However, we estimated that the regulation functions did not differ significantly between pegfilgrastim and Maxy‐G34. For both drugs, there is a large set of possible regulation functions resulting in a good fit of the model. Hence, we conclude that the present data base is not sufficient to make precise predictions with respect to regulation functions.
Unknown parameters of the model were determined by fitting the model to the data. We first fitted the PK model and then the more complex PD model, keeping already identified parameters of the PK model constant. We avoided fitting of individual time courses of the rats since we were mostly interested in a model that represents population median, in order to allow comparisons between different dosing and timing schedules (29, 30). We identified a single parameter setting for the combined PD and PK model, resulting in good agreement between model and data of ANC and G‐CSF serum concentrations of all experiments. ANC data of Maxy‐G34 administration in healthy rats were used only for model validation of the combined PK and PD model and also agreed well with model prediction.
Since the combined PK and PD model is fully data independent and is based on a single parameter set applicable for all dosing and timing schedules of the drugs, the model can be used to estimate time course of ANC and G‐CSF serum concentrations of new dosing and timing schedules after CP‐induced granulotoxicity, for which no experimental data are available. This is a major advantage of our model compared to other models (46). Hence, model simulations can be used to systematically explore different dosing and timing options of pegfilgrastim or Maxy‐G34 after chemotherapy, which can yield to preclinical selection of G‐CSF schedules optimized for reduction of neutropenia, or to predict remaining serum levels at certain time points after administration of G‐CSF, as has been demonstrated in the literature for pegfilgrastim alone (24).
We demonstrated this by simulating administration of 30, 100, 300 and 1000 μg/kg of pegfilgrastim and Maxy‐G34 timed at day 0 to day 2 after CP administration. We estimated that both dosing and timing have important impacts on AOC, DN and minimal ANC. Higher doses result in lower toxicity, except for pegfilgrastim 30 μg/kg given on day 2. Interestingly, the timing effect vanished for high doses. For pegfilgrastim, the latter fact has been closely confirmed in mouse experiments that we performed after this study (51), providing evidence that model predictions would be correct.
In general, we predict that timing of G‐CSF dosing on day 2 is better than on days 0 and 1 for both pegfilgrastim and Maxy‐G34. This can be explained by two competing processes. At first, amplification of mitotic precursors should be stimulated early after CP. On the other hand, G‐CSF increases release of the bone marrow reserve pool (by preventing early apoptosis), which we call post‐mitotic amplification (9, 10, 12, 52). Since this effect leads to a rapid increment in circulating ANC, it should happen best in the nadir phase of ANC. Hence, one can conclude that there is optimum of timing. This could also explain why pegfilgrastim 30 μg/kg might be slightly better than 100 μg/kg at day 2. It could happen that the higher dose reaches maximum stimulation of post‐mitotic amplification too early in comparison to the lower dose, which could favour the lower dose in absorbing the nadir phase. Finally, we predicted that for the same dosing and timing, Maxy‐G34 is superior to pegfilgrastim except for 30 μg/kg day 2 scheduling.
Summarizing the insights generated from both our experiments and model work, we conclude that Maxy‐G34 and pegfilgrastim considerably differ with respect to both dynamics of G‐CSF serum concentration and resulting ANC dynamics. Our modelling approach revealed that these differences could be explained by differences in a small subset of PK parameters rather than different mechanisms of action. We can perform model simulations in order to explore differences between the drugs and corresponding schedules in various situations. Experiments and model simulations showed that Maxy‐G34 is a promising candidate for further improvement of G‐CSF therapy, due to improved PK properties. Corresponding clinical phase II trials are ongoing and will reveal suitable fields of clinical application. Selection of optimal dose and timing schedules for Maxy‐G34 will be supported by our ongoing systems‐biology modelling approach which will be extended to the human situation.
Acknowledgement
This research has been funded by Maxygen Inc.
References
- 1. Blayney DW, McGuire BW, Cruickshank SE, Johnson DH (2005) Increasing chemotherapy dose density and intensity: phase I trials in non‐small cell lung cancer and non‐Hodgkin’s lymphoma. Oncologist 10, 138–149. [DOI] [PubMed] [Google Scholar]
- 2. Dale D (2003) Current management of chemotherapy‐induced neutropenia: the role of colony‐stimulating factors. Semin. Oncol. 30, 3–9. [DOI] [PubMed] [Google Scholar]
- 3. Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH (2006) Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer 106, 2258–2266. [DOI] [PubMed] [Google Scholar]
- 4. Schwenkglenks M, Jackisch C, Constenla M, Kerger JN, Paridaens R, Auerbach L, Bosly A, Pettengell R, Szucs TD, Leonard R (2006) Neutropenic event risk and impaired chemotherapy delivery in six European audits of breast cancer treatment. Support. Care Cancer 14, 901–909. [DOI] [PubMed] [Google Scholar]
- 5. Siena S, Secondino S, Giannetta L, Carminati O, Pedrazzoli P (2003) Optimising management of neutropenia and anaemia in cancer chemotherapy‐advances in cytokine therapy. Crit. Rev. Oncol. Hematol. 48, S39–S47. [DOI] [PubMed] [Google Scholar]
- 6. Wunderlich A, Kloess M, Reiser M, Rudolph C, Truemper L, Bittner S, Schmalenberg H, Schmits R, Pfreundschuh M, Loeffler M; German High‐Grade Non‐Hodgkin’s Lymphoma Study Group (DSHNHL) (2003) Practicability and acute haematological toxicity of 2‐ and 3‐weekly CHOP and CHOEP chemotherapy for aggressive non‐Hodgkin’s lymphoma: results from the NHL‐B trial of the German High‐Grade Non‐Hodgkin’s Lymphoma Study Group (DSHNHL). Ann. Oncol. 14, 881–893. [DOI] [PubMed] [Google Scholar]
- 7. Begley CG, Nicola NA, Metcalf D (1988) Proliferation of normal human promyelocytes and myelocytes after a single pulse stimulation by purified GM‐CSF or G‐CSF. Blood 71, 640–645. [PubMed] [Google Scholar]
- 8. Christopher MJ, Link DC (2007) Regulation of neutrophil homeostasis. Curr. Opin. Hematol. 14, 3–8. [DOI] [PubMed] [Google Scholar]
- 9. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A (1992) Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80, 2012–2020. [PubMed] [Google Scholar]
- 10. Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, Tosato G (2006) G‐CSF down‐regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood 108, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lord BI, Bronchud MH, Owens S, Chang J, Howell A, Souza L, Dexter TM (1989) The kinetics of human granulopoiesis following treatment with granulocyte colony‐stimulating factor in vivo . Proc. Natl Acad. Sci. USA 86, 9499–9503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mackey MC, Aprikyan AA, Dale DC (2003) The rate of apoptosis in post mitotic neutrophil precursors of normal and neutropenic humans. Cell Prolif. 36, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Souza LM, Boone TC, Gabrilove J, Lai PH, Zsebo KM, Murdock DC, Chazin VR, Bruszewski J, Lu H, Chen KK, Barendt J, Platzer E, Moore MAS, Mertelsmann R, Welte K (1986) Recombinant human granulocyte colony‐stimulating factor: effects on normal and leukemic myeloid cells. Science 232, 61–65. [DOI] [PubMed] [Google Scholar]
- 14. Borleffs JC, Bosschaert M, Vrehen HM, Schneider MM, Van Strijp J, Small MK, Borkett KM (1998) Effect of escalating doses of recombinant human granulocyte colony‐stimulating factor (filgrastim) on circulating neutrophils in healthy subjects. Clin. Ther. 20, 722–736. [DOI] [PubMed] [Google Scholar]
- 15. Bronchud MH, Potter MR, Morgenstern G, Blasco MJ, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, Testa NG, Dexter TM (1988) In vitro and in vivo analysis of the effects of recombinant human granulocyte colony‐stimulating factor in patients. Br. J. Cancer 58, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El Ouriaghli F, Fujiwara H, Melenhorst JJ, Sconocchia G, Hensel N, Barrett AJ (2003) Neutrophil elastase enzymatically antagonizes the in vitro action of G‐CSF: implications for the regulation of granulopoiesis. Blood 101, 1752–1758. [DOI] [PubMed] [Google Scholar]
- 17. Ericson SG, Gao H, Gericke GH, Lewis LD (1997) The role of polymorphonuclear neutrophils (PMNs) in clearance of granulocyte colony‐stimulating factor (G‐CSF) in vivo and in vitro . Exp. Hematol. 25, 1313–1325. [PubMed] [Google Scholar]
- 18. Hunter MG, Druhan LJ, Massullo PR, Avalos BR (2003) Proteolytic cleavage of granulocyte colony‐stimulating factor and its receptor by neutrophil elastase induces growth inhibition and decreased cell surface expression of the granulocyte colony‐stimulating factor receptor. Am. J. Hematol. 74, 149–155. [DOI] [PubMed] [Google Scholar]
- 19. Khwaja A, Carver J, Jones HM, Paterson D, Linch DC (1993) Expression and dynamic modulation of the human granulocyte colony‐stimulating factor receptor in immature and differentiated myeloid cells. Br. J. Haematol. 85, 254–259. [DOI] [PubMed] [Google Scholar]
- 20. Kotto‐Kome AC, Fox SE, Lu W, Yang BB, Christensen RD, Calhoun DA (2004) Evidence that the granulocyte colony‐stimulating factor (G‐CSF) receptor plays a role in the pharmacokinetics of G‐CSF and PegG‐CSF using a G‐CSF‐R. KO model. Pharmacol. Res. 50, 55–58. [DOI] [PubMed] [Google Scholar]
- 21. Tanaka H, Tokiwa T (1990) Influence of renal and hepatic failure on the pharmacokinetics of recombinant human granulocyte colony‐stimulating factor (KRN8601) in the rat. Cancer Res. 50, 6615–6619. [PubMed] [Google Scholar]
- 22. Crawford J (2002) Clinical uses of pegylated pharmaceuticals in oncology. Cancer Treat. Rev. 28(Suppl. A), 7–11. [DOI] [PubMed] [Google Scholar]
- 23. Tanaka H, Satake‐Ishikawa R, Ishikawa M, Matsuki S, Asano K (1991) Pharmacokinetics of recombinant human granulocyte colony‐stimulating factor conjugated to polyethylene glycol in rats. Cancer Res. 51, 3710–3714. [PubMed] [Google Scholar]
- 24. Yang BB, Lum PK, Hayashi MM, Roskos LK (2004) Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J. Pharm. Sci. 93, 1367–1373. [DOI] [PubMed] [Google Scholar]
- 25. Grigg A, Solal‐Celigny P, Hoskin P, Taylor K, McMillan A, Forstpointner R, Bacon P, Renwick J, Hiddemann W (2003) Open‐label, randomized study of pegfilgrastim vs. daily filgrastim as an adjunct to chemotherapy in elderly patients with non‐Hodgkin’s lymphoma. Leuk. Lymphoma 44, 1503–1508. [DOI] [PubMed] [Google Scholar]
- 26. Holmes FA, Jones SE, O’Shaughnessy J, Vukelja S, George T, Savin M, Richards D, Glaspy J, Meza L, Cohen G, Dhami M, Budman DR, Hackett J, Brassard M, Yang BB, Liang BC (2002) Comparable efficacy and safety profiles of once‐per‐cycle pegfilgrastim and daily injection filgrastim in chemotherapy‐induced neutropenia: a multicenter dose‐finding study in women with breast cancer. Ann. Oncol. 13, 903–909. [DOI] [PubMed] [Google Scholar]
- 27. Pinto L, Liu Z, Doan Q, Bernal M, Dubois R, Lyman G (2007) Comparison of pegfilgrastim with filgrastim on febrile neutropenia, grade IV neutropenia and bone pain: a meta‐analysis of randomized controlled trials. Curr. Med. Res. Opin. 23, 2283–2295. [DOI] [PubMed] [Google Scholar]
- 28. Schippinger W, Holub R, Dandachi N, Bauernhofer T, Samonigg H (2006) Frequency of febrile neutropenia in breast cancer patients receiving epirubicin and docetaxel/paclitaxel with colony‐stimulating growth factors: a comparison of filgrastim or lenograstim with pegfilgrastim. Oncology 70, 290–293. [DOI] [PubMed] [Google Scholar]
- 29. Engel C, Scholz M, Loeffler M (2004) A computational model of human granulopoiesis to simulate the hematotoxic effects of multicycle polychemotherapy. Blood 104, 2323–2331. [DOI] [PubMed] [Google Scholar]
- 30. Scholz M, Engel C, Loeffler M (2005) Modelling human granulopoiesis under poly‐chemotherapy with G‐CSF support. J. Math. Biol. 50, 397–439. [DOI] [PubMed] [Google Scholar]
- 31. Ziepert M, Schmits R, Trumper L, Pfreundschuh M, Loeffler M (2008) Prognostic factors for hematotoxicity of chemotherapy in aggressive non‐Hodgkin’s lymphoma. Ann. Oncol. 19, 752–762. [DOI] [PubMed] [Google Scholar]
- 32. Scholz M, Engel C, Loeffler M (2006) Model‐based design of chemotherapeutic regimens that account for heterogeneity in leucopoenia. Br. J. Haematol. 132, 723–735. [DOI] [PubMed] [Google Scholar]
- 33. Ihaka R, Gentleman R (1996) R. a language for data analysis and graphics. J. Comp. Graph. Stat. 5, 299–314. [Google Scholar]
- 34. Falanga A, Marchetti M, Evangelista V, Manarini S, Oldani E, Giovanelli S, Galbusera M, Cerletti C, Barbui T (1999) Neutrophil activation and hemostatic changes in healthy donors receiving granulocyte colony‐stimulating factor. Blood 93, 2506–2514. [PubMed] [Google Scholar]
- 35. Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ (2001) Vascular cell adhesion molecule‐1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony‐stimulating factor. Blood 98, 1289–1297. [DOI] [PubMed] [Google Scholar]
- 36. Shimazaki C, Uchiyama H, Fujita N, Araki S, Sudo Y, Yamagata N, Ashihara E, Goto H, Inaba T, Haruyama H, (1995) Serum levels of endogenous and exogenous granulocyte colony‐stimulating factor after autologous blood stem cell transplantation. Exp. Hematol. 23, 1497–1502. [PubMed] [Google Scholar]
- 37. Kuwabara T, Uchimura T, Takai K, Kobayashi H, Kobayashi S, Sugiyama Y (1995) Saturable uptake of a recombinant human granulocyte colony‐stimulating factor derivative, nartograstim, by the bone marrow and spleen of rats in vivo . J. Pharmacol. Exp. Ther. 273, 1114–1122. [PubMed] [Google Scholar]
- 38. Dale DC (2002) Colony‐stimulating factors for the management of neutropenia in cancer patients. Drugs 62(Suppl. 1), 1–15. [DOI] [PubMed] [Google Scholar]
- 39. Tanaka H, Okada Y, Kawagishi M, Tokiwa T (1989) Pharmacokinetics and pharmacodynamics of recombinant human granulocyte‐colony stimulating factor after intravenous and subcutaneous administration in the rat. J. Pharmacol. Exp. Ther. 251, 1199–1203. [PubMed] [Google Scholar]
- 40. Bodey GP, Buckley M, Sathe YS, Freireich EJ (1966) Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann. Intern. Med. 64, 328–340. [DOI] [PubMed] [Google Scholar]
- 41. Kuwabara T, Kato Y, Kobayashi S, Suzuki H, Sugiyama Y (1994) Nonlinear pharmacokinetics of a recombinant human granulocyte colony‐stimulating factor derivative (nartograstim): species differences among rats, monkeys and humans. J. Pharmacol. Exp. Ther. 271, 1535–1543. [PubMed] [Google Scholar]
- 42. Kuwabara T, Kobayashi S, Sugiyama Y (1996) Pharmacokinetics and pharmacodynamics of a recombinant human granulocyte colony‐stimulating factor. Drug Metab. Rev. 28, 625–658. [DOI] [PubMed] [Google Scholar]
- 43. Kuwabara T, Uchimura T, Kobayashi H, Kobayashi S, Sugiyama Y (1995) Receptor‐mediated clearance of G‐CSF derivative nartograstim in bone marrow of rats. Am. J. Physiol. 269, E1–E9. [DOI] [PubMed] [Google Scholar]
- 44. Layton JE, Hockman H, Sheridan WP, Morstyn G (1989) Evidence for a novel in vivo control mechanism of granulopoiesis: mature cell‐related control of a regulatory growth factor. Blood 74, 1303–1307. [PubMed] [Google Scholar]
- 45. Sarkar CA, Lauffenburger DA (2003) Cell‐level pharmacokinetic model of granulocyte colony‐stimulating factor: implications for ligand lifetime and potency in vivo . Mol. Pharmacol. 63, 147–158. [DOI] [PubMed] [Google Scholar]
- 46. Roskos LK, Lum P, Lockbaum P, Schwab G, Yang BB (2006) Pharmacokinetic/pharmacodynamic modeling of pegfilgrastim in healthy subjects. J. Clin. Pharmacol. 46, 747–757. [DOI] [PubMed] [Google Scholar]
- 47. Wang B, Ludden TM, Cheung EN, Schwab GG, Roskos LK (2001) Population pharmacokinetic‐pharmacodynamic modeling of filgrastim (r‐metHuG‐CSF) in healthy volunteers. J. Pharmacokinet. Pharmacodyn. 28, 321–342. [DOI] [PubMed] [Google Scholar]
- 48. Hayashi N, Aso H, Higashida M, Kinoshita H, Ohdo S, Yukawa E, Higuchi S (2001) Estimation of rhG‐CSF absorption kinetics after subcutaneous administration using a modified Wagner‐Nelson method with a nonlinear elimination model. Eur J. Pharm. Sci. 13, 151–158. [DOI] [PubMed] [Google Scholar]
- 49. Steinman RA, Tweardy DJ (1994) Granulocyte colony‐stimulating factor receptor mRNA upregulation is an immediate early marker of myeloid differentiation and exhibits dysfunctional regulation in leukemic cells. Blood 83, 119–127. [PubMed] [Google Scholar]
- 50. Tkatch LS, Rubin KA, Ziegler SF, Tweardy DJ (1995) Modulation of human G‐CSF receptor mRNA and protein in normal and leukemic myeloid cells by G‐CSF and retinoic acid. J. Leukoc. Biol. 57, 964–971. [DOI] [PubMed] [Google Scholar]
- 51. Scholz M, Ackermann M, Emmrich F, Loeffler M, Kamprad M (2009) Effectiveness of cytopenia prophylaxis for different Filgrastim and Pegfilgrastim schedules in a chemotherapy mouse model. Biologics: Targets & Therapy 3, 27–37. [PMC free article] [PubMed] [Google Scholar]
- 52. Mukae H, Zamfir D, English D, Hogg JC, Van Eeden SF (2000) Polymorphonuclear leukocytes released from the bone marrow by granulocyte colony‐stimulating factor: intravascular behavior. Hematol. J. 1, 159–171. [DOI] [PubMed] [Google Scholar]