

Abstract
Objectives: We investigated the antiproliferative effect of Myc down‐regulation via cell proliferation inhibition, cell cycle perturbation and apoptosis in two human astrocytoma models (T98G and ADF) steadily expressing an inducible c‐myc Anti‐sense RNA.
Materials and methods: Cell growth experiments were performed using the trypan blue dye exclusion test and cell cycle analysis was evaluated by flow cytometry. Cell cycle molecules were detected by Western blot analysis, co‐immunoprecipitation and reverse transcription–polymerase chain reaction assays.
Results: We showed that Myc down‐regulation in astrocytoma cells led to G1 accumulation and an inhibition of cell proliferation characterized by S phase delay. Co‐immunoprecipitation experiments detected formation of inactive cyclin D1/cdk4 complexes as evaluated by presence of an active unphosphorylated form of retinoblastoma protein, the best characterized target substrate for cyclin D1/cdk4 complex, in ADF pINDc‐myc anti‐sense 7 cells. We also found that either p57Kip2 “apice” or p27Kip1 “apice” inhibitors bound to cyclin D1/cdk4 complex, thus, suggesting that they cooperated to inhibit the activity of cyclin D1/cdk4. Moreover, c‐Myc down‐regulation led to activation of the apoptotic mitochondrial pathway, characterized by release of cytochrome c and Smac/Diablo proteins and by reduction of c‐IAP levels through activation of proteasome‐mediated protein degradation system.
Conclusions: Our results suggest that c‐Myc could be considered as a good target for the study of new approaches in anticancer astrocytoma treatment.
Introduction
Astrocytoma tumours are the most important causes of cancer‐related mortality in children. Even though chemotherapy is the best known treatment of astrocytoma (1), the commonly used antitumor therapeutic agents have provided disappointing results (2, 3, 4). The dismal outcome of standard therapy has led to exploration of new approaches for treatment of childhood astrocytoma cancer. Today, rational drug design, targeting proteins involved in regulation of cell proliferation and/or apoptosis, is one major focus for development of novel cancer therapies. Moreover, treatments with compounds interfering with cell signalling pathways used alone or in combination with conventional anticancer therapy, could be particularly promising. In this context, therapeutic interest in c‐Myc protein is undeniable; in fact, its expression and activity are closely related to cancer aggression. In particular, c‐Myc in astrocytoma is overexpressed and generally associated with an undifferentiated tumour state and poor prognosis (5, 6).
The c‐Myc protein, a basic helix–loop–helix/leucine zipper‐type transcription factor, is a master regulator in a number of cellular pathways, including for cell growth and proliferation, metabolism, and apoptosis (7). The Myc protein family influences expression of about 10% of all human genes, with either positive or negative effects on gene transcription (8). Cyclin‐dependent kinases (cdk) are cell cycle checkpoint regulators of downstream c‐Myc activity. In particular, orderly progression of cells through the G1 phase and G1/S transition is driven by association of cyclin D1 and cdk4, and cyclin E and cdk2. The cyclin D1/cdk4 complex regulates activity of retinoblastoma protein, the only known target of the cyclin D1/cdk4 complex. Activity of retinoblastoma protein depends on its phosphorylation state; the hypophosphorylated form is active in cell population growth suppression (G1 phase), whereas the hyperphosphorylated form is inactive in suppressing cell growth (S, G2 and M phases). In particular, during the G1 phase of the cell cycle, unphosphorylated retinoblastoma protein is associated with the E2F transcription factor family, inhibiting gene expression required for S transition (9).
Regulation of G1 cyclin D1/cdk4 activity is also dependent on cdk4 inhibitory protein, which can bind to and inactivate the cyclin D1/cdk4 complex (10). Cyclin D1/cdk4 complex sequesters p21Cip1 and p27Kip1, a Cip/Kip family of protein inhibitors, prevents them from interaction and consequent inhibition of cyclin E/cdk2 and cyclin A/cdk2 (which determine progression through the S phase) (9, 11). Although p21Cip1 and p27Kip1 clearly inhibit cdk2 activity, their effects on cdk4 are still unresolved. Some studies suggest that p21Cip1 and p27Kip1 are obligatory assembling factors for cyclin D/cdk4 complexes (12), and that cyclin D/cdk4 complexes, including cyclin D1/cdk4, containing a single p21Cip1 or p27Kip1, are active (13, 14, 15). In contrast, other studies have shown that cyclin D1/cdk4 complexes containing p21Cip1 or p27Kip1 are always inactive (16). Moreover, the cyclin D1/cdk4 complex binds to another inhibitor of Cip/Kip family: p57Kip2 (17).
It has also been reported that c‐Myc stimulates expression of cyclin A, cyclin D1, cyclin E, cdk2 and cdk4, and represses expression of p21Cip1 and p27Kip1 proteins (18, 19). On the contrary, Guo et al. have reported that transcription of cyclin D1 is repressed by c‐myc (19). In addition, array analyses and chromatin immunoprecipitation experiments suggest that p57Kip2 can also be repressed by Myc expression (20). On the whole, although c‐myc affects normal and neoplastic cell proliferation by altering gene expression, the precise pathways involved remain unclear (18).
It has been reported that c‐Myc also plays a key role in a number of apoptotic pathways (1). Experimental studies have recently shown that c‐Myc, by inducing apoptosis via the mitochondria‐mediated pathway, causes release of several pro‐apoptotic factors, including cytochrome c and apoptosis‐inducing factor, with consequent caspase‐3 activation (21, 22, 23). In particular, cytochrome c binds to apoptosis‐activating‐factor‐1 to activate caspase‐9 and subsequently caspase‐3, in caspase‐3‐dependent apoptosis (24). However, caspase‐3 activation is also regulated by expression of Second Mitochondria‐Derived Activator (Smac/Diablo). Smac/Diablo is released from the mitochondria along with cytochrome c during apoptosis, and promotes caspase‐3 activation by inhibiting the c‐IAP (inhibitors of apoptosis) family of proteins (25, 26).
Data reported here show that induction of c‐myc anti‐sense (As) RNA in both T98G and ADF astrocytoma cells led to the same extent of cell growth inhibition, and that Myc down‐regulation affected the cell cycle pathway by modulating several crucial target proteins, including cyclins, cdk and cyclin‐dependent kinase inhibitors (ckis). By switching off c‐myc expression, the G1/S checkpoint and S phase cell progression were affected. We also found that in Myc down‐regulated cells, although cyclin D1 and cdk4 were assembled, the complex was nonfunctional. Inhibition of cyclin D1/cdk4 complex activity could be ascribed to Cip/Kip family proteins, since we found that either p57Kip2 or p27Kip1 inhibitors were bound to the cyclin D1/cdk4 complex.
We have also shown that down‐regulation of c‐Myc promotes the mitochondrial intrinsic apoptotic pathway by rendering astrocytoma cells more prone to apoptosis, thanks to release of cytochrome c and Smac/Diablo and reduction of c‐IAP levels. Smac/Diablo negatively regulated c‐IAP1 and c‐IAP2 proteins and then directly activated caspase‐3, allowing ADF cells to overcome their defective mitochondrial apoptotic pathway caused by presence of a mutated form of caspase‐9 (27).
Materials and Methods
Culture conditions, transfection, treatments and cell population growth
The T98G human glioblastoma cell line (glioblastoma multiforme) was cultured in RPMI 1640 (Cambrex Corp., East Rutherford, NJ, USA) and ADF (Grade IV astrocytoma) in Dulbecco's modified Eagle's medium (DMEM; Cambrex) supplemented with foetal calf serum 10% (Life Technologies, Paisley, UK), penicillin (100 µg/ml), streptomycin (100 µg/ml) and l‐glutamine (2 mm) at 37 °C in a 5% CO2/95% air atmosphere. RPMI medium was also added with 1 mm sodium pyruvate and 1× nonessential amino acids.
T98G pINDc‐myc As and ADF pINDc‐myc As clones were generated as previously described (28). Both cell lines were also transfected with empty vector (pINDneo). Generation of stable c‐myc As (Anti‐sense) inducible expression clones was achieved by using the Ecdysone‐Inducible Expression System (Invitrogen, San Diego, CA, USA). Two micrograms of double‐stranded DNA of pINDc‐myc As and pVgRXR plasmids were mixed in Optimem (Gibco, Invitrogen, Grand Island, NY, USA) with 10 µl of lipofectamine reagent (Gibco) and incubated, for 6 h, with both 1 × 105 T98G and ADF cells the day after seeding, according to the manufacturer's instructions. RPMI and DMEM fresh media were supplemented with 10% fetal calf serum and after 48 h culturing, both the transfected cell lines were maintained for 4 weeks in the presence of either G418 (Gibco) or Zeocyn (Invitrogen). G418/Zeocyn‐resistant clones were isolated and selected from pINDc‐myc As and from pINDneo transfected cells.
To perform siRNA silencing experiments, 1 × 105 ADF cells were grown in complete DMEM; after 24 h culturing, cells were transfected with siRNA oligonucleotides for c‐myc with two thymidine residues (dTdT) at the 3′ end of the sequence (sense, 5′‐AACAGAAAUGUCCUGAGCAAU‐3′) (MWG Biotech AG, Ebersberg, Germany). Transfection was performed by incubating cells with 200 pmol of siRNAs in 1 ml of Optimem and with 10 µl of lipofectamine reagent. After 6 h of incubation, transfection medium was replaced with fresh complete DMEM and then the cells were continually exposed to bromodeoxyuridine (BrdUrd) for 48 h and subsequently processed. In the same experimental conditions, samples were processed for apoptosis and Western blot analysis, as described below.
To determine the optimal schedule of hormone treatment, pINDc‐myc As clones from both cell lines were exposed to different dosages (5 µm, 10 µm, 15 µm, 20 µm and 25 µm) of ponasterone A (Invitrogen) given every 24 h and scored for hormone‐inducible cell growth inhibition. At each time‐point, from day 2 (24 h after induction) to day 5 (96 h after induction), cells were harvested and counted by using the trypan blue dye exclusion test (ICN Biomedicals Inc., Aurora, OH, USA). Ponasterone treatment was also added to the pINDneo control cells.
In experiments with caspase‐3 inhibitor, Ac‐DEVD‐cho (Sigma‐Aldrich Logistik GmbH) was used at 50 µm concentration (which had no toxic effect on the cell growth) and added to the culture medium concomitantly with ponasterone treatment. The inhibitor was left in the culture medium until time of analysis.
In the experiments with proteasome inhibitor, cells were treated with 0.25 µm MG132 (Calbiochem, San Diego, CA, USA) for 4 h, 24 h after ponasterone induction or 24 h after siRNA c‐myc trasfection.
RNA extraction and semiquantitative reverse transcription–polymerase chain reaction (RT‐PCR)
Total RNA was prepared from 4 × 106 cells by using the SV Total RNA Isolation Kit (Promega Corp., Madison, WI, USA). From each sample, 0.5 µg of total RNA was reverse transcribed in a 20‐µl reaction volume using dNTP Mix (0.5 mm each dNTP), Oligo‐dt primer (1 µm) with 4 units Moloney murine leukaemia virus reverse transcriptase (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. PCR was carried out in 20‐µl volume containing 0.25 mm of deoxynucleotide triphosphate, 0.25 mm of oligonucleotide primer using 3 µl of reverse transcriptase reaction mixture and 2.5 units Amplitherm Hot Start DNA Polymerase (Fisher Molecular Biology, Rovigo, Italy). Mixtures were subjected to different PCR cycles, as indicated, including the first denaturation cycle at 94 °C for 5 min; denaturation at 94 °C for 30 s; annealing at 57 °C for 30 s; and extension at 72 °C for 30 s. Amplification products (10 µl) were analysed by electrophoresis on 2% agarose gel stained with ethidium bromide. Primes and annealing temperature used were as follows: c‐myc (Hmyc 01 for ATTCTCTGCTCTCCTCGA; Hmyc02 rev TCTTGGCAGCAGGATAGT, TA = 57 °C); c‐myc As (Hmyc 03 for CTCCTCGTCGCAGTAGAA; BGH rev TAGAAGGCACAGTCGAGG, TA = 57 °C); β‐actin (sense GCGCGGCGTAGCCCCCGTCAG; anti‐sense CGCGGCAGGAAGCCAGGCCCC, TA = 57 °C); p57Kip2, GenBank accession number U22398 (p57Kip2 for CACGATGGAGCGTCTTGTC; p57Kip2 rev CCT GCTGGAAGTCGTAATCC, TA = 59 °C); H‐GAPDH (H‐GAPDH for GCAGGGGGGAGCCAAAAGGG; H‐GAPDH rev CAGCGCCAGTAGAGGCAGGG, TA = 60 °C).
Western blot and co‐immunoprecipitation analysis
Samples were solubilized in lysis buffer [10% glycerol, 1% Tween 20, 150 mm NaCl, 50 mm HEPES (pH 7.6), 0.05% Nonidet P40, 1 mm CaCl2, 100 mm NaF, 1 mm phenylmethanesulphonylfluoride (PMSF), 2 mm Na3VO4, 10 mm Na4P2O7, protease inhibitors] on ice and then briefly sonicated. Mitochondria/cytosol protein separation was obtained by mitochondria/cytosol Fractionation Kit (MBL International Corporation, Woburn, MA) following the manufacturer's instructions. Protein content in the different samples was quantified by using the Bradford method (Bio‐Rad, Richmond, CA, USA). Aliquots (70 µg) of proteins were subjected to 10–12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) and the resolved proteins were blotted on a nitrocellulose membrane, then membranes were blocked in TBS buffer (20 mm tris Base, 137 mm NaCl, 1 m hydrochloric acid, pH 7.6) containing 5% nonfat dry milk (Bio‐Rad, Hercules, CA, USA) for at least 1 h. Blots were then incubated with primary antibodies: anti‐c‐Myc (clone 9E10, BD PharMingen, San Diego, CA, USA), anti‐cyclin D1 (clone G124‐326), anti‐cyclin E (clone HE12, BD PharMingen), anti‐cyclin A (clone BF683, BD PharMingen), anti‐cdk4 (clone DCS‐156, BD PharMingen), anti‐cdk2 (clone 55, BD PharMingen), anti‐underphosphorylated retinoblastoma protein (clone G99‐549, BD PharMingen), anti‐retinoblastoma protein (clone C‐15, Santa Cruz Biotechnology), anti‐cytochrome c (clone 7H8.2C12, BD PharMingen), anti‐Smac (clone FL‐239, Santa Cruz Biotechnology), anti‐c‐IAP1 (clone H‐83, Santa Cruz Biotechnology), anti‐c‐IAP2 (clone F‐20, Santa Cruz Biotechnology), anti‐caspase‐3 (clone N‐19, Santa Cruz Biotechnology), anti‐cleaved‐caspase‐3 (clone h176, Santa Cruz Biotechnology), anti‐β‐actin (clone AC‐15, Sigma, St. Louis, MO, USA) anti‐COX‐4 (clone 20E8, Santa Cruz Biotechnology), and anti‐β‐tubulin (clone 5H1, BD PharMingen). Peroxidase‐labelled anti‐mouse, anti‐goat and anti‐rabbit IgG (Sigma‐Aldrich) were used as secondary antibodies. Immunoblots were processed for enhanced chemiluminescence detection (Amersham Life Sciences, Buckinghamshire, UK). The relative amount of transferred protein in a given sample was quantified by scanning X‐ray films and by densitometry analysis (TotalLab image analysis solution, version 2003, Nonlinear Dynamics, Newcastle upon Tyne, UK). In the co‐immunoprecipitation experiments, samples were lysed in low stringency lysis buffer (1% Nonidet P40, 0.2 mm PMSF, 10 mm NaF, 0.7 µg/ml pepstatine, 25 µg/ml aprotinine, protease inhibitors). Co‐immunoprecipitation was carried out using 500 µg protein extracts and anti‐cyclin D1 (clone H‐295, Santa Cruz Biotechnology) and immunocomplexes were bound to protein A‐agarose (Sigma‐Aldrich). Resulting precipitates were analysed using 10% SDS‐PAGE and resolved proteins were blotted to a nitrocellulose membrane. Blots were incubated with anti‐cdk4 (clone DCS‐156, BD PharMingen), anti‐p57Kip2 (clone A120‐1, BD PharMingen), anti‐p27Kip1 (clone 57, BD PharMingen), and anti‐cyclin D1 (clone H‐295, Santa Cruz Biotechnology) antibodies and processed as described above.
Cell cycle analysis
Cell cycle analysis was performed after BrdUrd (Sigma) incorporation. Both BrdUrd continuous‐labelling and pulse‐chase experiments were carried out. For BrdUrd continuous‐labelling experiments, 10 µm BrdUrd was continuously added to the medium for 48 h starting 24 h after hormone induction (given every 24 h) or 24 h after the end of c‐myc siRNA trasfection. Pulse‐labelling experiments were performed at 24 h after hormone induction, by adding 10 µm BrdUrd to the medium for the last 30 min of Myc induction (chase 0). Then the medium was changed and analysis was performed at 2, 4, 6, 8, 10 and 12 h post‐labelling.
At the indicated times cells were harvested, washed once in phosphate‐buffered saline (PBS), fixed in 70% ethanol and stored at 4 °C until analysis. Then, cell suspensions were rinsed with cold PBS and incubated with 4 N HCl for 20 min at room temperature to partially denature DNA. Cells were washed twice with borax‐borate buffer (pH = 9.1) to neutralize the acid pH and once with PBS. Samples were incubated with mouse monoclonal antibody anti‐BrdUrd (Boehringer Mannheim, Monza, Italy) diluted 1 : 50 in complete medium containing 0.5% Tween‐20 (Calbiochem, San Diego, CA, USA) at 4 °C for 1 h. After washing twice in PBS, cells were exposed to FITC‐conjugated F(ab′) rabbit anti‐mouse (Dako SA, Glostrup, Denmark) at dilution 1 : 20 in PBS at 4 °C for 1 h. Finally, the cells were washed twice with PBS, and stained with a solution containing 5 mg/ml propidium podide (PI) and 75 KU/ml RNase in PBS for 30 min at room temperature. Samples that had not been incubated with anti‐BrdUrd monoclonal antibody were used as negative controls.
For both continuous and pulse‐labelling experiments, 3 × 104 events/sample were acquired using a FACScan cytofluorimeter (Becton Dickinson, Sunnyvale, CA, USA). The analysis was performed using a CellQuest software package (Becton Dickinson).
Apoptosis detection
For apoptosis, cells were fixed in 80% ethanol and stained with PI (50 mg/ml, MP Biomedicals, Eschwege, Germany) in PBS containing RNase A (75 KU/ml, Sigma Chemical). It was evaluated as percentage of apoptotic cells in the sub‐G1 region (sub‐G1 peak) of flow cytometric DNA content distribution.
Induction of apoptosis was evaluated by terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay (Roche Diagnostics GmbH, Mannheim, Germany) also by using flow cytometry. The TUNEL assay was performed as previously described (29) and experiments were assessed at the indicated times.
Statistical analysis
Statistical significance of differences between groups was tested using the paired Student's t‐test or, if there were more than two groups, by one‐way analysis of variance followed by the Tukey post‐hoc test.
Results
Inducible expression of c‐myc anti‐sense RNA inhibited cell population growth in astrocytomas
To investigate whether c‐myc anti‐sense RNA inhibits cell growth in human astrocytomas, we used two human astrocytoma cell lines expressing high levels of c‐Myc, which is considered to play an important role in aggression of growing astrocytomas. Protein levels of c‐Myc in T98G and ADF cell lines are shown in Fig. 1a (30, 31).
Figure 1.
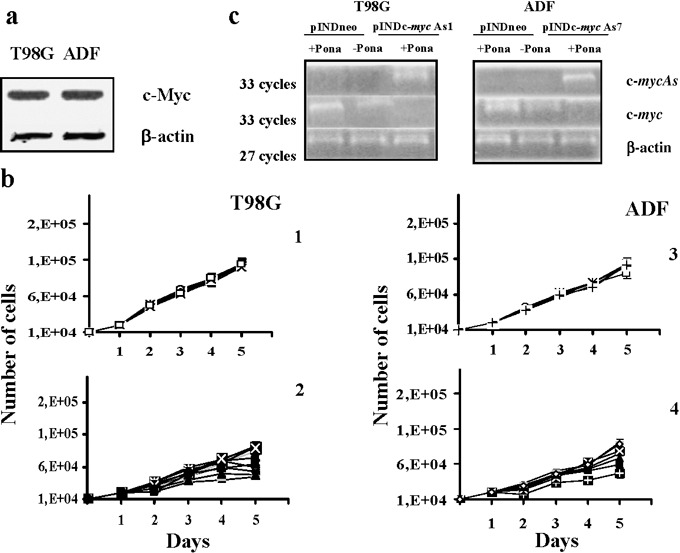
(a) Western blot analysis of c‐Myc protein expression on both T98G and ADF cell lines. Each lane was loaded with 70 µg of proteins from cell lysate. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results. (b) Growth curves of both T98G and ADF pINDneo control and pINDc‐myc As clones. pINDneo control clones were transfected with the empty vector (1, 3), and pINDc‐myc As clones (2, 4) were treated with 25 µm of ponasterone given every 24 h. At the indicated times, from day 2 (24 h after induction) to day 5 (96 h after induction), cells were harvested and counted by using the trypan blue dye exclusion test. All data are expressed as means ± standard deviation of three independent experiments with similar results. Statistical significance of data was evaluated by one‐way analysis of variance followed by Tukey post‐hoc test. (c) Representative RT‐PCR analysis of ectopic c‐myc As and endogenous c‐myc in pINDneo control and pINDc‐myc As transfected cells. Clones were exposed for 24 h to 25 µm of ponasterone. c‐myc (Hmyc 01 for ATTCTCTGCTCTCCTCGA; Hmyc02 rev TCTTGGCAGCAGGATAGT); c‐myc As (Hmyc 03 for CTCCTCGTCGCAGTAGAA; BGH rev TAGAAGGCACAGTCGAGG); β‐actin (sense GCGCGGCGTAGCCCCCGTCAG; anti‐sense CGCGGCAGGAAGCCAGGCCCC). β‐actin c‐DNA was used as an internal control. The experiment was repeated three times showing similar results.
To reduce levels of Myc protein expression, we established both T98G and ADF cells steadily expressing a hormone‐inducible c‐myc As RNA. Both cell lines were co‐transfected with either the pINDc‐myc As construct or the pINDneo empty vector, together with the pVgRXR plasmid, encoding the ecdysone receptor and carrying zeocyn resistance. G418/zeocyn‐resistant clones were isolated under hormone‐free conditions after a selection period of 4 weeks. Ten T98G pINDc‐myc As clones and nine ADF pINDc‐myc As clones were obtained. These clones were treated with 25 µm of hormone (ponasterone A) given every 24 h and scored for hormone‐inducible cell growth inhibition (Fig. 1b). The figure reports growth curves for both T98G and ADF pINDneo control clones transfected with the empty vector pINDneo (1, 3) and both the hormone‐treated T98G and ADF pINDc‐myc As cells (2, 4). All T98G and ADF pINDc‐myc As clones displayed cell growth reduction, after 1 day of induction with the hormone (24 h after treatment), ranging from 18% to 53% and 19% to 57%; in contrast, control clones from both cell lines did not show any significant inhibition on cell growth when exposed to the same ponasterone concentration.
To validate quality of the system and to verify whether ponasterone was really able to activate expression of our target gene, levels of total RNA transcripts of both ectopic c‐myc As and endogenous c‐myc were determined by semiquantitative RT‐PCR analysis, using suitable pairs of primers as previously described (28). Three pINDc‐myc As clones were chosen for T98G and ADF cell lines among the multiple independent clones obtained, since they showed the highest inhibition of cell growth upon 25 µm hormone‐exposure. Figure 1c shows RT‐PCR results relative to only T98G pINDc‐myc As1 and ADF pINDc‐myc As7 clones. The results obtained by using other T98G pINDc‐myc As and the ADF pINDc‐myc As clones were similar to those in the T98G pINDc‐myc As1 and ADF pINDc‐myc As7, respectively (data not shown).
As shown in Fig. 1c, PCR‐amplified c‐myc As cDNA was only detectable in ponasterone‐treated pINDc‐myc As clones in both cell lines; on the contrary, the PCR‐amplified c‐myc cDNA was not detectable in the same clones. The pINDc‐myc As cells not exposed to ponasterone and hormone‐treated control cells in both cell lines showed comparable levels of the PCR‐amplified c‐myc cDNA. β‐actin was used as an internal control for the amount of RNA used in RT‐PCRs.
To determine the optimal schedule of hormone treatment, the three pINDc‐myc As clones from T98G and ADF cell lines were exposed to different concentrations of ponasterone A (5, 10, 15, 20 and 25 µm). One of the pINDneo control clones for each cell line was used as a control (Fig. 2). Dosage of 25 µm ponasterone repeated every 24 h confirmed its maximum growth inhibitory effect in three T98G and ADF pINDc‐myc As clones compared to the control. A significant anti‐proliferative effect 24 h after hormone exposure (about 52–55% and 55–57% in T98G pINDc‐myc As clones and ADF pINDc‐myc As clones, respectively) was observed. This persisted until 96 h after Myc induction in all the clones. Similar cell growth inhibitory effects were observed after 20 µm ponasterone dosage in three T98G and ADF pINDc‐myc As clones. In contrast, after addition of 15 and 10 µm doses, we observed a modest anti‐proliferative effect (28% and 23%, respectively, either in T98G and in ADF pINDc‐myc As clones) at 24 h. These inhibitory effects were maintained after 96 h of hormone induction in all clones (16–18% and 30–32%). When pINDc‐myc As clones were exposed to the lower ponasterone dose of 5 µm, neither exposed cell line elicited significant inhibition of cell growth up to 96 h after induction. Considering the super‐imposable results in both cell lines, we have chosen the ADF pINDc‐myc As clones for further experiments.
Figure 2.
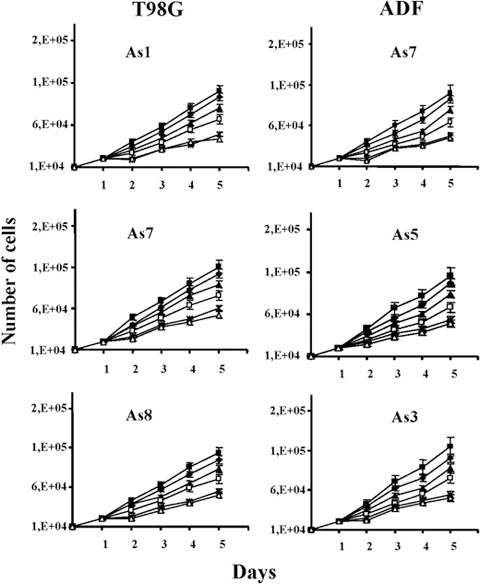
Determination of the optimal schedule of hormone treatment in both T98G and ADF pINDc‐myc As clones. Cell growth number effects in both T98G and ADF clones exposed to 5 ( ), 10 (
), 10 ( ), 15 (
), 15 ( ), 20 (*) and 25 µm (
), 20 (*) and 25 µm ( ) dosages of ponasterone given every 24 h. As control we used pINDneo () for each cell line. At the indicated times, from day 2 (24 h after induction) to day 5 (96 h after induction), cells were harvested and counted by using trypan blue dye exclusion. All data are expressed as means ± standard deviation of three independent experiments with similar results. Statistical significance of the data was evaluated by one‐way analysis of variance followed by Tukey post‐hoc test.
) dosages of ponasterone given every 24 h. As control we used pINDneo () for each cell line. At the indicated times, from day 2 (24 h after induction) to day 5 (96 h after induction), cells were harvested and counted by using trypan blue dye exclusion. All data are expressed as means ± standard deviation of three independent experiments with similar results. Statistical significance of the data was evaluated by one‐way analysis of variance followed by Tukey post‐hoc test.
Inhibition of cell proliferation observed after pINDc‐myc As cells induction with 5, 10, 15, 20 and 25 µm dosages was consistent with the reduction of Myc protein expression, as detected by Western blot analysis performed at 24 h after induction (Fig. 3). Indeed, we observed dose‐dependent Myc protein down‐regulation; amount of c‐Myc protein in the clones pINDc‐myc As7, As5 and As3 were about 8, 7 and 6.5 times lower than that of pINDneo control clone after 25 and 20 µm doses. On the contrary, no significant reduction in the level of Myc protein after exposure of pINDc‐myc As clones to 5 µm ponasterone was observed.
Figure 3.

Dose‐dependent c‐Myc protein down‐regulation. Western blot analysis of c‐Myc protein in both ADF pINDneo control cells and ADF pINDc‐myc As clones exposed to 5, 10, 15, 20 and 25 µm dosages 24 h after hormone induction. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
All the subsequent experiments based on these results were conducted using 20 µm ponasterone to efficiently down‐regulate Myc protein.
Cell cycle response to c‐myc down‐regulation
Cell cycle effects on Myc down‐regulation were investigated in the logarithmically expanding ADF pINDc‐myc As7, As5 and As3 clones by using BrdUrd incorporation and flow cytometry. Myc down‐regulation blocked the exit of cells from G1 phase. Indeed, 24 h after continuous BrdUrd labelling time, during which the entire cycling cell population was labelled, a high percentage of BrdUrd‐negative cells (65%, 60% and 62%, in ADF pINDc‐myc As7, As5 and As3, respectively) was still present in G1 (48 h after hormone induction), but in pINDneo control clone all cells were labelled by BrdUrd (Fig. 4).
Figure 4.
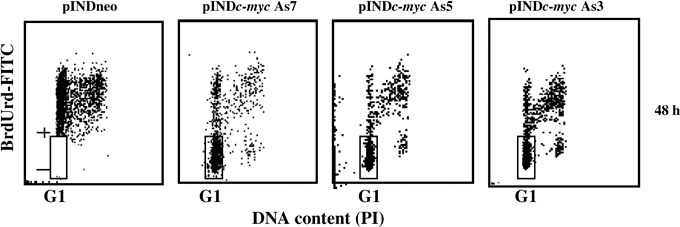
Cell cycle analysis after continuous bromodeoxyuridine (BrdUrd) incorporation in ADF pINDneo control and ADF pINDc‐myc As clones. The dosage of 10 µm BrdUrd was added continuously to the medium for 48 h starting from 24 h of hormone induction (dosage 20 µm given every 24 h). The regions represent BrdUrd‐negative G1 cells. Samples were acquired using a FACScan cytofluorimeter (Becton Dickinson). Biparametric flow cytometry analysis was performed using the CellQuest software package. The experiment was repeated three times showing similar results.
We then performed a pulse chase experiment giving BrdUrd during the last 30 min (0 h chase) of Myc induction (24 h of hormone treatment). Then, to follow the fate of S phase cells (BrdUrd‐positive cells) and their movement through cell cycle, the pulse chase experiment was performed at 2, 4, 6, 8, 10, and 12 h post‐labelling. Only the results obtained in pINDc‐myc As7 clone are shown in Fig. 5 but similar behaviour was elicited by the pINDc‐myc As5 and As3 clones (data not shown). Immediately after the pulse chase (0 h), percentage of BrdUrd‐positive cells was similar in pINDneo control clone and pINDc‐myc As7 cells (59% and 68%, respectively). During the chase, in pINDneo control clone, the BrdUrd‐positive cells, which were in S phase at the beginning of the pulse, proceeded through S phase. Indeed, in pINDneo, BrdUrd‐positive cells moved out of S through G2 and subsequently reached G1, completing their DNA replication, mitosis and cell division. These cells are recognized in the dot plot as BrdUrd‐positive cells with an evident G1 DNA content at 8 h pulse chase (36%). Conversely, such movement is almost absent in the 8 h plot of pINDc‐myc As7 (G1 +19%) indicating that the transition of cells out of S phase, into G2 and then into G1 was delayed. For BrdUrd‐positive cells the percentages in cell cycle distribution were estimated on linear PI histograms by using the MODFIT software.
Figure 5.

Representative bromodeoxyuridine (BrdUrd) pulse‐labelling experiment in ADF pINDneo control (a) and ADF pINDc‐myc As7 clone (b). The pulse‐labelling experiment was performed at 24 h after 20 µm hormone induction, by adding 10 µm BrdUrd to the medium during the last 30 min of Myc induction (chase 0 h). Afterwards, medium of the cells was changed and they were analysed by biparametric flow cytometry at 2, 4, 6, 8, 10 and 12 h post‐labelling. During the chase, in pINDneo control clone, the BrdUrd‐positive cells, which were in S phase at the beginning of the pulse (chase 0 h), proceed through S phase, moved out of S through G2, and subsequently reached G1, completing their DNA replication, mitosis and cell division. The upper region of the cytograms represents BrdUrd‐positive cells. For BrdUrd‐positive cells, the percentages of the cell cycle distribution were estimated on linear PI histograms by using the MODFIT software. All the experiments were repeated three times showing similar results.
pINDneo, BrdUrd‐negative cells had also partly moved into S phase and some had reached the G2; on the contrary, no BrdUrd‐negative cells progressed from G1 to S during the chase in pINDc‐myc As cells.
To strengthen our results, the continuous BrdUrd labelling experiment was also performed by using transient c‐myc transduction with inhibitory siRNA. As shown in Fig. 6a, at 48 h c‐myc siRNA cells showed the same cell cycle profile as the pINDc‐myc As clones (BrdUrd‐negative cells 61%), thus confirming the key role of c‐myc in ADF cell cycle G1 checkpoint. Effectiveness of c‐myc interference was assessed by Western blot analysis. As shown in Fig. 6b, the densitometry analyses revealed reduction in Myc protein of around 7.5‐fold at 48 h.
Figure 6.
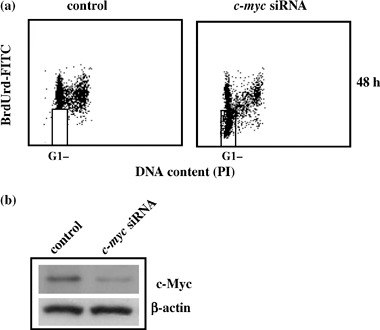
(a) c‐myc siRNA cell cycle profile. The dosage of 10 µm bromodeoxyuridine (BrdUrd) was continuously added to the medium for 48 h, after the end of transfection. c‐myc siRNA sequence (sense, 5′‐AACAGAAAUGUCCUGAGCAAU‐3′). The regions represent BrdUrd‐negative G1 cells. Samples were acquired using a FACScan cytofluorimeter (Becton Dickinson). Biparametric flow cytometry analysis was performed using the CellQuest software package. In the regions BrdUrd‐negative cells are localized. All the experiments were repeated three times showing similar results. (b) c‐myc siRNA Western blot analysis. c‐myc interference was assessed at 48 h. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
Myc down‐regulation determines the loss of cyclin E and cyclin A proteins and inhibits cyclin D1/cdk4 complex activity via p57Kip2 and p27Kip1 inhibitors
As a consequence, given the strong comparability of results obtained with the different ADF clones utilized, our further experiments were performed only on the pINDc‐myc As7 clone.
Since Myc has been widely recognized as an upstream regulator of cdk activity, we investigated whether down‐regulation of Myc protein levels could affect expression of the main molecules that control cell cycle progression, from one phase to the next in the astrocytoma ADF cell line. As shown in Fig. 7a, levels of cyclin D1, cyclin E, cyclin A, cdk4 and cdk2 in pINDc‐myc As7 cells were measured by Western blot analysis 24, 48 and 72 h after hormone exposure. As evident in the figure, 24 h after hormone induction, protein expression levels decreased in the order of 2.5‐fold in cyclin A and cdk2, and 1.5‐fold in cdk4 when compared to controls. On the other hand, levels of cyclin E were reduced to around 1.5‐fold with respect to controls 48 h after ponasterone exposure. A further decrease (about 6‐fold) of both cdk4 and cdk2 levels in the pINDc‐myc As7 clone was observed 72 h after ponasterone exposure. At the same time, loss of cyclin E and cyclin A protein expression was detected in the pINDc‐myc As clone.
Figure 7.
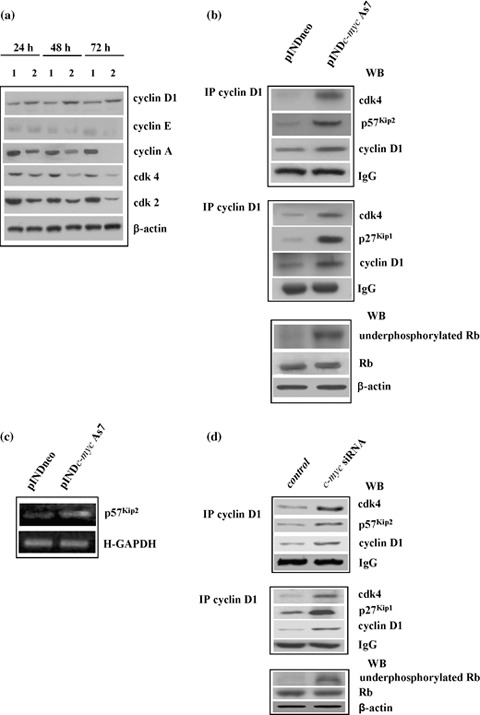
(a) Western blot analysis of cell cycle‐related proteins. ADF pINDneo control clone (1) and ADF pINDc‐myc As7 clone (2) were treated with 20 µm ponasterone (given every 24 h) 24, 48 and 72 h after treatment. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results. (b) Co‐immunoprecipitation analysis. Cyclin D1/cdk4/p57Kip2 “apice” (top panel) and cyclin D1/cdk4/p27Kip1 “apice” (medium panel) tertiary complexes were detected by co‐immunoprecipitation analysis using anti‐cyclin D1 antibody 48 h after 20 µm hormone induction in pINDneo control and ADF pINDc‐myc As7 clones. The bottom panel shows Western blot analysis of active unphosphorylated form and total retinoblastoma protein performed 48 h after 20 µm ponasterone treatment. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results. (c) RT‐PCR analysis of total RNA extracted from the control ADF pINDneo clone and the ADF pINDc‐myc As7. Clones were exposed for 24 h to 20 µm of ponasterone. p57Kip2, GenBank accession number U22398 (p57Kip2 for CACGATGGAGCGTCTTGTC; p57Kip2 rev CCT GCTGGAAGTCGTAATCC); H‐GAPDH (H‐GAPDH for GCAGGGGGGAGCCAAAAGGG; H‐GAPDH rev CAGCGCCAGTAGAGGCAGGG). Levels of transcription of p57Kip2 “apice” were normalized versus H‐GAPDH. All the experiments were repeated three times showing similar results. (d) Cyclin D1/cdk4/p57Kip2 “apice” and Cyclin D1/cdk4/p27Kip1 “apice” ternary complexes in siRNA c‐myc cells. Cyclin D1/cdk4/p57Kip2 “apice” (top panel) and cyclin D1/cdk4/p27Kip1 “apice” (medium panel) tertiary complexes were detected by co‐immunoprecipitation analysis using anti‐cyclin D1 antibody at 48 h. The bottom panel shows Western blot analysis of the active unphosphorylated form and total retinoblastoma protein. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
Nevertheless, G1 arrest is classically associated with decreased levels of either cdk4 or cyclin D1; on the contrary, in our experiments, cyclin D1 protein levels increased up to 3‐fold at 72 h in response to c‐Myc down‐regulation; the role of c‐myc in the regulation of cyclin D1 is indeed unclear: since it is well known that progression through G1 requires both formation and activation of the cyclin D1/cdk4 complex, we examined the interaction of these cell cycle molecules in Myc down‐regulated cells by co‐immunoprecipitation and Western blot assays. We also tested its activity by evaluating the status of retinoblastoma protein, the best characterized target substrate of cyclin D1/cdk4 complex, 48 h after c‐myc As RNA induction. As evident in Fig. 7b, we found that in the pINDc‐myc As7 clone, cyclin D1 physically interacted with cdk4 even though this binding was not functional. In fact, we observed the presence of an active unphosphorylated form of retinoblastoma protein in pINDc‐myc As cells compared to controls.
It is well known that cdk activity is further regulated by association with cdk inhibitors, such as those of the Cip/Kip families, and that c‐myc represses expression of p21Cip1 and p27Kip1 inhibitors (18). However, it is still unclear whether p57Kip2 is a c‐myc target gene. Considering these observations, we examined whether Myc down‐regulation would be able to inhibit the activity of cyclin D1/cdk4 complex via Cip/Kip inhibitors. Here, we have shown that either p57Kip2 or p27Kip1 were bound to cyclin D1/cdk4 complex in the Myc down‐regulated cells 48 h after ponasterone treatment, suggesting that they might participate in inhibition of the activity of cyclin D1/cdk4 complex.
We found that inducible down‐regulation of Myc led to up‐regulation of total p57Kip2 RNA levels (2.5‐fold), as compared to controls, evaluated at 48 h after hormone exposure by RT‐PCR (Fig. 7c). This suggests that when it is present at high levels, p57Kip2 promotes accumulation of cyclin D1 and association of cyclin D1/cdk4 complex, thus allowing stabilization of the binary complex and avoiding its rapid degradation. Formation of stable cyclin D1/cdk4/p57Kip2 tertiary complex hypophosphorylates retinoblastoma protein, thus preventing transcription of E2F responsive genes involved in entry of the cells into S phase.
In contrast, p27Kip1 assembled with cyclin D1/cdk4 complex, and inhibited complex activity by rendering the binary complex ineffective at activating cyclin E/cdk2 and cyclin A/cdk2 (32). Conversely, formation of the tertiary cyclin D1/cdk4/p21Cip1 complex was not detected in our cells (data not shown).
The data were also confirmed by using RNA interference for c‐myc. As shown in Fig. 7d, co‐immunoprecipitation and Western blot assays confirmed formation of inactive cyclin D1/cdk4 complex as evaluated by presence of an active unphosphorylated form of retinoblastoma protein. We also found that either p57Kip2 or p27Kip1 inhibitors bound to cyclin D1/cdk4 complex.
Myc down‐regulation induced apoptosis via the mitochondrial pathway
We also verified the contribution of apoptosis to decrease of cell number observed in pINDc‐myc As cells. Cell cycle analysis revealed that Myc down‐regulation was able to induce apoptosis and we detected the presence of a sub‐G1 peak (17%), which is typical of apoptosis, at 48 h after hormone exposure. Cell fraction of the sub‐G1 peak reached a maximum of 65% with a concurrent depletion of cells from G1 at 72 h after treatment (Fig. 8a). Induction of apoptosis was also confirmed by the TUNEL assay (Fig. 8b).
Figure 8.
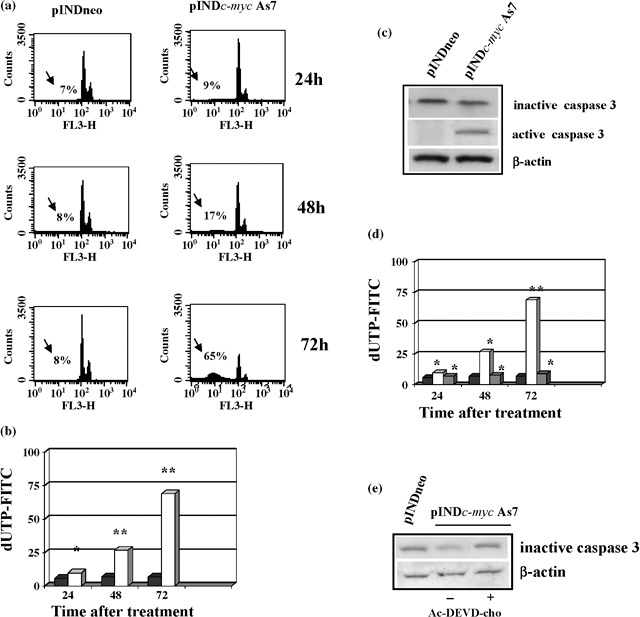
(a) Flow cytometry and apoptosis analysis. ADF pINDneo control cells and ADF pINDc‐myc As7 clone exposed to 20 µm of hormone treatment (given every 24 h) 24, 48 and 72 h after. The sub‐G1 peak (arrows) represents the percentage of apoptosis. (b) TUNEL assay. Percentages of apoptotic cells in ADF pINDc‐myc As7 (white) and pINDneo control cells (black) 24, 48 and 72 h after treatment. Statistical significance between different groups was evaluated by Student's t‐test (**P < 0.001; *P < 0.01). A P‐value < 0.001 was considered significant. (c) Western blot analysis of inactive precursor and active form of caspase‐3. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results. (d) Effect of Ac‐DEVD‐cho caspase‐3 inhibitor on apoptosis. Percentages of apoptotic cells in ADF pINDneo control (black), ADF pINDc‐myc As7 in the absence (white) or presence (grey) of 50 µm of Ac‐DEVD‐cho by using TUNEL assay. Statistical significance between the different groups was evaluated by Student's t‐test (**P < 0.001; *P < 0.01). A P‐value < 0.001 was considered significant. (e) The effect of 50 µm of Ac‐DEVD‐cho on caspase‐3 expression by Western blot. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
Several studies have demonstrated that Myc induces apoptosis via an intrinsic mitochondrial pathway. Apoptosis at the mitochondrial level occurs by release of cytochrome c into the cytosol, that in turn contributes to formation of a protein complex called apoptosome (33); it is this complex that allows activation of caspase‐9, which is directly responsible for activation of the central apoptosis regulator caspase‐3 (23). Our results showed that Myc down‐regulation was able to induce activation of caspase‐3 despite the presence of a mutated form of caspase‐9 in human astrocytoma ADF cells (27). Western blot analysis revealed 2.5‐fold reduction of the 32‐kDa inactive precursor of caspase‐3, 48 h after Myc induction with respect to controls. This was consistent with the presence of the 11‐kDa active form of caspase‐3 in pINDc‐myc As7 clone (Fig. 8c). To confirm the key role of caspase‐3 in Myc‐induced apoptosis, Ac‐DEVD‐cho (caspase‐3 inhibitor) was utilized to block caspase‐3 activity. When Ac‐DEVD‐cho was added to the pINDc‐myc As7 cells, the percentage of apoptotic cells was comparable with that of the control clone, thus demonstrating that caspase‐3 inhibitor scavenged Myc induced caspase‐3 apoptosis (Fig. 8d). The effect of Ac‐DEVD‐cho caspase‐3 inhibitor on caspase‐3 expression was also analysed (Fig. 8e) and caspase‐3 inhibitor, in the same conditions, was able to block caspase‐3 cleavage.
Reduction of inactive caspase‐3 was consistent with the strong increase in cytochrome c cytosolic levels in Myc‐treated cells compared to controls, while mitochondrial levels were reduced in the same experimental conditions. Since Smac/Diablo is also released from the mitochondria along with cytochrome c during apoptosis, and functions to promote caspase‐3 activation by inhibiting c‐IAPs proteins, we verified release of Smac/Diablo in the cytoplasmic compartment after Myc down‐regulation. We observed that Smac/Diablo levels were also enriched in the cytosolic compartment by Myc induction and concurrently depleted in the mitochondrial fraction (Fig. 9a). Moreover, we found that c‐Myc was able to regulate c‐IAPs proteins. Indeed, we detected a decrease in expression of c‐IAP1 and c‐IAP2 of about 2‐ and 3‐fold, respectively, 48 h after Myc induction (Fig. 9b). Since c‐IAP1 and c‐IAP2 are the most sensitive targets of the proteasome‐mediated protein degradation system, we verified involvement of the latter in reduced c‐IAP1 and c‐IAP2 levels. Four hours after 24‐h exposure of c‐Myc down‐regulated cells to 0.25 µm MG132 proteasome inhibitor, levels of c‐IAP1 and c‐IAP2 were significantly increased compared to pINDc‐myc As7 cells not exposed to MG132; thus, this demonstrates that inhibition of proteasome activity prevented degradation of c‐IAP1 and c‐IAP2 proteins (Fig. 9c).
Figure 9.
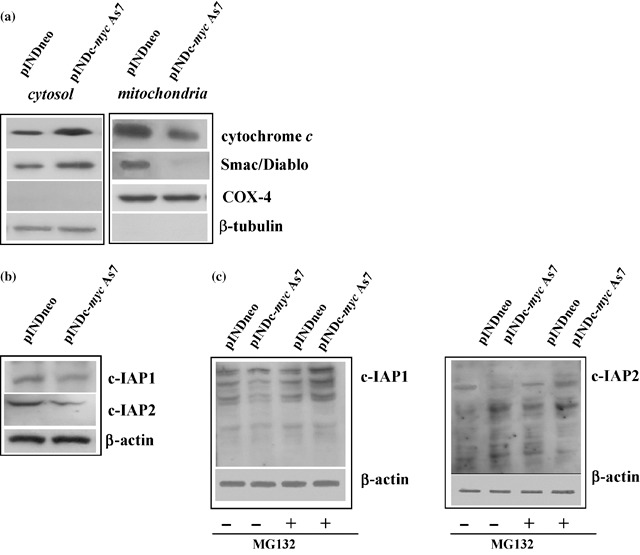
(a) Western blot analysis of cytochrome c and Smac/Diablo. Expression of cytochrome c and Smac/Diablo was analysed in the different compartments (mitochondria and cytosol) in ADF pINDneo and ADF pINDc‐myc As7 clone 48 h after hormone treatment. Samples were loaded with 30 µg of proteins from cell lysate. Blotting for COX‐4 and β‐tubulin were performed for control of quality and equal loading of the extracts and to exclude the contamination of mitochondrial with cytosolic components and viceversa. (b) Western blot analysis of c‐IAP1 and c‐IAP2 proteins. c‐IAP1 and c‐IAP2 levels in pINDneo control and in ADF pINDc‐myc As7 clones 48 h after of 20 µm hormone treatment (given every 24 h). Protein levels were quantified by densitometry (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results. (c) Effect of proteasome inhibition on c‐IAP1 and c‐IAP2 levels. ADF pINDneo and ADF pINDc‐myc As7 cells were treated or not with 0.25 µm proteasome inhibitor MG132 for 4 h 24 h after ponasterone induction. Protein levels were quantified by densitometric analysis (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
Apoptosis was also confirmed by c‐myc silencing experiments. c‐myc siRNA was able to induce apoptotic cell death at 48 and 72 h (22% vs. 2%, 68% vs. 4%, respectively) as shown in Fig. 10a. Induction of apoptosis was also detected by TUNEL assay (Fig. 10b). Moreover, we found that in siRNA c‐myc experiment, cells were also able to down‐regulate c‐IAP proteins by activation of the proteasome‐mediated protein degradation system. MG132 proteasome inhibitor increased levels of c‐IAP1 and c‐IAP2 when compared to c‐myc siRNA cells not exposed to MG132 (Fig. 10c). This suggests that down‐regulation of Myc overcame ADF resistance to apoptosis caused by a mutated mitochondrial apoptotic pathway, via release of Smac/Diablo protein, which reduced protein levels of c‐IAP1 and c‐IAP2 through activation of proteasome degradation and subsequently by directly promoting caspase‐3 activation.
Figure 10.
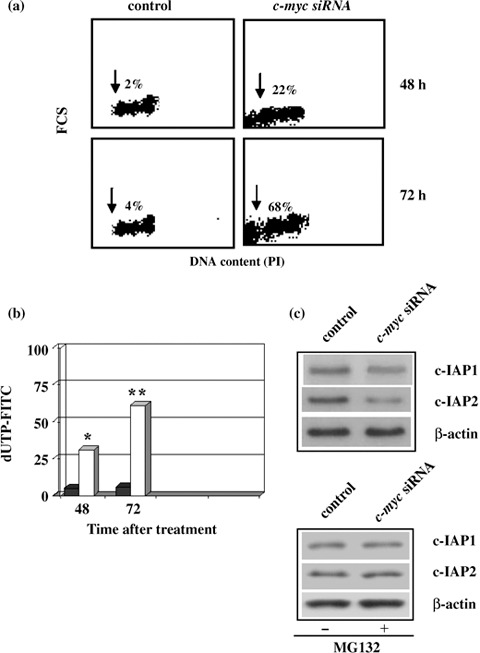
Apoptosis in siRNA c‐myc cells. Control and siRNA c‐myc cells evaluated at 48 and 72 h. The sub‐G1 peak (arrows) represents the percentage of apoptosis. (b) TUNEL assay. Percentages of apoptotic cells in control (black) and siRNA c‐myc (white) at 48 and 72 h. Statistical significance between the different groups was evaluated by Student's t‐test (**P < 0.001; *P < 0.01). A P‐value < 0.001 was considered significant. (c) Western blot analysis of c‐IAP1 and c‐IAP2 levels with or without proteasome inhibitor MG132. Control and siRNA c‐myc cells were treated or not treated with 0.25 µm proteasome inhibitor MG132 for 4 h 24 h after the trasfection. Protein levels were quantified by densitometry (TotalLab image analysis solution, version 2003) and normalized for β‐actin. The experiment was repeated three times showing similar results.
Discussion
In this paper, we have demonstrated that by switching off c‐myc expression, the G1/S checkpoint and S phase cell progression were affected in both human T98G and ADF astrocytoma cells.
Even though Mateyak et al. (34) reported a profound extension of both G1 and G2 phases by switching off c‐myc expression, our results showed a G1 arrest together with inhibition of DNA synthesis accompanied by a delay in S phase transition, in accordance with the findings of other studies (18, 28, 35, 36).
It has been investigated that Myc activates and represses target genes involved in cell cycle progression (37, 38). In the signal transduction cascade, cyclin‐dependent kinases are key regulators downstream of c‐myc, which drive cell cycle transition from one phase to the next. In addition, their activity is regulated by cyclins and cyclin‐dependent kinase inhibitors. It has been suggested that c‐myc stimulates expression of cyclin A, cyclin D1, cyclin D3, cyclin E, cdk2 and cdk4, and represses expression of p21Cip1 and p27kip1 inhibitors. In particular, it has been demonstrated that Myc activates the cyclin E/cdk2 kinase complex, promoting the G1/S progression in the cell cycle by inactivating inhibitors p27Kip1 and p21Cip1 (39, 40, 41, 42, 43, 44, 45, 46, 47).
In contrast, Daksis et al. (48) reported that cyclin D1 mRNA expression was enhanced in Rat1‐MycER cells after Myc induction, and Philipp et al. (49) showed that constitutive expression of c‐myc led to higher levels of cyclin A and cyclin E expression, while cyclin D1 was strongly suppressed in BALB/c3T3 fibroblasts. In addition, it has recently been reported that cyclin A, cyclin D1 and cyclin E are unaffected by c‐myc induction, whereas c‐myc involves both positive effects on cdk2 and negative effects on cdk4 in a mouse keratinocyte cell line (18). Nevertheless, it is not fully resolved whether expression of myc renders cell cycle progression independent from the functions of cdk4 in fibroblasts (40, 41, 50, 51, 52, 53).
Although c‐myc affects normal and neoplastic cell proliferation by altering gene expression, the precise pathways involved remain unclear (18).
We found that in Myc down‐regulated cells, cyclin D1 and cdk4 were assembled. Cyclin D1/cdk4 generally acts by phosphorylating and inactivating retinoblastoma protein by converting it from repressor to activator of E2F factors, leading to transcription of genes required for DNA synthesis. However, our data show that despite significant up‐regulation in expression of cyclin D1, the cyclin D1/cdk4 complex was nonfunctional, as evaluated by accumulation of the hypophosphorylated form of retinoblastoma protein in Myc down‐regulated cells and, thus, suggested a role of cyclin D1 in the myc‐mediated cell cycle regulation pathway. Even though Qi et al. (18) demonstrated no difference in cyclin D1 expression and no change in kinase activity of the cyclin D1/cdk4 complex in response to c‐Myc down‐regulation in A549 cells, Philipp et al. (49) reported that cyclin D1 could be a target gene for c‐myc and could be responsive to c‐myc under certain circumstances. The diversity of cell line types for the different studies could account for the variable levels of cyclin D1 expression and for diverse regulation of its activity in response to c‐myc.
In the present study, inhibition of cyclin D1/cdk4 complex depended on p57Kip2 or p27Kip1 inhibitors, which bound to cyclin D1/cdk4 complex; thus, this indicates that they cooperated to inhibit the activity of cyclin D1/cdk4. Although p27Kip1 and p21Cip1 clearly inhibit cdk2 activity, their effects on cdk4 activity are unresolved. Some studies suggest that p27Kip1 and p21Cip1 are obligatory assembling factors for cyclin D/cdk4 complexes (12) and that cyclin D/cdk4 complexes, including cyclin D1/cdk4, which contains a single p27Kip1 or p21Cip1, are active (14, 15). Other studies argue that cyclin D1/cdk4 complexes containing p27Kip1 or p21Cip1 are always inactive (16). In addition, Li et al. reported that the cyclin D1/cdk4 complex binds to another inhibitor of Cip/Kip family (p57Kip2) that, by promoting cyclin D1/cdk4 assembly, converts retinoblastoma protein to the active growth suppressing hypophosphorylated form, that sequesters E2F and, thus, prevents S phase entry (17). Conversely, Reynaud et al. found that p57Kip2 inhibits cyclin D1/cdk4 activity by repressing phosphorylation of retinoblastoma protein (54).
In addition, it is well known that c‐myc is an upstream regulator of p27Kip1 (18), while it is unclear whether p57Kip2 is a c‐myc target gene. Microarray analyses have shown that p57Kip2 is a target for regulation by the Myc‐Miz1 complex in fibroblasts and Northern blotting arrays showed that expression of p57Kip2 was repressed by Myc, but not by MycV394D, an allele of Myc that is unable to bind to Miz1 (55). Since it is possible that p57Kip2 could be a molecular target directly regulated by c‐myc, we analysed the levels of p57Kip2 mRNA by RT‐PCR. We found that the inducible down‐regulation of Myc led to an up‐regulation of p57Kip2 mRNA. This suggests that, when it is present at high levels, p57Kip2 promotes the accumulation of cyclin D1 and association of the cyclin D1/cdk4 complex and, therefore, allows stabilization of the binary complex, which converts retinoblastoma protein to an active growth suppressing hypophosphorylated form by preventing S phase entry. However, elevated levels of p57Kip2 resulted in a marked decrease of cyclin A, suggesting that p57Kip2 could be involved in regulation of several aspects of the cell cycle (17). This hypothesis is confirmed by our findings, even though we observed the loss of both cyclin A and E proteins in ADF Myc down‐regulated cells.
In addition, in accordance with Sugimoto et al. (32), we found that p27Kip1 also assembled with cyclin D1/cdk4 complex, and inhibited complex activity by rendering the binary complex ineffective to activate cyclin E/cdk2 and cyclin A/cdk2. Conversely, we did not find evidence that formation of tertiary cyclin D1/cdk4/p21Cip1 complex after Myc down‐regulation. In truth, even though p21Cip1 is considered a c‐myc target gene, and that it could generally promote assembly of cyclin D1/cdk4, presence of a p53 altered form renders c‐myc unable to regulate p53 itself and consequently p21Cip1 protein levels (56).
Myc down‐regulation also decreased the level of cdk4 total enzyme, suggesting that inhibition of cyclin D1/cdk4 complex activity could also be due to reduction of cdk4 levels, indicating that a temporal pattern of events could have acted in concert to inhibit cyclin D1/cdk4 complex activity.
In previous reports, we have demonstrated that reduction of Myc protein expression inhibited cell proliferation and led to apoptosis (28); moreover, several studies have demonstrated that Myc induces apoptosis via the intrinsic mitochondrial pathway through modulation of crucial biochemical parameters, such as release of cytochrome c and subsequent processing of caspase‐9 (57).
Notwithstanding, Hotti et al. (58) demonstrated that in c‐Myc‐induced apoptosis, cleavage of caspase‐9 is, however, minimal and a much later event than processing/activation of caspase‐3, suggesting that caspase‐9 cleavage it is not an apical event. Even though Ceruti et al. (27) demonstrated that human astrocytoma ADF cells are resistant to apoptosis, because they bear a defective mitochondrial pathway of apoptosis, we found caspase‐3‐dependent apoptosis in ADF Myc down‐regulated cells. We demonstrated that caspase‐3 activation is essential for c‐Myc‐induced apoptosis since caspase‐3 inhibitor was able to revert the apoptotic phenotype.
Similar to the findings of Ceruti et al. (27), we observed accumulation of cytochrome c despite presence of the mutant caspase‐9 form, suggesting that there were no alterations in mechanism(s) of protein release from mitochondria. Even though it is well known that cytochrome c triggers activation/autocleavage of caspase‐9, which in turn actives caspase‐3, presence of mutant caspase‐9 in ADF cells indicates that it is not responsible for processing of caspase‐3. Since it has been demonstrated that caspase‐3‐dependent cell death requires inhibition of c‐IAP proteins (25, 59), our hypothesis is that down‐regulation of Myc overcomes ADF resistance to apoptosis caused by a mutated mitochondrial apoptotic pathway via release of Smac/Diablo protein. In fact, we found an increase in levels of Smac/Diablo associated with a decrease in expression of c‐IAP1 and c‐IAP2 proteins through activation of a proteasome‐mediated protein degradation system, suggesting that Smac/Diablo selectively caused rapid degradation of c‐IAP1 and c‐IAP2, by overcoming caspase‐3 inhibition.
In conclusion, our results suggest that c‐Myc could be considered a good target to study new approaches in anticancer astrocytoma treatment.
Acknowledgement
We thank Elisabetta Perotti for her language assistance.
References
- 1. Shervington A, Cruickshanks N, Wright H, Atkinson‐Dell R, Lea R, Roberts G, Shervington L (2006) Glioma: what is the role of c‐Myc, hsp90 and telomerase? Mol. Cell. Biochem. 283, 1–9. [DOI] [PubMed] [Google Scholar]
- 2. Feun LG, Savaraj N, Landy HJ (1994) Drug resistance in brain tumors. J. Neurooncol. 20, 165–176. [DOI] [PubMed] [Google Scholar]
- 3. Henson JW, Cordon‐Cardo C, Posner JB (1992) P‐glycoprotein expression in brain tumors. J. Neurooncol. 14, 37–43. [DOI] [PubMed] [Google Scholar]
- 4. Phillips PC (1991) Antineoplastic drug resistance in brain tumors. Neurol. Clin. 9, 383–404. [PubMed] [Google Scholar]
- 5. Hoshimaru M, Ray J, Sah DW, Gage FH (1996) Differentiation of the immortalized adult neuronal progenitor cell line HC2S2 into neurons by regulatable suppression of the v‐myc oncogene. Proc. Natl. Acad. Sci. USA 93, 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Felsher DW, Bishop JM (1999) Transient excess of Myc activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 96, 3940–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pelengaris S, Khan M (2003) The many faces of c‐Myc. Arch. Biochem. Biophys. 416, 129–136. [DOI] [PubMed] [Google Scholar]
- 8. Levens DL (2003) Reconstructing Myc. Genes Dev. 17, 1071–1077. [DOI] [PubMed] [Google Scholar]
- 9. Zygmunt A, Tedesco VC, Udho E, Krucher NA (2002) Hypoxia stimulates p16 expression and association with cdk4. Exp. Cell Res. 278, 53–60. [DOI] [PubMed] [Google Scholar]
- 10. Chen JH, Tseng TH, Ho YC, Lin HH, Lin WL, Wang CJ (2003) Gaseous nitrogen oxides stimulate cell cycle progression by retinoblastoma phosphorylation via activation of cyclins/cdks. Toxicol. Sci. 76, 83–90. [DOI] [PubMed] [Google Scholar]
- 11. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach R. (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366, 701–704. [DOI] [PubMed] [Google Scholar]
- 12. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ (1999) The p21 (Cip1) and p27 (Kip1) cdk ‘inhibitors’ are essential activators of cyclin d‐dependent kinases in murine fibroblasts. EMBO J. 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blain SW, Montalvo E, Massague J (1997) Differential interaction of the cyclin‐dependent kinase (cdk) inhibitor p27Kip1 with cyclin A‐cdk2 and cyclin D2‐cdk4. J. Biol. Chem. 272, 25863–25872. [DOI] [PubMed] [Google Scholar]
- 14. Labaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E (1997) New functional activities for the p21 family of cdk inhibitors. Genes Dev. 11, 847–862. [DOI] [PubMed] [Google Scholar]
- 15. Zhang H, Hannon GJ, Beach D (1994) p21‐containing cyclin kinases exist in both active and inactive states. Genes Dev. 8, 1750–1758. [DOI] [PubMed] [Google Scholar]
- 16. Bagui TK, Mohapatra S, Haura E, Pledger WI (2003) p27Kip1 and p21Cip1 are not required for the formation of active D cyclin‐cdk4 complexes. Mol. Cell. Biol. 23, 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li G, Domenico J, Lucas JJ, Gelfand EW (2004) Identification of multiple cell cycle regulatory functions of p57Kip2 in human T lymphocytes. J. Immunol. 173, 2383–2391. [DOI] [PubMed] [Google Scholar]
- 18. Qi Y, Tu Y, Yang D, Chen Q, Xiao J, Chen Y, Fu J, Xiao X, Zhou Z. (2007) Cyclin A but not cyclin D1 is essential for c‐myc‐modulated cell‐cycle progression. J. Cell. Physiol. 210, 63–71. [DOI] [PubMed] [Google Scholar]
- 19. Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee NH, Dang CV, Liu ET (2000) Identification of c‐myc responsive genes using rat cDNA microarray. Cancer Res. 60, 5922–5928. [PubMed] [Google Scholar]
- 20. Dauphinot L, De Oliveira C, Melot T, Sevenet N, Thomas V, Weissman BE, Delattre O (2001) Analysis of the expression of cell cycle regulators in Ewing cell lines: EWS‐FLI‐1 modulates p57Kip2 and c‐Myc expression. Oncogene 20, 3258–3265. [DOI] [PubMed] [Google Scholar]
- 21. Juin P, Evan G (2000) Caspase 8: the killer you can't live without. Nat. Med. 6, 498–500. [DOI] [PubMed] [Google Scholar]
- 22. Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D, Penninger J, Kroemer G (2000) Mitochondrio‐nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14, 729–739. [PubMed] [Google Scholar]
- 23. Kagaya S, Kitanaka C, Noguchi K, Mochizuki T, Sugiyama A, Asai A, Yasuhara N, Eguchi Y, Tsujimoto Y, Kuchino Y (1997) A functional role for death proteases in s‐Myc‐ and c‐Myc‐mediated apoptosis. Mol. Cell. Biol. 17, 6736–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15, 2922–2933. [PubMed] [Google Scholar]
- 25. Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c‐dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42. [DOI] [PubMed] [Google Scholar]
- 26. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz. RL , Simpson RJ, Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53. [DOI] [PubMed] [Google Scholar]
- 27. Ceruti S, Mazzola A, Abbracchio MP (2005) Resistance of human astrocytoma cells to apoptosis induced by mitochondria‐damaging agents: possible implications for anticancer therapy. J. Pharmacol. Exp. Ther. 314, 825–837. [DOI] [PubMed] [Google Scholar]
- 28. D’Agnano I, Valentini A, Fornari C, Bucci B, Starace G, Felsani A, Citro G (2001) Myc down‐regulation induces apoptosis in M14 melanoma cells by increasing p27 (Kip1) levels. Oncogene 20, 2814–2825. [DOI] [PubMed] [Google Scholar]
- 29. D’Agnano I, Antonelli A, Bucci B, Marcucci L, Petrinelli P, Ambra R, Zupi G, Elli R. (1998) Effects of poly(ADP‐ribose) polymerase inhibition on cell death and chromosome damage induced by VP16 and bleomycin. Environ. Mol. Mutagen. 32, 56–63. [PubMed] [Google Scholar]
- 30. Yomori H, Yasunaga K, Takahashi C, Tanaka A, Takashima S, Sekijima M (2002) Elliptically polarized magnetic fields do not alter immediate early response genes expression levels in human glioblastoma cells. Bioelectromagnetics 23, 89–96. [DOI] [PubMed] [Google Scholar]
- 31. Hirvonen HE, Salonen R, Sandberg MM, Vuorio E, Västrik I, Kotilainen E, Kalimo H (1994) Differential expression of myc, max and RB1 genes in human gliomas and glioma cell lines. Br. J. Cancer 69, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugimoto M, Martin N, Wilks DP, Tamai K, Huot TJ, Pantoja C, Okumura K, Serrano M, Hara E (2002) Activation of cyclin D1‐kinase in murine fibroblasts lacking both p21 (Cip1) and p27 (Kip1). Oncogene 21, 8067–8074. [DOI] [PubMed] [Google Scholar]
- 33. Igney FH, Krammer PH (2002) Death and anti‐death: tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288. [DOI] [PubMed] [Google Scholar]
- 34. Mateyak MK, Obaya AJ, Sedivy JM (1999) c‐Myc regulates cyclin d‐cdk4 and – cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell. Biol. 19, 4672–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heikkila R, Schwab G, Wickstrom E, Loke SL, Pluznik DH, Watt R, Neckers LM (1987) A c‐myc antisense oligodeoxynucleotide inhibits entry into S phase but not progress from G0 to G1. Nature 328, 445–449. [DOI] [PubMed] [Google Scholar]
- 36. Holt JT, Redner RL, Nienhuis AW (1988) An oligomer complementary to c‐myc mRNA inhibits proliferation of HL‐60 promyelocytic cells and induces differentiation. Mol. Cell. Biol. 8, 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pelengaris S, Khan M, Evan G (2002) c‐Myc: more than just a matter of life and death. Nat. Rev. Cancer 2, 764–776. [DOI] [PubMed] [Google Scholar]
- 38. Prendergast GC (1999) Mechanisms of apoptosis by c‐Myc. Oncogene 18, 2967–2987. [DOI] [PubMed] [Google Scholar]
- 39. Steiner P, Philipp A, Lukas J, Godden‐Kent D, Pagano M, Mittnacht S, Bartek J, Eilers M (1995) Identification of a Myc‐dependent step during the formation of active G1 cyclin‐cdk complexes. EMBO J. 14, 4814–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berns K, Hijmans ME, Bernards R. (1997) Repression of c‐Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/cdk2 kinase activity. Oncogene 15, 1347–1356. [DOI] [PubMed] [Google Scholar]
- 41. Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL (1998) c‐Myc or cyclin D1 mimics estrogen effects on cyclin E‐cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 18, 4499–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amati B, Alevizopoulos K, Vlach J (1998) Myc and the cell cycle. Front. Biosci. 3, 250–268. [DOI] [PubMed] [Google Scholar]
- 43. Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B (1996) Growth arrest by the cyclin‐dependent kinase inhibitor p27Kip1 is abrogated by c‐Myc. EMBO J. 15, 6595–6604. [PMC free article] [PubMed] [Google Scholar]
- 44. Perez‐Roger I, Solomon DL, Sewing A, Land H (1997) Myc activation of cyclin E/cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27 (Kip1) binding to newly formed complexes. Oncogene 14, 2373–2381. [DOI] [PubMed] [Google Scholar]
- 45. Perez‐Roger I, Kim SH, Griffiths B, Sewing A, Land H (1999) Cyclins D1 and D2 mediate myc‐induced proliferation via sequestration of p27 (Kip1) and p21 (Cip1). EMBO J. 18, 5310–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bouchard C, Thieke K, Maier A, Saffrich R, Hanley‐Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 18, 5321–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hermeking H, Funk JO, Reichert M, Ellwart JW, Eick D (1995) Abrogation of p53‐induced cell cycle arrest by c‐Myc: evidence for an inhibitor of p21WAF1/CIP1/SDI1. Oncogene 11, 1409–1415. [PubMed] [Google Scholar]
- 48. Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ (1994) Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen‐stimulated signal transduction to the cell cycle. Oncogene 9, 3635–3645. [PubMed] [Google Scholar]
- 49. Philipp A, Schneider A, Vasrik I, Finke K, Xiong Y, Beach D, Alitalo K, Eilers M (1994) Repression of cyclin D1: a novel function of Myc. Mol. Cell. Biol. 14, 4032–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alevizopoulos K, Vlach J, Hennecke S, Amati B (1997) Cyclin E and c‐Myc promote cell proliferation in the presence of p16INK4a and of hypophosphorylated retinoblastoma family proteins. EMBO J. 16, 5322–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haas K, Staller P, Geisen C, Bartek J, Eilers M, Möröy T (1997) Mutual requirement of cdk4 and Myc in malignant transformation: evidence for cyclin D1/cdk4 and p16INK4A as upstream regulators of Myc. Oncogene 15, 179–192. [DOI] [PubMed] [Google Scholar]
- 52. Pusch O, Bernaschek G, Eilers M, Hengstschläger M (1997) Activation of c‐Myc uncouples DNA replication from activation of G1‐cyclin‐dependent kinases. Oncogene 15, 649–656. [DOI] [PubMed] [Google Scholar]
- 53. Obaya AJ, Mateyak MK, Sedivy JM (1999) Mysterious liaisons: the relationship between c‐Myc and the cell cycle. Oncogene 18, 2934–2941. [DOI] [PubMed] [Google Scholar]
- 54. Reynaud EG, Guillier M, Leibovitch MP, Leibovitch SA (2000) Dimerization of the amino terminal domain of p57Kip2 inhibits cyclin D1‐cdk4 kinase activity. Oncogene 19, 1147–1152. [DOI] [PubMed] [Google Scholar]
- 55. Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, Syvaoja J, Saluz. HP , Haenel F, Eilers M (2002) Negative regulation of the mammalian UV response by Myc through association with Miz‐1. Mol. Cell 10, 509–521. [DOI] [PubMed] [Google Scholar]
- 56. Ceruti S, Mazzola A, Abbracchio MP (2006) Proteasome inhibitors potentiate etoposide‐induced cell death in human astrocytoma cells bearing a mutated p53 isoform. J. Pharmacol. Exp. Ther. 319, 1424–1434. [DOI] [PubMed] [Google Scholar]
- 57. Juin P, Hueber AO, Littlewood T, Evan G (1999) c‐Myc‐induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 13, 1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hotti A, Järvinen K, Siivola P, Hölttä E (2000) Caspases and mitochondria in c‐Myc‐induced apoptosis: identification of ATM as a new target of caspases. Oncogene 19, 2354–2362. [DOI] [PubMed] [Google Scholar]
- 59. Morrish F, Giedt C, Hockenbery D (2003) c‐Myc apoptotic function is mediated by NRF‐1 target genes. Genes Dev. 17, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]