

Abstract
Abstract. Syringolin A is a new plant elicitor produced by the plant pathogen Pseudomonas syringae pv. syringae. The goal of this study was to investigate whether syringolin A exhibits anti‐proliferative properties in cancer cells. The treatment of human neuroblastoma (NB) cells (SK‐N‐SH and LAN‐1) and human ovarian cancer cells (SKOV3) with syringolin A (0–100 µm) inhibited cell proliferation in a dose‐dependent manner. The IC50 (50% inhibition) for each cell line ranged between 20 µm and 25 µm. In SK‐N‐SH cells, the treatment with 20 µm syringolin A led to a rapid (24 h) increase of the apoptosis‐associated tumour suppressor protein p53. In addition, we found that the treatment of SK‐N‐SH cells caused severe morphological changes after 48 h such as rounding of cells and loss of adherence, both conditions observed during apoptosis. The induction of apoptosis by syringolin A was confirmed by both poly (ADP‐ribose) polymerase (PARP) cleavage and annexin V assay. Taken together, we show for the first time that the natural product syringolin A exhibits anti‐proliferative activity and induces apoptosis. Syringolin A and structurally modified syringolin A derivatives may serve as new lead compounds for the development of novel anticancer drugs.
INTRODUCTION
Neuroblastoma (NB) is an embryonic malignancy of the sympathetic nervous system arising from neuroblasts and is considered the most common extracranial tumour of childhood with approximately 700 new cases per year in the United States of America (Brodeur 2003). Whereas many infants experience complete regression of primary tumours and even of metastatic disease, older children are often confronted with NB metastases that are aggressive and respond poorly to even the most intense multicomponent drug regimens. Hence, the identification of new drug therapeutics effectively targeting tumour formation and metastasis continues to be pivotal in an effort to improve patient care and survival rate.
Programmed cell death (apoptosis) often malfunctions in many cancer cells and is characterized by cell shrinkage, membrane blebbing, DNA fragmentation and detachment of cells from the extracellular matrix. Apoptosis eliminates unwanted and potentially dangerous cancer cells and is controlled by regulatory proteins such as p53 and the poly (ADP‐ribose) polymerase (PARP). The protein p53 is a tumour suppressor and a key component in the regulation of apoptosis as well as cell cycle progression (Harris & Levine 2005). Activation of p53 in response to DNA damage and genotoxic stress induces the transcription of Bax, Noxa and p53‐upregulated modulator of apoptosis (PUMA) which leads to cytochrome C release and caspase‐9‐mediated apoptosis. The DNA repair enzyme PARP is important for cells to maintain their viability, and cleavage of PARP by activated caspases facilitates cellular disassembly and serves as a marker of cells undergoing apoptosis (Li & Darzynkiewicz 2000).
Certain strains of the phytopathogenic bacterium Pseudomonas syringae pv. syringae secrete a family of structurally closely related peptide derivatives named syringolins, of which syringolin A is the major variant (Wäspi et al. 1998a; Wäspi et al. 1999; Amrein et al. 2004). The function of syringolins in the interaction of P. syringae pv. syringae with their host plants is presently unknown. It is hypothesized that they may constitute virulent factors; however, syringolins are determinants recognized and reacted to by nonhost plant species, and syringolin A has been shown to induce death of hypersensitive cells colonized by powdery mildew in wheat, and reprograms a compatible interaction into an incompatible one (Amrein et al. 2004; Wäspi et al. 2001). Syringolin A is an unusual derivative of a tripeptide that contains a 12‐membered ring consisting of the amino acids 5‐methyl‐4‐amino‐2‐hexenoic acid and 3,4‐dehydrolysine, two nonproteinogenic amino acids (Fig. 1). The genes involved in the biosynthesis of syringolin A encode proteins, which consist of modules typical for nonribosomal peptide synthetases and type I polyketide synthetases as well as proteins likely involved in the transcriptional regulation of syringolin A biosynthesis and export (Amrein et al. 2004).
Figure 1.
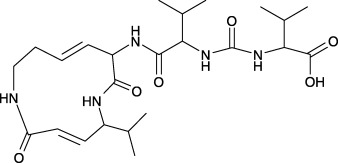
Chemical structure of syringolin A. The compound is produced and secreted by the plant pathogen Pseudomonas syringae pv. syringae and its ring structure is formed by the two nonproteinogenic amino acids 5‐methyl‐4‐amino‐2‐hexenoic acid and 3,4‐dehydrolysine. The α‐amino group of the latter is joined by a peptide bond to a valine residue, which is linked to another valine residue via a urea moiety. Molecular weight: 493 amu.
It is well known that a variety of terrestrial and marine natural products exhibit anticancer activities (Mayer & Gustafson 2004; El Sayed 2005; Hamblin 2005), and many drugs, which are clinically useful today (for example, paclitaxel/Taxol) are natural products or derivatives thereof (Derry et al. 1997; Mullins et al. 1997). We have previously shown that the P. syringae pv. phaseolicola‐produced phaseolotoxin inhibits cell proliferation of leukaemic cells and pancreatic cells (Bachmann et al. 2004). In this study, we demonstrate for the first time that pure syringolin A inhibits the proliferation of several mammalian cell lines including NB and ovarian cancer cells in a dose‐dependent manner and induces apoptosis. The syringolin A molecule is a new natural product with potential for further development into an effective anticancer drug.
MATERIALS AND METHODS
Purification of syringolin A
P. syringae pv. syringae strain B 301D‐R was grown as previously described (Gross 1985; Mo & Gross 1991). Isolation of syringolin A from conditioned medium was also performed as previously described (Wäspi et al. 1999). The purity of syringolin A was confirmed by reverse‐phase HPLC using a syringolin A standard, which itself had been previously purified in an identical manner and verified by mass spectrometry. Lyophilized syringolin A was dissolved in ultra‐pure distilled water (Invitrogen, Carlsbad, CA) at a concentration of 2.5 mm (stock solution), sterile‐filtered with a 0.2 µm filter, aliquoted into sterile tubes and was stored frozen at −80 °C. Aliquots were thawed and used for cell culture studies at different concentrations as indicated below.
Mammalian cell cultures and reagents
Human NB cell lines were provided by Dr Robert Seeger (LAN‐1; University of California, Los Angeles, CA) and the American Type Culture Collection (SK‐N‐SH; ATCC, Manassas, VA) and were maintained in RPMI 1640 (Biosource, Rockeville, MD) containing 10% (v/v) heat‐inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), penicillin (100 IU/ml), and streptomycin (100 µg/ml) as previously reported (Wallick et al. 2005). SK‐N‐SH is a p53 wild‐type cell line (Davidoff et al. 1992; McKenzie et al. 1999; Tweddle et al. 2001a) whereas LAN‐1 is a p53 mutated cell line (cys to stop at residue 182) (Davidoff et al. 1992; Tweddle et al. 2001b; Tweddle et al. 2003). The human ovarian cancer cell line SKOV3 was available through collaboration with Dr Bonnie Warn‐Cramer (Cancer Research Center of Hawaii) and was maintained in McCoys 5 A medium (Sigma Chemical Co., St. Louis, MO) supplemented with 5% (v/v) FBS and 100 µg/ml gentamicin. SKOV3 cells are p53‐deficient and over‐express activated protein kinase Akt/PKB (Wang et al. 2005). Immortalized fibroblast cells Rat‐1 were kindly provided by Dr A. Lau (Cancer Research Center of Hawaii, HI) and were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma Chemical Co., St. Louis, MO) supplemented with 10% (v/v) FBS (Goldberg & Lau 1993). Cell numbers were determined using a haemacytometer in the presence of trypan blue (Fisher Scientific, Pittsburgh, PA).
Cell proliferation assay
Cells (0.25–0.5 × 105 cells per ml) were seeded in 96‐well microtiter plates containing 100 µl of culture medium per well. After 24 h, syringolin A (0.5 µm to 100 µm final concentration) was added to wells. Cells in the absence of syringolin A served as controls. Cells were incubated for 48 h and the viability of cells (percentage of control) was determined using the sulphorhodamine B (SRB) assay as previously reported (Skehan et al. 1990). In brief, cell culture medium was removed from the microtiter plate and adherent cells were fixed with 10% (w/v) trichloracetic acid (TCA) for 30 min at room temperature. Following four washes with tap water, 100 µl of SRB (Sigma Chemical Co., St. Louis, MO) (0.4 g/100 ml 1% (v/v) glacial acetic acid in water) was added and the plate was then incubated for 30 min at room temperature, and rinsed four times with 3% (v/v) glacial acetic acid. After addition of 200 µl of 10 mm Tris base (not pH adjusted) to each well, the plate was incubated on an orbital shaker for 30 min until the SRB was uniformly dissolved. The absorption at a wavelength of 560 nm was read using an HTS 7000 Plus Bioassay Reader or a Victor 3, 1420 Multilabel Counter (PerkinElmer, Inc. Boston, MA).
Annexin V assay
Cells (1.0 × 105 cells per ml) were seeded in 12‐well plates containing 1 ml of culture medium per well. After 24 h, 10 µl of syringolin A (25 µm final concentration) or an equal volume of sterile water (control) was added per well. Cells were incubated for 48 h, trypsinized, washed twice in phosphate‐buffered saline (PBS) and were counted; 1–2 × 105 cells suspended in 0.1 ml of 1 × assay buffer per vial according to the manufacturer's instructions (BD Biosciences, Palo Alto, CA). Cells were stained with annexin V‐FITC (5 µl) and propidium iodide (5 µl) for 15 min in the dark, at room temperature. Assay buffer (0.4 ml) was added, and 5000 cells were analysed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA). The cellquest program was used for data analysis.
Protein extraction and Western blot analysis
Cells (1.0 × 105 cells per ml) were seeded in six‐well plates containing 2 ml of culture medium per well. After 24 h, 16 µl of syringolin A (20 µm final concentration) or an equal volume of sterile water (control) was added per well. Cells were incubated for 0, 24 and 48 h and cell lysates were prepared in radioimmuno‐precipitation assay (RIPA) buffer (20 mm Tris‐HCl, pH 7.5, 0.1% sodium lauryl sulphate, 0.5% sodium deoxycholate, 135 mm NaCl, 1% Triton X‐100, 10% glycerol, 2 mm EDTA), supplemented with a protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN) and phosphatase inhibitors sodium fluoride (NaF; 20 mm) and sodium vanadate (Na3VO4; 0.27 mm). The total protein concentration was determined using the protein assay dye reagent from Bio‐Rad Laboratories (Hercules, CA) and bovine serum albumin (BSA) as a standard. Cell lysates in Laemmli sample buffer (Bio‐Rad Laboratories, Hercules, CA) containing 5% (v/v) β‐mercaptoethanol were boiled for 5 min, and equal amounts of total protein were analysed by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (PAGE) and Western blotting using PVDF membranes. The antibodies used in this study were: mouse monoclonal anti‐p53 (sc‐126) (1 : 250) from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit polyclonal anti‐poly (ADP‐ribose) polymerase (PARP) (#9542) (1 : 1000), rabbit monoclonal anti‐caspase‐3 (#9665) (1 : 1000), rabbit polyclonal anti‐Akt (#9272) (1 : 1000) from Cell Signalling Technology, Inc. (Beverly, MA), and mouse monoclonal anti‐β‐actin (A5316) 1 : 1000 from Sigma Chemical Co. (St. Louis, MO). Secondary anti‐mouse (1 : 5000) and anti‐rabbit (1 : 5000) antibodies coupled to horseradish peroxidase (HRP) were from Amersham Biosciences (Piscataway, NJ). Membranes were stripped at 50 °C for 30 min with ECL stripping buffer (62.5 mm Tris‐HCL, pH 6.7, 2% sodium dodecyl sulphate, 100 mmβ‐mercaptoethanol) as previously described (Fo et al. 2006) and sequentially probed with a variety of different antibodies. Proteins were detected using the ECL Plus reagents (Amersham Biosciences, Piscataway, NJ) and Kodak BioMax XAR film (Fisher Scientific, Pittsburgh, PA). The results are representative of three (p53, procaspase‐3) or four (Akt/PKB, PARP) independent experiments.
Microscopy
Photomicrographs were taken of cells in 12‐well plates or 96‐well plates, in the absence or presence of 25 µm syringolin A, using a Nikon Diaphot inverted microscope (Nikon Corp., Tokyo, Japan) and a Carl Zeiss Axiovert 200m inverted microscope (Carl Zeiss, Goettingen, Germany), both equipped with a digital camera and computer software for image processing. Power of magnification: 20x (SK‐N‐SH, LAN‐1, SKOV3) or 10x (Rat‐1).
RESULTS
Anti‐proliferative effects and morphological changes in syringolin A‐treated neuroblastoma and ovarian cancer cells
To examine the effect of syringolin A on the proliferation rate of mammalian cancer cells, we used the p53‐wild type NB cell line SK‐N‐SH and the p53‐mutant NB cell line LAN‐1. As illustrated in Fig. 2, the cell viability of both cell lines SK‐N‐SH (a) and LAN‐1 (d) was reduced in a dose‐dependent manner after addition of syringolin A (0–100 µm) for 48 h. The IC50 (inhibitor concentration at which cell proliferation rate is reduced by 50% relative to control) for both syringolin A‐treated NB cell lines was determined between 20 and 25 µm. Concomitant to its inhibitory effects, syringolin A also induced significant morphological changes in both SK‐N‐SH cells (Fig. 2c) and LAN‐1 cells (Fig. 2f). In contrast with their typical triangular cell shape (Fig. 2b and e), nearly all cells now appeared round and were detached from the cell culture dish (floater cells) 48 h after treatment with syringolin A (Fig. 2c and f). The detachment was more pronounced in SK‐N‐SH cells (black arrows). The observed morphological changes are characteristic for cells that undergo apoptosis.
Figure 2.
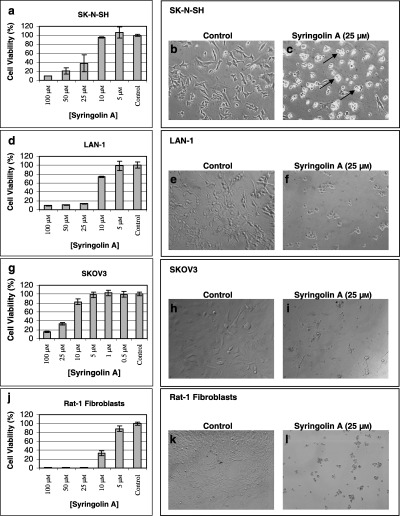
Effect of syringolin A on the proliferation rate and morphology of mammalian cells. (a–c) SK‐N‐SH cells (d–f) LAN‐1 cells (g–i) SKOV3 cells, and (j–l) Rat‐1 cells were seeded and syringolin A (0–100 µm) was added after 24 h. Cell viability was measured 48 h after the drug was added using the sulphorhodamine B (SRB) assay. Data represent mean of two (SK‐N‐SH and LAN‐1) or three (SKOV3 and Rat‐1) determinations ± SD. Photomicrographs of (b,c) SK‐N‐SH cells (e,f) LAN‐1 cells (h,i) SKOV3 cells, and (k,l) Rat‐1 cells in the absence or presence of 25 µm syringolin A show significant morphological changes 48 h after treatment. Black arrows indicate individual apoptotic floating cells, most prominently found in SK‐N‐SH cells (c). Magnification: 20× (SK‐N‐SH, LAN‐1, SKOV3) or 10× (Rat‐1).
To establish whether the anti‐proliferative effect of syringolin A is specific to NB cells, identical experiments were performed with p53‐deficient ovarian cancer cells SKOV3 and immortalized fibroblast cells Rat‐1. Syringolin A reduced the cell proliferation rate of SKOV3 cells (Fig. 2g) and Rat‐1 cells (Fig. 2j) in a dose‐dependent manner, thus suggesting that syringolin A is also active in other types of mammalian cell lines at comparable concentrations. Syringolin A also affected the cell morphology of both SKOV3 cells and Rat‐1 cells, however, neither cell detachment nor floating cells were observed (Fig. 2i and l).
Syringolin A treatment leads to rapid increase in p53 protein levels
To examine the mechanism by which syringolin A exhibits its activity, we selected the cell line SK‐N‐SH for further study, based on its strong apoptotic response in the presence of syringolin A (Fig. 2c). Because p53 is a key regulator and mediator of apoptosis, we measured the total protein levels of p53 in the p53‐wild type cell line SK‐N‐SH. As a control, the p53‐deficient cell line SKOV3 was included. Both cell lines were cultured in the absence or presence of 20 µm syringolin A for 0, 24, and 48 h and cell lysates were probed for p53 by Western blotting. We found that p53 in syringolin A‐treated SK‐N‐SH cells rapidly increased as early as 24 h after treatment (Fig. 3a). The decline of p53 after 48 h suggests that the induction of p53 is an early response and precedes the onset of apoptosis. A second p53‐reactive band at 40 kDa (p40) appeared and was previously observed in SK‐N‐SH cells (Heiligtag et al. 2002). As expected, p53 was not detected in p53‐deficient SKOV3 cells (Fig. 3b). Together, these data suggest that syringolin A activates p53 in SK‐N‐SH cells.
Figure 3.
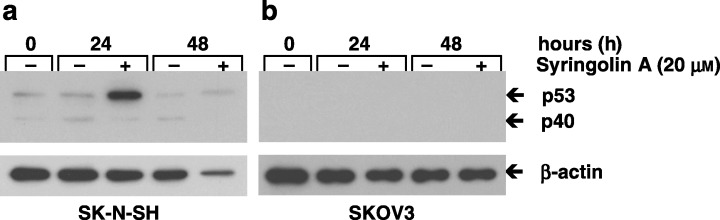
Effect of syringolin A on p53 protein levels in human neuroblastoma cells. (a) The p53‐wild type cell line SK‐N‐SH was cultured in the absence or presence of 20 µm syringolin A. (b) The p53‐deficient cell line SKOV3 served as control. Cells were harvested after 0, 24 and 48 h, lysed, and cell lysates probed for p53 by Western blot analysis. A strong increase of p53 protein levels after 24 h was observed in SK‐N‐SH cells. As expected, no p53 was detected in the p53‐deficient SKOV3 cells. Equal amounts of total protein were loaded in all experiments (10 µg per lane) as judged by protein assay using the protein dye reagent from Bio‐Rad. β‐actin was used as a loading control. The data are representative of three independent experiments (n = 3).
The observed decrease of p53 in syringolin A‐treated samples at 48 h may in part be the result of a general shutdown of protein synthesis, as we noticed that the levels of several other proteins including β‐actin, procaspase‐3 and protein kinase Akt/PKB decreased in a similar manner despite the loading of equal amounts of total protein in each lane (3, 4). This observation was reproduced in replicate experiments (n = 3). Visualization of protein bands, by the Coomassie blue membrane staining method, further revealed that several proteins are down‐regulated in syringolin A‐treated cells after 48 h (Fig. 4c), with the exception of one protein at approximately 72 kDa, which was strongly up‐regulated (black arrow) (n = 3). Because the same amount of total protein (10 µg) was loaded in each lane, it is possible that the observed general decrease in proteins is compensated by the strong increase of the 72 kDa protein, thus resulting in a protein sample that overall contains the same amount of total protein. Therefore, the differences observed for antibody staining at 48 h are the result of syringolin A treatment and not from differences in loading.
Figure 4.
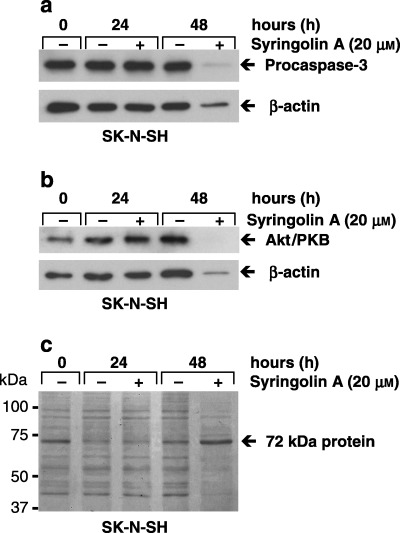
Effect of syringolin A on procaspase‐3 and Akt/PKB protein levels in human neuroblastoma cells. SK‐N‐SH cells were cultured in the absence or presence of 20 µm syringolin A. Cells were harvested after 0, 24 and 48 h, lysed, and cell lysates probed for (a) procaspase‐3 and (b) protein kinase Akt/PKB by Western blot analysis. Both procaspase‐3 and Akt/PKB protein levels and also β‐actin protein levels decreased in syringolin A‐treated cells after 48 h, thus suggesting a general shutdown of protein synthesis in these cells. (c) The Coomassie‐stained membrane depicts total amounts of protein in each lane and the black arrow indicates a prominent protein band at approximately 72 kDa. Equal amounts of total protein were loaded in all experiments (10 µg per lane) as judged by protein assay using the protein dye reagent from Bio‐Rad. The data are representative of 3 (procaspase‐3) or 4 (Akt/PKB) independent experiments.
Syringolin A treatment results in PARP cleavage
The cleavage of PARP is a common response during apoptosis. To further verify our foregoing results, we cultured SK‐N‐SH cells in the absence or presence of 20 µm syringolin A for 0, 24 and 48 h and probed the cell lysates for the presence of PARP by Western blotting. As shown in Fig. 5(a), noncleaved PARP (116 kDa) was detected in cell lysates of untreated cells after 0, 24 and 48 h, whereas cleaved PARP (89 kDa) and reduced levels of the noncleaved PARP were observed in syringolin A‐treated cells after 24 h. The treatment of cells for 48 h further increased the levels of cleaved PARP, whereas noncleaved PARP disappeared. In a similar manner, syringolin A induced PARP cleavage in SKOV3 cells, but noncleaved PARP was still detected 48 h after treatment (Fig. 5b). These results suggest that syringolin A induces apoptosis‐associated PARP cleavage in both SK‐N‐SH and SKOV3 cells.
Figure 5.
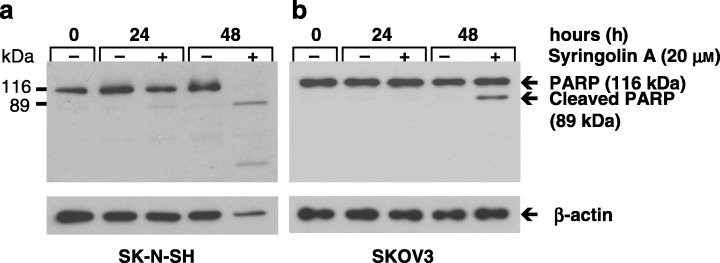
Cleavage of PARP after treatment with syringolin A. (a) SK‐N‐SH cells and (b) SKOV3 cells cultured in the absence or presence of 20 µm syringolin A for 0, 24 and 48 h were harvested, lysed, and cell lysates probed for PARP by Western blot analysis. The distinct presence of cleaved PARP (89 kDa) in treated samples of both SK‐N‐SH and SKOV3 cells shows that syringolin A induces apoptosis. Equal amounts of total protein were loaded in all experiments (10 µg per lane) as judged by protein assay using the protein dye reagent from Bio‐Rad. β‐actin was used as a loading control. The data are representative of four independent experiments (n = 4).
Induction of apoptosis in syringolin A‐treated cells
Our foregoing results indicated that syringolin A induces apoptosis by activation of p53 and cleavage of PARP, which ultimately leads to cell detachment and the formation of rounded cell bodies in SK‐N‐SH cells. To further verify whether syringolin A induces apoptosis, SK‐N‐SH cells were analysed by flow cytometry using the annexin V assay. This sensitive method is used to detect apoptotic cells. Annexin V detects phosphatidyl serine, an intracellular cell membrane component, which is extraverted in apoptotic cells and displayed on the cell surface. As shown in Fig. 6, SK‐N‐SH cells treated with 25 µm syringolin A for 48 h strongly bound annexin V, whereas cells in the absence of syringolin A did not bind annexin V. A shift to the right on the x‐axis (FL1‐H) indicates the specific binding of annexin V to the cell surface of treated cells, and is compared to control cells by superimposition (overlay) of both histograms (n = 3). This result further confirms that syringolin A induces apoptosis in SK‐N‐SH cells.
Figure 6.
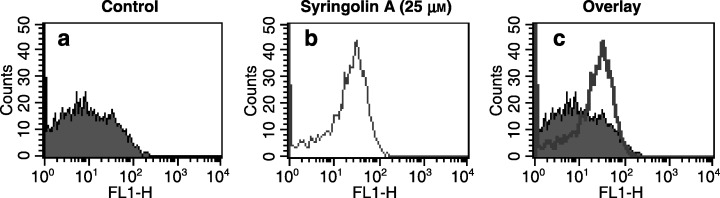
Syringolin A treatment induces apoptosis. Annexin V assay with SK‐N‐SH cells in the (a) absence or (b) presence of 25 µm syringolin A, 48 h after treatment. The superimposition (overlay) of both histograms shown in (a) and (b) clearly reveals a right‐shift of the annexin V peak (FL1‐H), which is indicative of apoptotic cells (c). Cells were stained with annexin V and propidium iodide (PI), analysed by flow cytometry, and data interpreted using the cellquest software program. FL1‐H, log fluorescence intensity. The results are representative of three independent determinations (n = 3).
DISCUSSION
As the role of syringolin A has never been studied in a mammalian cell system, its role in plants has been previously examined. In cultured rice cells, syringolin A, at nanomolar concentrations, causes a transient accumulation of the Pir7b gene transcript, which encodes an esterase (EC 3.1.1.1) of the large family of ‘α/β hydrolase fold’ proteins (Wäspi et al. 1998b; Hassa et al. 2000). This accumulation can be prevented by cycloheximide, indicating the involvement of de novo protein synthesis. Furthermore, calyculin and okadaic acid, two protein phosphatase inhibitors, block Pir7b gene induction, whereas the serine/threonine protein kinase inhibitors staurosporine and K‐252a have no effect on Pir7b transcript levels. These results implied that dephosphorylation of a phosphoprotein is an important step in the syringolin A‐triggered signal transduction pathway in rice plant cells.
The aim of the present study was to investigate the effects of syringolin A in cancer cells. Our results showed that syringolin A, at micromolar concentrations, exhibits strong anti‐proliferative activity with typical signs of apoptosis in NB and ovarian cancer cells. To gain more insight into the mechanisms by which syringolin A induces apoptosis we analysed the pro‐apoptotic signalling proteins p53 and PARP. We found that syringolin A rapidly increased p53 levels in p53 wild‐type SK‐N‐SH cells and cleaved PARP in both SK‐N‐SH and p53‐deficient SKOV3 cells. It is well known that proteolytic cleavage of procaspase‐3 by caspase‐8 and caspase‐9 results in activated caspase‐3, which is a critical regulator of the signalling cascade that triggers apoptosis. In turn, activated caspase‐3 cleaves PARP, ultimately resulting in DNA fragmentation (Fulda & Debatin 2003). Thus, it is possible that syringolin A induces apoptosis in SK‐N‐SH cells in a p53‐dependent manner that involves caspase activation and PARP cleavage, whereas in p53‐deficient SKOV3 cells, syringolin A induces PARP cleavage and apoptosis through p53‐independent mechanisms. The induction of apoptosis by p53‐independent mechanisms has previously been reported (Michieli et al. 1994; Wu et al. 2002). This observation is of importance and suggests that syringolin A may kill cancer cells that are refractory to other drugs because of their p53 deficiency. The mutation of p53 occurs in up to 60% of human cancers and is an important mechanism of chemoresistance (Levine 1997). Interestingly, p53 mutations are rare in primary NB tumours and cell lines, but develop frequently in relapsed NB (Tweddle et al. 2001b). The p53‐mutant cell line LAN‐1 was established from a relapsed NB tumour after chemotherapeutic treatment (Tweddle et al. 2001b). Because syringolin A inhibited LAN‐1 cells in a dose‐dependent manner (Fig. 2d), it is possible that syringolin A is able to overcome the p53‐dependent mechanism of resistance to therapy, and therefore, might be more effective against resistant NB disease at relapse.
Although our results have shown that syringolin A strongly reduced proliferation of immortalized fibroblast cells Rat‐1, we also found in earlier experiments that comparable doses of syringolin A had more moderate effects on human embryonic kidney cells (HEK‐293), even at several‐fold higher concentrations (250 µm) (not shown). Additional comparative studies with other cancer cells as well as primary normal cells will be necessary to further examine the differential effects and to assess the value of syringolin A as a potential therapeutic compound. Anti‐proliferative activities in normal cells are a common problem in the development of cancer therapeutics. Strategies including the generation of structurally modified syringolin A derivatives and the use syringolin A conjugates that specifically target tumour cells may be employed to optimize its efficacy in cancer cells while reducing the effect on normal cells. Also of importance will be to elucidate the precise mode of action of syringolin A and the mechanism by which it regulates cellular signalling events in mammalian cells.
ACKNOWLEDGEMENTS
The following colleagues at the Cancer Research Center of Hawaii are gratefully thanked: Dr Joe Ramos for helpful scientific advice, Dr Alan Lau for providing the Rat‐1 cells, Mohana Valmiki and Anne Hernandez for technical support. Dr Carl‐Wilhelm Vogel and the Cancer Research Center of Hawaii are thanked for the provided support during the course of this study. Funds for this work were in part provided by the NIH‐supported CURE Summer Student Program and a Career Development grant from the Cancer Research Center of Hawaii to Dr André S. Bachmann, Research and Training Revolving Funds of the Cancer Research Center of Hawaii to Dr Bonnie J. Warn‐Cramer, and by a Swiss National Science Foundation Grant (# 3100A0‐100046) to Dr Robert Dudler.
REFERENCES
- Amrein H, Makart S, Granado J, Shakya R, Schneider‐Pokorny J, Dudler R (2004) Functional analysis of genes involved in the synthesis of syringolin A by Pseudomonas syringae pv. syringae B301 D-R. Mol. Plant Microbe Interact. 17, 90–97. [DOI] [PubMed] [Google Scholar]
- Bachmann AS, Xu R, Ratnapala L, Patil SS (2004) Inhibitory effects of phaseolotoxin on proliferation of leukemia cells HL‐60, K‐562 and L1210 and pancreatic cells RIN‐m5F. Leuk. Res. 28, 301–306. [DOI] [PubMed] [Google Scholar]
- Brodeur GM (2003) Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer 3, 203–216. [DOI] [PubMed] [Google Scholar]
- Davidoff AM, Pence JC, Shorter NA, Iglehart JD, Marks JR (1992) Expression of p53 in human neuroblastoma‐ and neuroepithelioma‐derived cell lines. Oncogene 7, 127–133. [PubMed] [Google Scholar]
- Derry WB, Wilson L, Khan IA, Luduena RF, Jordan MA (1997) Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta‐tubulin isotypes. Biochemistry 36, 3554–3562. [DOI] [PubMed] [Google Scholar]
- El Sayed KA (2005) Natural products as angiogenesis modulators. Mini Rev. Med. Chem. 5, 971–993. [DOI] [PubMed] [Google Scholar]
- Fo CS, Coleman CS, Wallick CJ, Vine AL, Bachmann AS (2006) Genomic organization, expression profile, and characterization of the new protein PRA1 domain family, member 2 (PRAF2). Gene 371, 154–165. [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM (2003) Apoptosis pathways in neuroblastoma therapy. Cancer Lett. 197, 131–135. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Lau AF (1993) Dynamics of connexin43 phosphorylation in pp60v‐src transformed cells. Biochem. J. 295, 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DC (1985) Regulation of syringomycin synthesis in Pseudomonas syringae pv. syringae and defined conditions for its production. J. Appl. Bacteriol. 58, 167–174. [DOI] [PubMed] [Google Scholar]
- Hamblin T (2006) Natural products and the treatment of leukemia. Leuk Res 30, 649–650. [DOI] [PubMed] [Google Scholar]
- Harris SL, Levine AJ (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24, 2899–2908. [DOI] [PubMed] [Google Scholar]
- Hassa P, Granado J, Freydl E, Wäspi U, Dudler R (2000) Syringolin‐mediated activation of the Pir7b esterase gene in rice cells is suppressed by phosphatase inhibitors. Mol. Plant Microbe Interact. 13, 342–346. [DOI] [PubMed] [Google Scholar]
- Heiligtag SJ, Bredehorst R, David KA (2002) Key role of mitochondria in cerulenin‐mediated apoptosis. Cell Death Differ. 9, 1017–1025. [DOI] [PubMed] [Google Scholar]
- Levine A (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Li X, Darzynkiewicz Z (2000) Cleavage of poly (ADP‐ribose) polymerase measured in situ in individual cells: relationship to DNA fragmentation and cell cycle position during apoptosis. Exp. Cell Res. 255, 125–132. [DOI] [PubMed] [Google Scholar]
- Mayer AM, Gustafson KR (2004) Marine pharmacology in 2001–2: antitumour and cytotoxic compounds. Eur J. Cancer 40, 2676–2704. [DOI] [PubMed] [Google Scholar]
- McKenzie PP, Guichard SM, Middlemas DS, Ashmun RA, Danks MK, Harris LC (1999) Wild‐type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage‐induced G1 checkpoint function is attenuated. Clin. Cancer Res. 5, 4199–4207. [PubMed] [Google Scholar]
- Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D (1994) Induction of WAF1/CIP1 by a p53‐independent pathway. Cancer Res. 54, 3391–3395. [PubMed] [Google Scholar]
- Mo YY, Gross DC (1991) Plant signal molecules activate the syrB gene, which is required for syringomycin production by Pseudomonas syringae pv. syringae . J. Bacteriol. 173, 5784–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins DW, Alleva DG, Burger CJ, Elgert KD (1997) Taxol, a microtubule‐stabilizing antineoplastic agent, differentially regulates normal and tumor‐bearing host macrophage nitric oxide production. Immunopharmacology 37, 63–73. [DOI] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer‐drug screening. J. Natl. Cancer Inst. 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Tweddle DA, Malcolm AJ, Cole M, Pearson AD, Lunec J (2001a) p53 Cellular localization and function in neuroblastoma: evidence for defective G (1) arrest despite WAF1 induction in MYCN‐amplified cells. Am. J. Pathol 158, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweddle DA, Malcolm AJ, Bown N, Pearson DJ, Lunec J (2001b) Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 61, 8–13. [PubMed] [Google Scholar]
- Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming C, Lunec J (2003) The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 197, 93–98. [DOI] [PubMed] [Google Scholar]
- Wallick CJ, Gamper I, Thorne M, Feith DJ, Takasaki KY, Wilson SM, Seki JA, Pegg AE, Byus CV, Bachmann AS (2005) Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor‐induced G1 cell cycle arrest in MYCN‐amplified human neuroblastoma cells. Oncogene 24, 5606–5618. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR (2005) Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 24, 3574–3582. [DOI] [PubMed] [Google Scholar]
- Wäspi U, Blanc C, Winkler C, Ruedi P, Dudler R (1998a) Syringolin, a novel peptide elicitor from Pseudomonas syringae pv. syringae that induces resistance to Pyricularia oryzae in rice. Mol. Plant Microbe Interact. 11, 727–733. [Google Scholar]
- Wäspi U, Misteli B, Hasslacher M, Jandrositz. A, Kohlwein SD, Schwab H, Dudler R (1998b) The defense‐related rice gene Pir7b encodes an alpha/beta hydrolase fold protein exhibiting esterase activity towards naphthol AS‐esters. Eur J. Biochem. 254, 32–37. [DOI] [PubMed] [Google Scholar]
- Wäspi U, Hassa P, Staempfli AA, Molleyres LP, Winker T, Dudler R (1999) Identification and structure of a family of syringolin variants: unusual cyclic peptides from Pseudomonas syringae pv. syringae that elicit defense responses in rice. Microbiol. Res. 154, 89–93. [Google Scholar]
- Wäspi U, Schweizer P, Dudler R (2001) Syringolin reprograms wheat to undergo hypersensitive cell death in a compatible interaction with powdery mildew. Plant Cell 13, 153–161. [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Kirschmeier P, Hockenberry T, Yang TY, Brassard DL, Wang L, McClanahan T, Black S, Rizzi G, Musco ML et al. (2002) Transcriptional regulation during p21WAF1/CIP1‐induced apoptosis in human ovarian cancer cells. J. Biol. Chem. 277, 36329–36337. [DOI] [PubMed] [Google Scholar]