

Abstract
Objectives
Conventional isolation of epidermis from the dermis and disruption of epidermal sheets to liberate the cells, are performed using proteolytic enzymes such as thermolysin or collagenase. Selective population expansion of melanocytes is achieved by suppressing proliferation of keratinocytes and fibroblasts in epidermal cell suspensions, using phorbol esters and cholera toxin. Here, we introduce a new procedure for isolation of epidermal cells, using proteolytic activity of kiwi fruit actinidin, and also an improved growth medium for melanocytes in the presence of leukaemia inhibitory factor (LIF) and forskolin.
Materials and methods
Dermo‐epidermal separation and epidermal sheet cell dispersion were performed using actinidin compared to conventional proteases including collagenase, thermolysin or trypsin. Thereafter, melanocyte culture was performed in two common media and one modified medium to discover optimization for these cells.
Results
We found that dermo‐epidermal separation and epidermal sheet cell dispersion using kiwi fruit actinidin were considerably better than previously used methods, both from the aspect of less fibroblast and keratinocyte contamination, and of more viable native cells. Also, melanocytes proliferated better in phorbol ester‐ and cholera toxin‐free proliferation medium supplemented with LIF and forskolin.
Conclusion
Less contamination and higher numbers of viable cells were actinidin preferential for separation of epidermis and isolation of epidermal cells. Supplementation of LIF and forskolin to new medium increased proliferation potential of melanocytes in comparison to exogenous mitogens.
Introduction
Melanocytes are relatively large, clear, dendritic and pigment‐producing differentiated cells that develop from embryonic neural crest progenitors. They share certain traits with other neural crest derived cells, such as those found in the adrenal medulla and peripheral nervous system 1. Neural crest cells migrate along lateral pathways at an outer periphery between epidermal ectoderm and dermatomyotome of the embryo. Of the neural crest progenitors, a proportion are are committed to differentiate into melanocytes 2, 3. Melanoblasts, the lineage‐restricted precursors, enter the epidermis through the dermis, from there they migrate and secondarily invade epidermis to colonize and differentiate into new melanocytes 4.These cells are important to the human body as they provide protection from sunlight, but also are involved in many abnormalities and diseases such as malignant melanoma and vitiligo. They also generate distinct pigment patterns in different species of vertebrates, which play important ecological roles 5. In the basal layer of the epidermis, each melanocyte, via its dendritic arms, is in contact with approximately 36 surrounding keratinocytes 6. Melanocytes, after production of melanin and transfer of these granules to neighbouring keratinocytes, give rise to a special structure for skin protection, called the epidermal melanin unit 7. Because of the important role of these cells, isolation and primary culture of melanocytes and optimization of their culture have been attempted since 1957 8. Melanocytes in vitro are sought, as a tool for studying normal processes of melanogenesis, as well as of disorders of pigmentation and malignant melanoma. Many techniques have been devised to isolate melanocytes 9, 10 and the aim of this studies was to culture melanocytes with the lowest, or no contamination, by fibroblasts and keratinocytes. Selective population expansion of melanocytes and having pure melanocyte cultures were initially achieved by using specific mitogens to enhance melanocyte proliferation and to suppress keratinocyte and fibroblast proliferation in epidermal cell suspensions. This was achieved using tumour promoter 12‐O‐tetradecanoyl phorbol‐13‐acetate (TPA) or its derivates, as well as with intracellular cyclic adenosine 3′,5′ monophosphate (cAMP) enhancer such as cholera toxin 11. As the mitogens alter physiological responses of the melanocyte, attempts have been made to define culturing techniques in the absence of these mitogens 12, 13, 14. Also, it is known that phorbol esters are metabolically stable and have prolonged effects on multiple cell responses 9. In the present study, growth factors were used, that melanocytes normally take naturally from their neighbouring cells. Also, actinidin was used instead of common enzymes such as collagenase, thermolysin or trypsin for dermo‐epidermal separation and epidermal sheet cell dispersion. Actinidin (EC 3.4.22.14) is a cysteine (thiol) protease, first characterized by Arcus 15 and is the major protein in most Actinidia fruits 16; it is well known as a good meat‐tenderizing enzyme 17. However, there are few studies on collagenolytic activity of this protease 18, 19 and furthermore, it has been used previously, successfully for isolation and culture of three different cell types, by our group 19. In the study described here, after dermo‐epidermal separation and epidermal cell dispersion, we used three different media to be able to optimize conditions for culture of melanocytes.
Materials and methods
Ethical statement
Protocols used here conform to the National Health and Medical Research Council of the Iranian Ministry of Health and Medical Education, and guidelines for the use of human tissue, and were approved by the Kermanshah University Medical Ethics Committee.
Tissue sources
Human foreskin specimens were obtained from neonates undergoing circumcision in Hazrat e Maesoumeh hospital. Fifteen foreskin specimens were used, 5 for each culture medium. Fresh tissue samples were immersed in Hanks balanced salt solution (HBSS) without Ca2+ or Mg2+, were supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), gentamicin (100 μg/ml) and amphotrycin B (0.25 μg/ml) and kept at 4 °C until processing. Time interval between sample collection and processing averaged 4 h.
Tissue preparation
After soaking skin specimens in 70% ethanol for 1 min, specimens were transferred to Petri dishes containing HBSS to rinse off the ethanol. Excess fatty tissue and blood were carefully removed and foreskins were cut into pieces (approximately 5 × 5 mm2), using a surgical scalpel blade. The pieces were transferred into tubes containing epidermal isolation solution. In our study, we used three main procedures for preparing epidermal isolation solutions using actinidin, trypsin and thermolysin.
Dermo‐epidermal separation and epidermal cell dispersion using proteolytic enzymes
Actinidin was extracted and purified using a method we have described previously 19. To discover best concentration of actinidin, we used 0.25–10 mg/ml over a time course from 6 to 24 h, for dermo‐epidermal separation at 4 °C, and 2–10 mg/ml over a time course from 15 min to 2 h, for epidermal cell dispersion. In both mentioned steps, actinidin was used together with 0.25% EDTA. All of these experiments were performed in comparison to common methods using trypsin and thermolysin, that have been described and used previously 9, 10. After separation of epidermis from the dermis, epidermal sheets were immersed in 0.25% trypsin‐EDTA (15 min), collagenase H (30, 60 and 90 min), actinidin (30, 60 and 90 min) and a mixture of actinidin and trypsin (3:1) for 20 min at 37 °C. Following this step, the epidermis was mechanically torn apart and vigorously washed to release epidermal cells.
Melanocyte growth medium preparation
Cell suspensions, which may contain remaining stratum corneum, were filtered through an 80‐μm cell strainer mesh and resulting suspensions were centrifuged for 5 min at 800 g at room temperature. Cell pellets were re‐suspended in 1 ml growth medium and cultured in a T25 cell‐culture vessels at 5 × 105 cells/ml, for all media, as described below.
Medium 1 contained PMA (Sigma, Saint Louis, Missouri, USA, cat. no. p1585) a common phorbol ester used for melanocyte culture, and preparation and culture were performed as per Eisinger's method 9. Fresh culture medium was changed three times a week.
Medium 2 contained main growth factors for survival, proliferation and differentiation of melanoblasts and melanocytes, MCDB153 (Sigma, cat. no. M7403), recombinant human stem‐cell factor (10 ng/ml) (rhSCF; R&D systems, Minneapolis, MN, USA cat. no. 255‐SC‐050), recombinant human endothelin‐3 (100 nm) (rhET‐3, Sigma, cat. no. E9173), recombinant human basic fibroblast growth factor (1.14 ng/ml) (rhbFGF; Reliatech, Wolfenbüttel, Germany, cat. no. 300‐003), heat‐inactivated foetal bovine serum (FBS), chelated FBS, L‐glutamine (2 mm), heparin (1 ng/ml) and cholera toxin (20 pm) (Sigma, cat. No. C3021). This method was originally proposed by Hsu et al. 11. Fresh culture medium was changed three times a week.
Medium 3 was new for melanocyte culture, based on a modification of the Hsu et al. method 11. We used LIF (10 ng/ml) (Sigma, cat. no. L5283) and forskolin (100 nm) (Sigma, cat. No. F3917) instead of cholera toxin. Fresh culture medium was changed three times a week.
Measurement of cell viability
To measure cell viability, trypan blue (Gibco, Canada) exclusion assay was used. Melanocytes from all culture media were incubated, in triplicate, in 0.25% (w⁄v) trypan blue stain in phosphate‐buffered saline (PBS). Total cell number and number of trypan blue positive cells were determined by counting at least 200 cells per sample, using a Neubauer lam. Cell viability (percentage) was the ratio of trypan blue‐impermeable cells to total cell count.
Measurement of cell expansion/proliferation
Cell counts were performed in triplicate using a Neubauer lam, after trypsinization of culture flasks, centrifugation and re‐suspension of cell pellets, in 1 ml buffer, on days 1, 4, 7, 9, 12 and 14. Only viable cells – those excluding trypan blue, were counted. On day 3 of all cultures, used medium which contained floating and non‐adherent cells include but are not limited to keratinocytes and fragments of stratum corneum was removed and a volume of 4 ml of fresh medium was added instead.
Reverse transcription polymerase chain reaction (RT‐PCR)
Total RNA was extracted from 1 × 106 melanocytes with 1 ml RNX TM (‐Plus) reagent (Sinagen, Tehran, Iran) following the manufacturer's instructions. After incubation at room temperature for 5 min, 200 μl ice cold chloroform was added and cells were incubated for 5 min on ice; the mixture was then centrifuged at 12 000 g for 15 min. Aqueous phase was transferred to new 1.5 ml tubes and 500 μl of isopropanol was added. Te resultant mixture was incubated for 15 min on ice, centrifuged at 12 000 g and pellets washed in 1 ml ethanol (75%). RNA pellets were dried at room temperature for 30 min and resuspended in 50 μl RNase‐free water. Then extracted RNA was verified by electrophoresis in 1% agarose gel. Quantity and quality of RNA were assayed by spectrophotometery at 260 and 280 nm. cDNA was synthesized from total RNA in 20 μl final volumes using oligo dt primers according to cDNA Synthesis Kit (RT; Qiagen, Hilden, Germany). For all cDNA synthesis, minus RT control was performed. After reverse transcription, amplification was carried out by PCR, primers used were as follows: F: 5′‐ctgtgccagcctgtgctac‐3′ and R: 5′‐caccaatgggacaagagcag‐3′ for Homo sapiens premelanosome protein (PMEL), F: 5′‐gtctttatgcaatggaacgc‐3′ and R: 5′‐gctatcccagtaagtggact‐3′ for Tyrosinase and F: 5′‐ttatagtaccttctctttgcc‐3′ and R: 5′‐gcttgctgtatgtggtacttg‐3′ for Microphthalmia transcription factor (MITF). PCR was carried out in 25 μl of reaction. Following incubation at 94 °C for 3 min, 40 PCR cycles were performed; each PCR cycle consisted of a denaturation step at 94 °C for 45 s, annealing step for 45 s (at 58 °C for MITF and PMEL and 60 °C for tyrosinase) and an extension step at 72 °C for 1 min. The PCR reaction for GAPDH as internal control was 30 cycles.
Immunocytochemistry
Cells in 96‐well plates were fixed in 4% paraformaldehyde for 15 min at room temperature then washed three times in PBS. Cells were permeabilized using PBS containing 1% Triton X‐100 for 15 min (for two of the three markers) and washed three times in PBS. Non‐specific binding was blocked by 1‐h treatment with 6% normal goat serum (Gibco) and 0.1% (w⁄v) BSA (Gibco), then cells were incubated with primary antibodies at 4 °C overnight. Primary antibodies to MITF (M3621), tyrosinase (M3623) and Human Melanoma Black (HMB‐45) (M0634) were bought from Dako (Glostrup, Denmark). Cells were then washed three times in PBS, and incubated with secondary antibody (goat anti‐mouse IgG conjugated to FITC) containing 3% normal goat serum, for 1 h at room temperature. Then they were washed five times in PBS and visualized against controls using an optical immunofluoresence microscope (TS100; Nikon, Tokyo, Japan) with appropriate fluorescence filters; images were captured using an appropriate camera (ELWD 0.3 ⁄OD75; Nikon).
Statistical analysis
All analyses were performed using SPSS statistical software 16.0 (Chicago, IL, USA) and data are presented as mean ± SD. Tukey's test was used to determine statistical proliferation differences between groups. Data were evaluated by one‐way analysis of variance (ANOVA) to assess effects of average percentage differences between experimental groups. Results were considered significant when P‐values were <0.05. All experiments were performed in triplicate unless otherwise noted.
Results
Dermo‐epidermal separation
Isolation of epidermis from dermis was successful at many concentrations and time courses, although best results were obtained with 4 mg/ml of kiwi fruit actinidin at 4 °C overnight compared to the conventional procedure using thermolysin (0.4 mg/ml) for dermo‐epidermal separation 10. Epidermal sheets isolated by actinidin were completely intact with no dermal contamination (Fig. 1a) and thus, there was no need to further separate epidermis from dermis using mechanical help. In comparison, when skin samples were treated with thermolysin (Fig. 1b) or trypsin, it was necessary to use mechanical force to separate epidermis from dermis, which caused dermal cell contamination. When skin tissues were treated with actinidin, the epidermal sheets had no contamination by fibroblasts, as the epidermis was easily detached from the dermis. When epidermal sheets were disintegrated in the next (second) step, treatment of skin samples with actinidin yielded statistically higher numbers of fresh cells.
Figure 1.
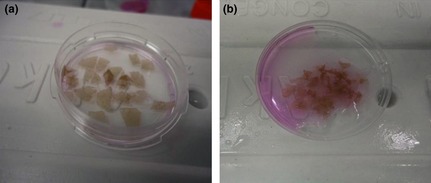
Separation of epidermis from dermis using kiwifruit actinidin. 4 mg/ml (a); epidermal sheets, isolated by actinidin are completely intact with no dermal fibroblast contamination. Separation of epidermis from dermis using thermolysin 0.4 mg/ml. (b); Separation of epidermis from dermis by thermolysin needs extra mechanical forces by forceps, which causes more dermal cell contamination.
In spite of successful epidermal disintegration in trypsin solution, considerable numbers of fibroblasts as well as keratinocytes was observed after separating epidermal sheets, in comparison to the two other enzymatic means; it was often needed to use geneticin for selective elimination of fibroblasts after trypsin treatment 20.
Epidermal cell dispersion
Isolation of epidermal cells by actinidin at different concentrations and time courses showed that the best conditions were for 90 min in 8 mg/ml of protease at 37 °C, in a shaker incubator. In comparison, epidermal disintegration by collagenase H at 2 mg/ml for 90 min at 37 °C (commonly recommended) had weaker results, although isolation with trypsin‐EDTA (2.5 mg/ml) for 15 min and 37 °C in the shaker incubator was completely successful. Isolated cells from the different methods are shown in Fig. 2 and as shown, best results were achieved by mixture of actinidin (8 mg/ml) and trypsin‐EDTA (2.5 mg/ml), 3 and 1 ratio, respectively (Fig. 3). Final concentrations of actinidin and trypsin in this mixture were 6 and 0.62 mg/ml, respectively; thus, a mixture of actinidin and trypsin was used for perfect isolation of epidermal cells. Results of viability testing indicated that mixture of the two proteases provided best results with respect to isolated cell number and viability (Fig. 4).
Figure 2.
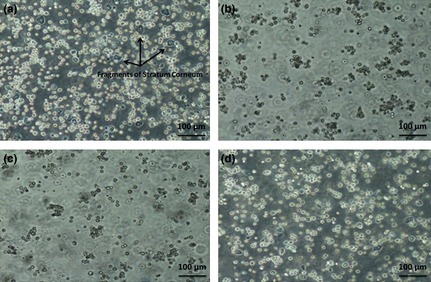
Disintegration of epidermal sheets to liberate epidermal cells using different enzymes. Trypsin‐EDTA (a), provided the high yield of isolated cells, but also high numbers of dead cells; kiwi fruit actinidin (b), which is weaker than trypsin in disintegration of epidermal sheets, but stronger than collagenase‐H, Collagenase‐H (c), the weakest of these enzymes in cell isolation from epidermal sheets, and a mixture of actinidin and trypsin (d), which provided the highest yield of isolated cells like trypsine‐EDTA, but with lower numbers of dead cells.
Figure 3.
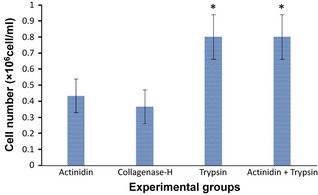
Cell numbers isolated from similar epidermal sheets using different proteolytic enzymes (* is significant when P < 0.05). As results show, best were achieved by mixture of actinidin (8 mg/ml) and trypsin‐EDTA (2.5 mg/ml) with 3:1 ratio.
Figure 4.
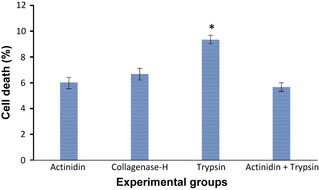
Analysis of cell viability by trypan blue exclusion testing of isolated epidermal cells by proteolytic enzymes. Percentage of dead cells ± standard error of the mean is presented in the histogram (* is significant when P < 0.05). Results of viability testing indicated that mixture of actinidin and trypsin was more convenient for isolation of cells from epidermal sheets due to low numbers of dead cells besides high number of isolated cells.
Melanocyte culture
As mentioned above, dermo‐epidermal separation, 4 mg/ml actinidin overnight, prior to epidermal digestion with the mixture of actinidin and trypsin‐EDTA, proved most reliable in isolating melanocytes from the skin samples, with high cell yield and viability. This method was used for isolation of epidermal sheets and cells, for all culture media. The three different media for culture of melanocytes were the following: medium 1 (containing phorbol ester and cholera toxin), medium 2 (containing cholera toxin only) and medium 3 (without phorbol ester and cholera toxin). Melanocytes grown in all three media (1, 2 and 3), in triplicate, were counted to allow calculation of mean values. Figure 5 shows averages from triplicate experiments for up to 2 weeks of culture.
Figure 5.
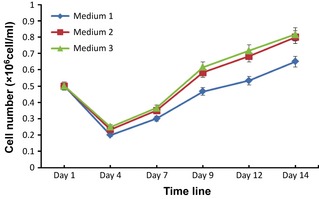
Analysis of melanocyte proliferation in three different media over a 14‐day period: medium 1 (containing phorbol ester and cholera toxin), medium 2 (containing cholera toxin alone) and medium 3 (without phorbol ester and cholera toxin). As the result for cell proliferation indicates, medium 3 provided better results than the other two media. Results are average ± standard error of the mean of three independent experiments.
Results show that melanocytes in medium 3 were more confluent and healthier with higher proliferation rates compared to those of media 1 and 2. Confluence of cells in pure melanocyte in medium 3 (Fig. 6c) after 2 weeks was in the order of 80%, whereas confluence of cells in media 1 and 2 were 40% and 75% after 3 and 2 weeks, respectively (Fig. 6a and 6b).
Figure 6.

Human melanocytes in three different culture media. Melanocyte confluence in medium 1 (a), medium 2 (b) and medium 3 (c) after 14 days. Results show that melanocytes in medium 3 were more confluent and morphologically healthier, with higher proliferation rates than in media 1 and 2.
Cell identification
Melanocytes from all cultured media were identified by immunocytochemistry, for the three most specific markers, HMB‐45, tyrosinase and MITF (Fig. 7a–c). RT‐PCR was also performed for PMEL, tyrosinase and MITF (Fig. 7d).
Figure 7.
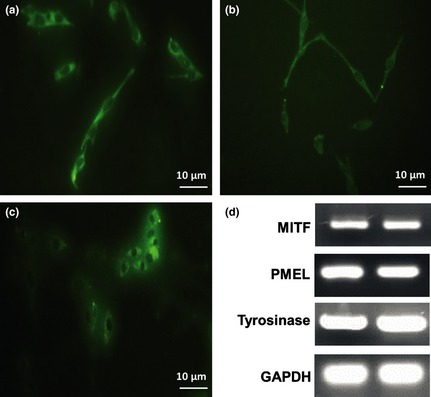
Immunofluorescence staining and reverse transcription–polymerase chain reaction analysis of human melanocytes. Immunofluorescence staining for HMB‐45 (a), tyrosinase (b) and MITF (c). Reverse transcription–polymerase chain reaction analysis for expression of mRNAs for PMEL, tyrosinase and MITF, and GAPDH as internal control (d). Analysis of PCR products showed single‐band amplification products with anticipated sizes, on 2% agarose gel. Data shown for this gel are representative of three independent experiments.
Discussion
Human epidermal melanocytes comprise an important integumentary cell population, protecting the skin from damage by solar irradiation, are responsible for production of the epidermal melanin unit. The first practical method for culture of human melanocytes was proposed by Eisinger and Marko 9. They found that in the culture medium, TPA (12‐O‐tetradecanoyl‐phorbol‐13‐acetate) and cholera toxin were effective additives to serum. These two substances are clearly not natural mitogens for melanocytes, yet induce population expansion of slow‐growing, melanocytes, into cells that proliferate at rates similar to those of their malignant counterparts 21. Because elimination of these two exogenous factors is important for achieving more native melanocytes, some researchers have tried to eliminate one 11 or both of them 22 in their culture medium.
In the initial step of the present study, actinidin, a cysteine protease with collagenolytic activity 19, was used for dermo‐epidermal separation and also epidermal sheet disaggregation, instead of conventional proteolytic enzymes, to achieve higher numbers of viable melanocytes. In conventional methods for skin cell isolation 9, 23, some fibroblasts may also become isolated. However, our results indicated that using actinidin for dermo‐epidermal separation and a combination of actinidin and trypsin for epidermal cell dispersion, provides better results than the other tested methods, in respect of cell yield and contamination of other cell types. Actinidin is more specific than other proteases for digestion of types I and II collagen 19, thus we found this enzyme to be a suitable candidate for dermo‐epidermal separation.
In addition, our results showed that a mixture of actinidin and trypsin was superior for epidermal sheet disintegration compared to the other procedures. Trypsin is a serine protease with broad‐spectrum substrates, but potentially damages cells if conditions of tissue disintegration are not controlled precisely 24. Trypsin is used as a cell surface protein cleavage agent 25, and changes cell surface properties 26, thus, it is not an aproppriate protease for isolation of intact and viable cells from tissues. Although Eisinger's method is capable of producing significant numbers of neonatal melanocytes, only a few of our tissue specimens using this procedure yielded successful heterogenous primary cultures. The cultures had a great deal of fibroblast contamination together with keratinocytes that needed treatment by geneticin 20 plus selective trypsinization. Actinidin, the most abundant protein of kiwi fruit, can be purified using a fast and simple method. Furthermore, it incurs no microbial risk – actinidin is a mild plant protease increasingly it is used for isolation of cells from tissues 19.
Finally, three culture media were compared for maintenance and culture of melanocytes after their isolation. Melanocytes have close physical association to keratinocytes in vivo, and interact with keratinocytes when cultured together in vitro 27, 28, 29. Keratinocytes, however, are able to support proliferation and differentiation of melanocytes in culture 30, 31 and, together with fibroblasts, produce various growth factors, including bFGF, endothelins, SCF and LIF, which are essential for normal melanocyte proliferation and differentiation 32, 33. TPA or other non‐physiological mitogens substitute for keratinocyte‐derived factors partially and support long‐term culture of human melanocytes. However, the mitogens affect melanocyte behaviour, reduce numbers of melanosomes and inhibit terminal differentiation 22, 34, 35, 36. Due to all these limitations, it is better to use culture medium which is free of these agents. While many other teams have used different combinations of factors for this purpose 11, SCF, bFGF, ET‐3, heparin, FCS, chelated FCS, L‐glutamine and MCDB‐153 were used together with LIF and forskolin instead of phorbol esters and cholera toxin in the study reported here. LIF, a 20 kDa protein with 180 amino acid residues, is a multifunctional glycoprotein involved in regulating functions of various types of cells and tissues. LIF has been reported to stimulate proliferation and differentiation of various types of mammalian cells 37 and also differentiation of neonatal mouse epidermal melanocytes, in culture 38. Forskolin is a known α‐melanocyte‐stimulating hormone (α‐MSH) agonist, a cAMP enhancer, and induces melanogenesis by activating MITF 39; also, it has been shown that forskolin can induce MITF 40. Thus, here we used forskolin instead of cholera toxin that is a cAMP enhancer. Although melanocyte microenvironments in media 1 and 2 were not as in vivo (bearing in mind presence of other skin cells there, and their secretions), simulation of in vivo conditions for isolated melanocytes was tried by adding common growth factors to compensate for absence of keratinocytes/fibroblast growth factors. Results show that LIF and forskolin in the presence of SCF, ET‐3 and bFGF stimulated proliferation of melanocytes successfully. Thus, it is suggested that LIF, one of the keratinocyte‐derived factors which is involved in regulating proliferation and differentiation of neonatal mouse epidermal melanoblasts and melanocytes in culture 38, in co‐operation with forskolin, a cAMP elevator, provided the best results for melanocyte culture compared to the other tested methods, which use exogenous mitogens.
In conclusion, less contamination and higher numbers of viable cells in comparison to other common methods and enzymes, indicated actinidin to be preferential for separation of epidermis from dermis and isolation of epidermal cells. Supplementation of LIF and forskolin, as two main natural growth factors, to new medium, instead of exogenous mitogens, increased proliferation of melanocytes more conveniently in comparison to common media, which contain mitogens.
Acknowledgements
We thank Dr. KirollahYari, Dr Maryam Mehrabi and Mr Kaikawoos Gholami (Medical Biology Research Center, Kermanshah University of Medical Sciences) for their technical help and for reviewing the manuscript. We also thank Deputy University for Research of Kermanshah Medical University for financial support.
References
- 1. Tolleson WH (2005) Human melanocyte biology, toxicology, and pathology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 23, 105–161. [DOI] [PubMed] [Google Scholar]
- 2. Teillet MA, Kalcheim C, Le Douarin NM (1987) Formation of the dorsal root ganglia in the avian embryo: segmental origin and migratory behavior of neural crest progenitor cells. Dev. Biol. 120, 329–347. [DOI] [PubMed] [Google Scholar]
- 3. Lee HO, Levorse JM, Shin MK (2003) The endothelin receptor‐B is required for the migration of neural crest‐derived melanocyte and enteric neuron precursors. Dev. Biol. 259, 162–175. [DOI] [PubMed] [Google Scholar]
- 4. Mayer TC (1973) The migratory pathway of neural crest cells into the skin of mouse embryos. Dev. Biol. 34, 39–46. [DOI] [PubMed] [Google Scholar]
- 5. Graham A (2009) Melanocyte production: dark side of the Schwann cell. Curr. Biol. 19, R1116–R1117. [DOI] [PubMed] [Google Scholar]
- 6. Santiago‐Walker A, Li L, Haass NK, Herlyn M (2009) Melanocytes: from morphology to application. Skin Pharmacol. Physiol. 22, 114–121. [DOI] [PubMed] [Google Scholar]
- 7. Nordlund JJ (2007) The melanocyte and the epidermal melanin unit: an expanded concept. Dermatol. Clin. 25, 271–281. [DOI] [PubMed] [Google Scholar]
- 8. Hu F, Staricco RJ, Pinkus H, Fosnaugh RP (1957) Human melanocytes in tissue culture. J. Invest. Dermatol. 28, 15–32. [DOI] [PubMed] [Google Scholar]
- 9. Eisinger M, Marko O (1982) Selective proliferation of normal human melanocytes in vitro in the presence of phorbol ester and cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 79, 2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McLeod SD, Mason RS (1995) Isolation of enriched human melanocyte cultures from fetal, newborn and adult skin. Methods Cell Sci. 17, 187–193. [Google Scholar]
- 11. Hsu MY, Li L, Herlyn M (2005) Cultivation of normal human epidermal melanocytes in the absence of phorbol esters. Methods Mol. Med. 107, 13–28. [DOI] [PubMed] [Google Scholar]
- 12. Falabella R, Escobar C, Borrero I (1989) Transplantation of in vitro‐cultured epidermis bearing melanocytes for repigmenting vitiligo. J. Am. Acad. Dermatol. 21, 257–264. [DOI] [PubMed] [Google Scholar]
- 13. Szabad G, Kormos B, Pivarcsi A, Szell M, Kis K, Kenderessy Szabo A et al (2007) Human adult epidermal melanocytes cultured without chemical mitogens express the EGF receptor and respond to EGF. Arch. Dermatol. Res. 299, 191–200. [DOI] [PubMed] [Google Scholar]
- 14. Kormos B, Belso N, Bebes A, Szabad G, Bacsa S, Szell M et al (2011) In vitro dedifferentiation of melanocytes from adult epidermis. PLoS ONE 6, e17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arcus AC (1959) Proteolytic enzyme of Actinidia chinensis. Biochim. Biophys. Acta 33, 242–244. [DOI] [PubMed] [Google Scholar]
- 16. Boyes S, Strubi P, Marsh H (1997) Actinidin Levels in Fruit of Actinidia Species and Some Actinidia arguta Rootstock‐Scion Combinations. Lebenson Wiss Technol. 30, 379–389. [Google Scholar]
- 17. Lewis DA, Luh BS (1988) Application of actinidin from kiwifruit to meat tenderization and characterization of beef muscle protein hydrolysis. J. Food Biochem. 12, 147–158. [Google Scholar]
- 18. Morimoto K, Kunii S, Hamano K, Tonomura B (2004) Preparation and structural analysis of actinidain‐processed atelocollagen of yellowfin tuna (Thunnus albacares). Biosci. Biotechnol. Biochem. 68, 861–867. [DOI] [PubMed] [Google Scholar]
- 19. Mostafaie A, Bidmeshkipour A, Shirvani Z, Mansouri K, Chalabi M (2008) Kiwifruit actinidin: a proper new collagenase for isolation of cells from different tissues. Appl. Biochem. Biotechnol. 144, 123–131. [DOI] [PubMed] [Google Scholar]
- 20. Halaban R, Alfano FD (1984) Selective elimination of fibroblasts from cultures of normal human melanocytes. In vitro 20, 447–450. [DOI] [PubMed] [Google Scholar]
- 21. Halaban R (1988) Responses of cultured melanocytes to defined growth factors. Pigm. Cell Res. 1, 18–26. [Google Scholar]
- 22. Donatien P, Surleve‐Bazeille JE, Thody AJ, Taieb A (1993) Growth and differentiation of normal human melanocytes in a TPA‐free, cholera toxin‐free, low‐serum medium and influence of keratinocytes. Arch. Dermatol. Res. 285, 385–392. [DOI] [PubMed] [Google Scholar]
- 23. Rheinwald JG, Green H (1975) Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331–343. [DOI] [PubMed] [Google Scholar]
- 24. Letko G, Falkenberg B, Boschmann M (1990) Differences in time course of hepatocyte and pancreocyte damage after incubation with trypsin, chymotrypsin and 2.4‐dinitrophenol. Exp. Pathol. 40, 105–109. [DOI] [PubMed] [Google Scholar]
- 25. Piirainen L, Airaksinen A, Hovi T, Roivainen M (1996) Selective cleavage by trypsin of the capsid protein VP1 of type 3 poliovirus results in improved sorting of cell bound virions. Arch. Virol. 141, 1011–1020. [DOI] [PubMed] [Google Scholar]
- 26. Ortolani G, Patricolo E, Mansueto C (1979) Trypsin‐induced cell surface changes in ascidian embryonic cells: regulation of differentiation of a tissue‐specific protein. Exp. Cell Res. 122, 137–147. [DOI] [PubMed] [Google Scholar]
- 27. Wolff K (1973) Melanocyte‐keratinocyte interactions in vivo: the fate of melanosomes. Yale J. Biol. Med. 46, 384–396. [PMC free article] [PubMed] [Google Scholar]
- 28. Joshi PG, Nair N, Begum G, Joshi NB, Sinkar VP, Vora S (2007) Melanocyte‐keratinocyte interaction induces calcium signalling and melanin transfer to keratinocytes. Pigment Cell Res. 20, 380–384. [DOI] [PubMed] [Google Scholar]
- 29. Seiberg M (2001) Keratinocyte‐melanocyte interactions during melanosome transfer. Pigment Cell Res. 14, 236–242. [DOI] [PubMed] [Google Scholar]
- 30. Gordon PR, Mansur CP, Gilchrest BA (1989) Regulation of human melanocyte growth, dendricity, and melanization by keratinocyte derived factors. J. Invest. Dermatol. 92, 565–572. [DOI] [PubMed] [Google Scholar]
- 31. De Luca M, D'Anna F, Bondanza S, Franzi AT, Cancedda R (1988) Human epithelial cells induce human melanocyte growth in vitro but only skin keratinocytes regulate its proper differentiation in the absence of dermis. J. Cell Biol. 107, 1919–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halaban R, Langdon R, Birchall N, Cuono C, Baird A, Scott G et al (1988) Basic fibroblast growth factor from human keratinocytes is a natural mitogen for melanocytes. J. Cell Biol. 107, 1611–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirobe T (2005) Role of keratinocyte‐derived factors involved in regulating the proliferation and differentiation of mammalian epidermal melanocytes. Pigment Cell Res. 18, 2–12. [DOI] [PubMed] [Google Scholar]
- 34. Donatien PD, Hunt G, Pieron C, Lunec J, Taieb A, Thody AJ (1992) The expression of functional MSH receptors on cultured human melanocytes. Arch. Dermatol. Res. 284, 424–426. [DOI] [PubMed] [Google Scholar]
- 35. Payette R, Biehl J, Toyama Y, Holtzer S, Holtzer H (1980) Effects of 12‐O‐tetradecanoylphorbol‐13‐acetate on the differentiation of avian melanocytes. Cancer Res. 40, 2465–2474. [PubMed] [Google Scholar]
- 36. Weinstein IB, Lee LS, Fisher PB, Mufson A, Yamasaki H (1979) Action of phorbol esters in cell culture: mimicry of transformation, altered differentiation, and effects on cell membranes. J. Supramol. Struct. 12, 195–208. [DOI] [PubMed] [Google Scholar]
- 37. Auernhammer CJ, Melmed S (2000) Leukemia‐inhibitory factor‐neuroimmune modulator of endocrine function. Endocr. Rev. 21, 313–345. [DOI] [PubMed] [Google Scholar]
- 38. Hirobe T (2002) Role of leukemia inhibitory factor in the regulation of the proliferation and differentiation of neonatal mouse epidermal melanocytes in culture. J. Cell. Physiol. 192, 315–326. [DOI] [PubMed] [Google Scholar]
- 39. Busca R, Ballotti R (2000) Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 13, 60–69. [DOI] [PubMed] [Google Scholar]
- 40. Lekmine F, Salti GI (2008) Induction of microphthalmia transcription factor (Mitf) by forskolin and stimulation of melanin release in UISO‐Mel‐6 cells. J. Surg. Res. 149, 27–30. [DOI] [PubMed] [Google Scholar]