

Abstract
Abstract. The vascular network is a series of linked conduits of blood vessels composed of the endothelium, a monolayer of cells that adorn the vessel lumen and surrounding layer(s) of mesenchymal cells (vascular smooth muscle, pericytes and fibroblasts). In addition to providing structural support, the mesenchymal cells are essential for vessel contractility. The extracellular matrix is a major constituent of blood vessels and provides a framework in which these various cell types are attached and embedded. The composition and organization of vascular extracellular matrix is primarily controlled by the mesenchymal cells, and is also responsible for the mechanical properties of the vessel wall, forming complex networks of structural proteins which are highly regulated. The extracellular matrix also plays a central role in cellular adhesion, differentiation and proliferation. This review examines the cellular and extracellular matrix components of vessels, with specific emphasis on the regulation of collagen type I and implications in vascular disease.
INTRODUCTION
Blood vessels deliver nutrients as well as oxygen to all the tissues of the body, and export toxic metabolites for detoxification in the liver and excretion via the kidney. The vascular network branches in hierarchical fashion and is organized spatially to provide access to all organs of the body. Thus blood vessels also serve as the ‘highways’ by which immune cells traverse the body and ‘search’ for pathogens in tissues as a mechanism of immunological surveillance. There are three main types of blood vessels: arteries, veins and capillaries. These have been characterized in a number of ways including by anatomical location, size, physiological function and structural composition (Table 1). Blood vessels exhibit specialized structural features at distinct sites of the vascular tree, which reflect the required function of the vessel at that location. In general, similar primary components are found within all blood vessels, but their organization and relative abundance are uniquely distributed. The walls of the vessels are composed of an endothelial cell layer in direct contact with flow through the vessel lumen. This single cell layer is surrounded by layer(s) of extramural cells (pericytes/smooth muscle cells) which share a common basement membrane, and are embedded in a complex extra‐cellular matrix, which is in turn enveloped by an outer layer composed of resident fibroblasts embedded in connective tissue. In addition to blood vessels, a network of vessels forming the lymphatic system develops in parallel, shares many properties and is as extensive as the blood circulation but less well studied. This review focuses on the extra‐cellular matrix of blood vessels and highlights the use of transgenic technology to examine the expression of extra‐cellular matrix components, primarily collagen type I, and to identify the cell types secreting matrix proteins in the normal state and in changes that take place in pathological conditions.
Table 1.
Types and composition of vessels
| Type of vessel (wall and lumen size) | Composition (cell types) | Extra‐cellular matrix | Reference |
|---|---|---|---|
| Elastic artery (largest arteries, 30 mm in aorta to 5 mm) | Endothelial cells (tunica intima) | Elastin, fibronectin, fibrillin,fibulin, collagen type I, II, III,IV, V, VI, proteoglycans | Rich (1993) |
| Smooth muscle cells (tunica media) | Zhang (1996) | ||
| Fibroblasts (adventitia) | Glukhova (1993) | ||
| Muscular artery (∼5 mm) | Endothelial cells | Elastin, fibronectin, fibullin,collagen type I, III, IV, V,VI, proteoglycans | Jerdan (1986) |
| Smooth muscle cells | Zhang (1996) | ||
| Fibroblasts | |||
| Vein (∼1–5 mm) | Endothelial cells | Elastin, fibronectin, collagentype I, II, III, IV, VI, XII,XIV proteoglycans | Lethias (1996) |
| Smooth muscle cells | Jerdan (1986) | ||
| Maurel (1990) | |||
| Arteriole (> 50 µm) | Endothelial cells | Elastin, collagen I, III, fibrillin | Sterzel 2000 |
| Smooth muscle cells | Baumbach (1993) | ||
| Venule (∼20–100 µm) | Endothelial cells | Laminin, collagen IV, fibronectin | Amselgruber (1999) |
| Smooth muscle cells | Ninomiya (1999) | ||
| Perivascular | Matsubara (1987) | ||
| Capillary (< 20 µm) | Endothelial cells pericytes | Collagen IV, laminin, fibronectin, HSPG | Nerlich (1991) |
| Lymph vessel a (small ∼20–100 µmlarge ∼0.5–5 mm) | Endothelial cells | Collagen IV, laminin, HSPG, fibronectin | Jussila (2002) |
| Smooth muscle cells | Nerlich (1991) | ||
| Aukland (1993) | |||
| Pauli (1983) | |||
| Witte (2001) |
The smaller lymph vessels are thought not to have fibronectin and heparan sulfate proteoglycan (HSPG) (Nerlich 1991).
BLOOD VESSELS
Cellular component of vessels
Large vessels, such as muscular and elastic arteries, are composed of three structural layers known as tunicae. The innermost layer surrounding the lumen, the tunica intima, contains a longitudinally arranged endothelial cell lining, basement membrane and a subendothelial cell connective tissue region with sporadic smooth muscle cells. The middle layer, known as the tunica media, is composed mainly of circumferentially arranged smooth muscle cells (SMC) embedded in a collagen, elastin/fibrillin and proteoglycan‐rich extra‐cellular matrix (ECM). The outer layer, the tunica adventitia, is made up primarily of fibroblasts arranged in a longitudinal direction within a collagen‐ and elastin‐containing ECM separated from the outer media by an external elastic lamina.
Vascular SMC (vSMC) are phenotypically and functionally diverse and their origin remains the subject of ongoing research. Although vSMC are largely mesenchymally derived, some populations are ectodermally derived and have the ability to differentiate to mesenchymal cells (Hungerford & Little 1999). In addition, reports have been made claiming that some vSMC populations originate from vascular endothelial cells through a transdifferentiation process (DeRuiter et al. 1997). Whatever the origin, the vSMC phenotype changes from ‘fibroblast‐like’ in appearance during the embryonic stages of development to a more filamentous, ‘contractile’ phenotype in mature and adult tissues. The ‘fibroblast‐like’ SMC phenotype, often referred to as ‘synthetic’, is characterized by a lack of basement membrane, increased protein synthesis, including ECM synthesis, and a lack of organized contractile apparatus within the cytoplasm. In contrast, the mature vSMC phenotype is characterized by the presence of a basement membrane, reduced synthetic capacity for ECM proteins, an increase of contractile myofilaments and the expression of α‐smooth muscle actin. Vascular SMC can be migratory and proliferative, can produce cytokines and growth factors as well as ECM proteins when required, and are essential in maintaining the vascular tone (Shanahan & Weissberg 1998; Hungerford & Little 1999).
The dated view that the function of the adventitia is as a supporting tissue for the media, is being replaced by evidence that the adventitia plays an active role in vascular remodelling in response to vascular injury and in vascular pathology (Sartore et al. 2001). Central to this function is the adventitial fibroblast, which can be stimulated to an ‘active’ myofibroblastic phenotype by various stimuli including cytokines and growth factors such as transforming growth factor‐β (TGF‐β), often observed in wound healing and repair (Gabbiani 2003). The adventitial fibroblast is involved in mechanisms of apoptosis and cell proliferation and even migration towards the subendothelial space in vascular lesions. The myofibroblast is probably responsible for ECM deposition and adventitial thickening in various vascular pathologies (Sartore et al. 2001). Myofibroblasts and vSMC are phenotypically similar and the question of whether vSMC and myofibroblasts are indeed distinct cell types or represent different phases within a differentiation spectrum is still an open one. It has therefore been a challenge to evaluate the contribution in neointima formation or medial scarring of each cell type. Differentiated myofibroblasts, such as vSMC, consistently express α‐smooth muscle actin but may also express, under specific conditions, other markers of vSMC, such as smooth muscle (SM) myosin heavy chains and caldesmon (Serini & Gabbiani 1999). It has been reported that vSMC and myofibroblasts utilize different elements of the α‐smooth muscle actin promoter to regulate gene expression (Roy, Nozaki & Phan 2001). More recently the observation that the vSMC differentiation marker, smoothelin is not expressed by myofibroblasts may allow a distinction between vSMC and differentiated myofibroblasts to be made (Christen et al. 2001). Differentiated myofibroblasts can also be characterized by their expression of Extra‐Domain A (ED‐A) fibronectin (Tomasek et al. 2002), therefore a more detailed temporal and spatial analysis of these markers may help to clarify the relationship between vSMC and myofibroblasts and identify their respective contribution to a fibrotic lesion.
In small blood vessels (microvessels or capillaries), where a proper adventitia is absent and where there are no SMC, other perivascular cells, the pericytes, are found adjacent to the vascular endothelium. These cells are also present in continuous and fenestrated capillaries, venules and arterioles less than 30 µm in diameter (Rhodin 1968). Pericytes (sometimes known as cells of Rouget) may not represent a homogeneous population of cells (Sims 2000). Developmentally, pericytes are mesenchymally derived, embedded within a basement membrane and are intimately associated through cell‐to‐cell contacts and interdigitating cellular processes with endothelial cells (Fig. 1). Although it is apparent that pericytes and endothelial cells are functionally interdependent, it is believed that they act as a functional unit to regulate blood vessel phenotype (Kalluri 2003). Pericyte ultrastructure reveals many similarities to SMC (Nehls & Drenckhahn 1991) leading to proposals for a contractile function analogous to that of SMC in larger vessels. Immunohistochemistry has shown that they contain actin, myosin, tropomysin and desmin, and are therefore believed to be crucial regulators of microvessel integrity, controlling vessel contraction and permeability (Diaz‐Flores et al. 1991). In addition, their shape suggests a tight grip on the capillary and a mechanical supporting role. Pericytes have also been proposed as progenitors for other mesenchymal cells such as osteoblasts and adipocytes (Sims et al. 2000) and even that some pericytes are a source of new endothelial cells to replace any that are damaged in the endothelium. Studies using knock‐out technology indicate that the absence of pericytes results in severe microvascular abnormalities such as microangiopathy and dilatation (Suri et al. 1996; Hellstrom et al. 1999).
Figure 1.
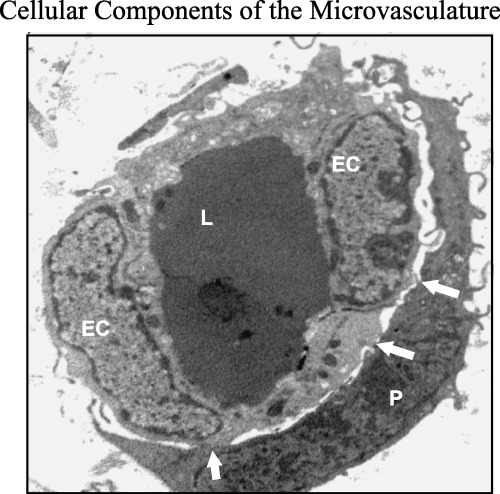
Transmission electron micrograph of dermal microvessel showing the endothelial–pericyte functional unit. The pericyte (P) envelops a capillary lined with two endothelial cells (EC). Note the numerous cytoplasmic interdigitations and adhesion complexes between pericyte and endothelium (arrow). Magnification ×5600.
ECM components
Major constituents of the vessel wall are the extra‐cellular materials, collectively known as the stroma or matrix. In arteries and veins the ECM constitutes more than half of the mass of the wall and is mainly composed of collagens and elastin (Hungerford & Little 1999). Other components, such as fibronectin, and microfibrils (mainly fibrillins) (Arteaga‐Solis et al. 2001) and abundant amorphous or soluble materials such as proteoglycans and small leucine‐rich glycoproteins (Williams 2001), are all present within the extra‐cellular spaces of the vessel wall and are crucial to vessel wall function and integrity.
The extra‐cellular material of the media, including collagen and elastic fibres, is produced primarily by the SMC during development. The turnover of these components is relatively slow compared with that of other tissues and the synthetic versus catabolic processes are also controlled by the SMC of the media. In the adventitia, collagen is synthesized and secreted by the adventitial fibroblasts, as in other connective tissues. As blood vessels develop during postnatal life, the vessel wall increases in diameter and thickness, accompanied by a parallel increase in ECM and both elastin and collagen content, as well as organization of the tunicae. There are also changes in vessel structure, with an increase in the ratio of collagen to elastin, and a reduction in vessel elasticity. Individual elastic fibres (0.1–1.0 µm in diameter) anastomose with each other forming net‐like structures which spread predominantly in a circumferential direction (Fauvel‐Lafeve 1999). A more extensive degree of fusion occurs amongst SMC‐producing lamellae of muscle cells, thus forming lamellar units, which allow the vessel to recoil after distension. An outer elastic lamina similar in appearance but markedly less well developed than the inner elastic lamina is situated at the outer aspect of the media at the boundary with the adventitia.
Elastic fibres are less abundant in the surrounding adventitia. Collagen fibrils (transversely banded cables 30–50 nm in diameter) are found in all three tunicae and especially around the muscle cells of the media. Collagen is also abundant in the adventitia where it forms large bundles of fibrils which increase in size from the innermost region near the media to the outermost component.
In general terms, collagen and elastic fibres in the media run parallel at a small angle to those of the muscle cells and therefore they are predominantly arranged in a circumferential fashion. In contrast, the arrangement of collagen fibres in the adventitia is longitudinal and thus imposes constraints on the elongation of large vessels under pressure. While the outer sheet of collagen in the adventitia limits the distension of the vessel, the collagen network of the media primarily provides attachment to the muscle cells and transmits force around the circumference of the vessels. Thus, by combining the relative rigidity and inextensibility of the collagen fibres with the properties of highly extensible elastic fibres, ample attachment is provided for the muscle cells, favouring a uniform spread of muscle tension around the vessel wall.
EXTRA‐CELLULAR MATRIX SYNTHESIS AND DEGRADATION
The regulation of the production of each vessel ECM component is beyond the scope of this review. Other major ECM proteins in vessels have been studied during development and disease and have been extensively reviewed elsewhere (Arteaga‐Solis et al. 2000; Brooke & Bayes‐Genis 2003).
Although many studies have looked at the formation of the multilayered vessel walls where the ECM is critical for proper formation, the mechanism by which the cells within the vessel wall regulate ECM composition has been largely overlooked. However, genetic manipulation of mice, whether by naturally occurring mutation or designed intervention, through deletion or disruption of ECM components in the early embryo, has either been lethal, such as for collagen type I (Schnieke et al. 1983; Lohler et al. 1984) and elastin (Li et al. 1998), or has resulted in deleterious effects, such as for fibronectin (George et al. 1993). Other components such as perlecan (Weiser et al. 1996) (expressed in vessel walls during development) and endothelium have been suggested to play a role in modulating cytokines found in the local environment and of the expression of transcription factors. Fibulin is another constituent of the core of the elastic microfibrils. The expression of fibulin in mesodermal cells committed to the vSMC lineage, suggests that it is the first ECM component to be expressed in the aortic wall and may act as a precursor to elastin deposition (Dettman et al. 1998).
Transcriptional regulation of collagen type I synthesis in vessels
Recently, several pieces of evidence from transgenic mouse data using promoter analysis of collagen type I from the human, mouse and rat have suggested that separate cis‐acting sequences, often referred to as ‘elements’, are involved in the regulation of collagen type I expression in different cell lineages (Slack et al. 1991; Sokolov et al. 1993; Bogdanovic et al. 1994; Liska et al. 1994; Rossert J. 1995; Krempen et al. 1999). Indeed, evidence from the human pro‐collagen type Iα1 has revealed that a partially deleted human promoter sequence, when used to generate transgenic mice, expressed levels of reporter gene corresponding to the endogenous gene in tail, bone, skin, muscle, heart, lung, intestine, kidney and eye (Sokolov et al. 1993), and vascular smooth muscle (Bedalov et al. 1994). The pro‐collagen type Iα2 gene has also been investigated in rat, chick, mouse and human. In the mouse, a minimal promoter between −350 and +54 was shown to drive a low level of expression in transgenic mice (Niederreither 1992). Such expression was enhanced by the identification of an enhancer region about −17 kilobases (kb) away from the transcription site (Bou‐Gharios et al. 1996).
This far‐upstream enhancer is expressed at a high level in most collagen‐producing cells, including in the vasculature (Fig. 2a–c). Moreover, it has been shown that this enhancer is conserved in humans and is located between −18.8 to −21.1 kb, overlapping three DNase hypersensitive sites, similar to its murine homologue (Antoniv et al. 2001), suggesting that important regulatory elements are phylogenically and evolutionarily conserved. This enhancer was shown to be involved in the activation of the collagen gene in pathological conditions in which deposition of excess collagen type I in skin leads to scarring, such as in the tight skin mouse model (Denton et al. 2001), kidney sclerosis (Chatziantoniou et al. 1998), liver cirrhosis (Inagaki et al. 1998) and lung fibrosis (Ponticos et al. 2004).
Figure 2.
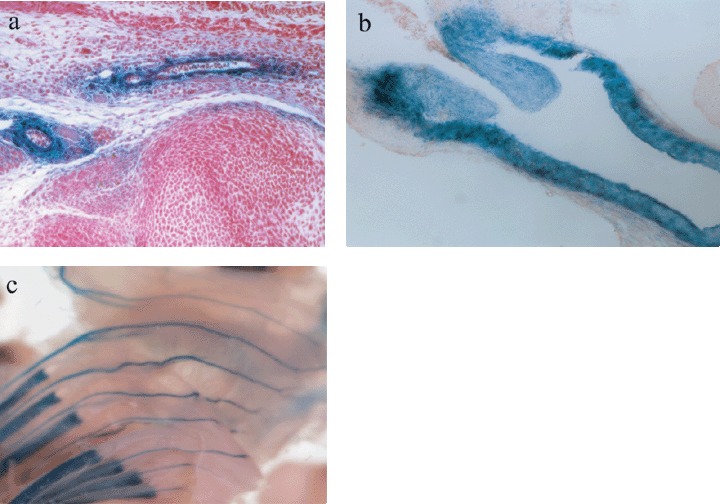
Transgenic mouse tissues showing expression of collagen type I (blue staining of Lacz reporter gene) in different types of blood vessels. (a) Cross section of lower limb in blood vessels of a newborn mouse. (b) Collagen type I expression in the wall and value of the aorta during mouse embryonic development (E15.5). (c) Collagen type I expression in intercostals arteries and ribs during mouse embryonic development (E15.5).
The demonstration that separate cis‐acting elements within the collagen promoter/enhancer can direct expression of collagen type I in a cell lineage‐specific or in an organ‐specific manner, provides the basis for the identification of a SMC‐specific element which can activate transcription of collagen type I within the vessel wall. The identification of such a sequence which regulates collagen expression exclusively in SMC and the delineation of transcription factors which regulate this sequence, can ultimately be used to interfere in the regulation of collagen type I in vascular diseases in which collagen type I production is altered.
It is notable that the activation of collagen is also regulated by cytokines and growth factors such as TGF‐β, connective tissue growth factor, platelet‐derived growth factor, epidermal growth factor, or fibroblast growth factor produced by different cell types present in inflammatory sites. Cytokines, most notably interleukin‐1, interleukin‐4 and tumour necrosis factor‐α, are also capable of modulating fibroblast proliferation and collagen deposition locally. Hormones such as aldosterone and angiotensin II stimulate collagen deposition (Burlew & Weber 2000; Lijnen et al. 2000). Myofibroblasts do not only produce collagen, but also contract the fibrotic area, prevent dilatation by cell–cell and cell–matrix interactions, and play an important role in the architectural control of scar tissue formation and further pathologies (Willems et al. 1994,, 1995, 1999; Blankesteijn et al. 1997).
Matrix degradation in vessels
The degradation of matrix involves a host of enzymes known as matrix metalloproteinases (MMP) which include collagenases, gelatinases, stromolysins, metalloelastases and membrane type‐matrix metalloproteinases (MT‐MMP), all of which can degrade specific matrix components (reviewed by Galis & Khatri 2002; Visse & Nagase 2003; Chase & Newby 2003). These enzymes are produced by the vascular cell component, by endothelial cells, SMC and fibroblasts, and are stored in a latent form within the ECM until they are required. They are capable of completely degrading the matrix and therefore, there must be tightly controlled mechanisms in place to regulate their activities. Matrix‐degrading enzyme activities are regulated on the transcriptional level, by growth factors and cytokines, via processes involving activation (plasminogen/plasmin, MT‐MMP) and processes involving inhibition (tissue inhibitors of matrix metalloproteinases, TIMP1–4) (Galis & Khatri 2002).
MMPs are believed to have a crucial role in vascular matrix remodelling and are essential in the processes of cell migration, novel synthesis of connective tissue and the regulation of growth factors. In addition to vascular remodelling, their involvement has been implicated with, and underlies the mechanisms of pathogenesis of, many vascular diseases including atherosclerosis (Gali & Khatri 2002; Heeneman et al. 2003), restenosis, pulmonary hypertension, and blood vessel abdominal aortic aneurysms (Thompson & Parks 1996).
Following vascular injury SMC migrate through the surrounding matrix to areas of remodelling where they are required for producing and laying down new matrix. This migration process requires degradation of collagen and gelatin by MMP‐1, MMP‐2, MMP‐3, MMP‐9 and MT1‐MMP (Zempo et al. 1996; Bendeck et al. 2002; Lijnen 2002). Furthermore, collagen type I has been shown to contribute to the regulation of SMC migration in vitro, suggesting complex and finely regulated mechanisms of remodelling of cellular ECM components and cell migration (Sierevogel et al. 2002).
Histological studies in normal vessels have revealed the constitutive and uniform expression of MMP‐2 and TIMP1 and TIMP2 (Galis et al. 1995). In diseased vessels a localized increase in expression of other MMPs such as MMP‐2, MMP‐7 (Halpert et al. 1996), MMP‐9 (Galis et al. 2002) and MT1‐MMP (Rajavashisth et al. 1999) is observed. Collagenases (MMP‐1, ‐2, ‐3, ‐9) cleave collagens at a single site within the triple helix and produce specific degradation products which can be detected in tissues and have distinct effects. For example, collagen type I degradation products stimulate the expression of tenascin C, a matrix glycoprotein itself important in controlling SMC proliferation, in remodelling arteries (Jones et al. 1999).
Growth factors that are involved in vessel remodelling are also believed to regulate proteolytic processes. In addition, transcription factors which are involved in the plasminogen/plasmin and MT‐MMP proteolytic mechanisms are often also involved in the activation of the genes expressed in vascular remodelling or in organization of diseased vessels (Bobik & Tkachuk 2003). However, it is worth bearing in mind that MMPs also regulate the biological activity of growth factors and their receptors (Gearing et al. 1995; Fernandez‐Patron et al. 1999).
VASCULAR DISEASES
During pathological processes major changes take place in the arterial wall structure. In this review, atherosclerosis is used as an example of vascular disease to illustrate aspects of ECM dysregulation within large blood vessels. Atherosclerosis is characterized by the accumulation of lipids, cells and ECM in the vessel wall (Ross 1999; Libby et al. 2002). Arterial lesions are rich in matrix, with collagen constituting up to 60% of the total plaque protein (Rekhter 1999). Recent research suggests that the collagen matrix is not simply an inert scaffold but that, instead, after injury there is a dynamic interplay involving matrix degradation and synthesis, with collagens regulating many cellular responses that contribute to arterial occlusion.
Advanced stable atherosclerotic plaques are characterized by a thick fibrous cap, rich in collagen (Fig. 3) and SMC, while unstable or ruptured plaques contain a thin fibrous cap with a dense inflammatory infiltrate, a large lipid core, a luminal thrombus or plaque erosion, and intraplaque haemorrhage (Virmani et al. 2000). Adult human atherosclerotic lesions start off as pre‐existing intimal thickening (Virmani et al. 2000), which is defined by the accumulation of SMC in the intima in the absence of lipid or macrophage foam cells (Virmani et al. 2000). Studies of human aortic intima have shown that type I collagen is the major type of collagen present. In addition, type III and smaller amounts of types IV, V and VI collagen are present (Stary et al. 1992). The importance of matrix proteins for intimal maturation was recently confirmed by gene expression profiling of the maturing neo‐intima and the underlying media. Thirteen genes were significantly up‐regulated in the neo‐intima. Seven of these coded for matrix proteins (six collagens and one versican) and two genes encoded known inducers of matrix synthesis (Core binding factor a1 (Cbfa1) and connective tissue growth factor) (Geary et al. 2002). These data confirm previous studies which showed that although connective tissue growth factor is not expressed in normal blood vessels, it is over‐expressed in atherosclerotic lesions, suggesting an important role in the development and progression of atherosclerosis (Oemar & Luscher 1997).
Figure 3.
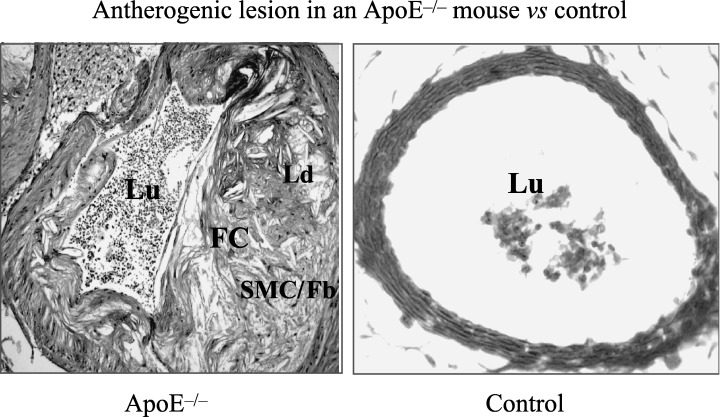
Histochemical staining of an atherogenic lesion in the apolipoprotein E (ApoE) knock‐out mouse and a control blood vessel. FC, fibrous cap; Lu, lumen; SMC/Fb, smooth muscle cells and fibroblasts; Ld, lipid deposits.
Upon progression of atherosclerosis, fibrous tissue remains a major component of atherosclerotic lesions. In a study of sudden cardiac death cases, it has been shown that the major constituent of coronary artery lesions was fibrous tissue, 10% of total plaque area (Schmermund et al. 2001). As in intimal lesions, collagen type I (and to a lesser extent collagen type III and proteoglycans) is the main extra‐cellular component of advanced atherosclerotic lesions and the fibrous cap (Stary et al. 1995). This fibrous cap has been emphasized as a strong determinant of plaque rupture (Newby & Zaltsman 1999). Fibrous cap formation depends on matrix deposition, migration, and proliferation of vSMC, while recently, stem cells or circulating progenitor cells have also been shown to contribute to lesion progression and/or fibrous cap formation (Sata et al. 2002). As for the matrix content of the fibrous cap, it was demonstrated that collagen and proteoglycans show differential accumulation in stable versus ruptured plaques. The fibrous cap of stable lesions was enriched in versican and biglycan and had abundant accumulation of collagen type I. In contrast, intense staining for hyaluronan and versican was found in erosions and at the plaque/thrombus interface with a relative predominance of type III collagen. This was accompanied by a clear reduction in β‐actin‐positive SMC and matrix content (mainly collagen) at rupture sites (Kolodgie et al. 2002). This study underscored the hypothesis that not only matrix content but also its composition is important for the stability of the advanced atherosclerotic lesion. Although the above‐mentioned studies would advocate fibrosis of the cap to reduce the risk of rupture, Moreno et al. recently showed that medial fibrosis was increased (up to 65%) in lesions with a thrombus. Medial fibrosis, characterized by increased collagen deposition, was also associated with rupture of the internal elastic lamina at the intima–medial interface. It was suggested that medial fibrosis and atrophy may reduce arterial compliance, increasing vessel wall circumferential stress. This may be related to reduced fibrous cap thickness and increased risk of rupture (Moreno et al. 2002). This study emphasized that, as in the myocardium, it may not always be beneficial to have more collagen in the vascular wall.
In the arterial wall, collagen has a half‐life of 60–70 days (Nissen et al. 1978). SMC exist in different phenotypes, with the ‘contractile’, quiescent, myofilament‐rich SMC at one end of the spectrum and the ‘synthetic’, proliferative, ECM‐synthesizing and endoplasmic reticulum‐rich SMC at the other. SMC at the more synthetic end of the phenotype spectrum are assumed to be responsible for ECM synthesis in both the media and the intima (Shanahan & Weissberg 1998). Of interest, several studies showed that SMC located in both intimal thickenings and advanced atherosclerotic lesions display more synthetic features, suggesting that also in atherosclerosis synthetic SMC are responsible for ECM synthesis (Mosse et al. 1985; Mosse et al. 1986; Shanahan & Weissberg 1998).
Although it is clear that adventitial myofibroblasts can move to the neointima, the lack of specific markers for myofibroblasts and vSMC complicates the evaluation of the relative contribution of adventitial cells to neointima formation and more advanced atherosclerotic lesions (Sartore et al. 2001). During atherosclerosis, a mechanistic link between lipid accumulation and loss of collagen has been suggested. Rekhter et al. recently showed that lipid accumulation in rabbit atherosclerotic plaques was associated with loss of SMC and collagen breakdown. Although the remaining SMC showed increased collagen synthesis, this was not sufficient to counterbalance collagen degradation and cell loss (Rekhter et al. 2000). The breakdown of collagen was also associated with the accumulation of foamy macrophages, which have been shown to express MMPs in situ (Galis 2000). As atherosclerosis progresses, local collagen degradation can also cause mechanical weakening of the atherosclerotic plaque and lead to plaque rupture (Rekhter et al. 2000). Indeed it has been possible to detect neoepitopes produced as a result of collagen cleavage in atherosclerotic plaques (Sukhova et al. 1999).
CONCLUDING REMARKS
The connective tissue, consisting of specialized cells and ECM, is a dynamic system integral to the structure and function of vessels. Regulation of the components of the ECM is paramount for normal vessel function with dysregulation of expression of proteins such as collagen leading to detrimental pathologies such as atherosclerosis and pulmonary arterial hypertension. Major advances have been made in our understanding of the role of the cellular components in the formation and maintenance of blood vessels and the regulatory mechanism(s) that these cell types control in the homeostasis of the ECM. Key cell types, the smooth muscle cells/pericytes and fibroblasts have been studied in some detail and their role in these processes is becoming clear. Moreover, in recent years the study of vessel pathologies has progressed and steadily advanced our understanding of the cellular and molecular mechanism(s) underlying these diseases. The use of animal models of human diseases (such as the atherogenic and hypertensive models) is now recognized as suitable in combination with transgenic and knock‐out (floxed) animal approaches. These are beginning to reveal the critical factors in pathogenesis of these diseases, and perhaps more importantly new targets for therapeutic intervention.
REFERENCES
- Amselgruber WM, Schafer M, Sinowatz F (1999) Angiogenesis in the bovine corpus luteum: an immunocytochemical and ultrastructural study. Anat. Histol. Embryol. 28, 157. [DOI] [PubMed] [Google Scholar]
- Antoniv TT, De Val S, Wells D, Denton CP, Rabe C, De Crombrugghe B, Ramierez F, Bou‐Gharious G (2001) Characterization of an evolutionary conserved far‐upstream enhancer in the human alpha 2(I) collagen (COL1A2) gene. J Biol Chem. 276, 21754. [DOI] [PubMed] [Google Scholar]
- Arteaga‐Solis E, Gayraud B, Ramirez F (2000) Elastic and collagenous networks in vascular diseases. Cell Struct. Funct. 25, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga‐Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F (2001) Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 154, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aukland K, Reed RK (1993) Interstitial‐lymphatic mechanisms in the control of extracellular fluid volume. Physiol. Rev. 73, 1. [DOI] [PubMed] [Google Scholar]
- Baumbach GL, Hajdu MA (1993) Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension 21, 816. [DOI] [PubMed] [Google Scholar]
- Bedalov A, Breault DT, Sokolov BP, Lichtler AC, Bedalov I, Clark SH, Mack K, Khillan JS, Woody CO, Kream BE, Rowe DW (1994) Regulation of the alpha 1 (I) collagen promoter in vascular smooth muscle cells. Comparison with other alpha 1(I) collagen‐producing cells in transgenic animals and cultured cells. J. Biol. Chem. 269, 4903. [PubMed] [Google Scholar]
- Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM (2002) Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am. J. Pathol. 160, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankesteijn WM, Essers‐Janssen YP, Verluyten MJ, Daemen MJ, Smits JF (1997) A homologue of Drosophila tissue polarity gene frizzled is expressed in migrating myofibroblasts in the infarcted rat heart. Nat. Med. 3, 541. [DOI] [PubMed] [Google Scholar]
- Bobik A, Tkachuk V (2003) Metalloproteinases and plasminogen activators in vessel remodeling. Curr. Hypertens Report 5, 466. [DOI] [PubMed] [Google Scholar]
- Bogdanovic Z, Bedalov A, Krebsbach PH, Pavlin D, Woody CO, Clark SH, Thomas HF, Rowe DW, Kream BE, Lichtler AC (1994) Upstream regulatory elements necessary for expression of the rat COL1A1 promoter in transgenic mice. J. Bone Miner. Res. 9, 285. [DOI] [PubMed] [Google Scholar]
- Bou‐Gharios G, Garrett LA, Rossert J, Niederreither K, Eberspaecher H, Smith C, Black C, De Crombrugghe B (1996) A potent far‐upstream enhancer in the mouse pro alpha 2(I) collagen gene regulates expression of reporter genes in transgenic mice. J. Cell Biol. 134, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke BS, Bayes‐Genis A, Li DY (2003) New insights into elastin and vascular disease. Trends Cardiovasc. Med. 13, 176. [DOI] [PubMed] [Google Scholar]
- Burlew BS, Weber KT (2000) Connective tissue and the heart. Functional significance and regulatory mechanisms. Cardiol. Clin. 18, 435. [DOI] [PubMed] [Google Scholar]
- Chase AJ, Newby AC (2003) Regulation of matrix metalloproteinase (matrixin) genes in blood vessels: a multi‐step recruitment model for pathological remodelling. J. Vasc. Res. 40, 329. (Epub 2003 July 29). [DOI] [PubMed] [Google Scholar]
- Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC (1998) Nitric oxide inhibition induces early activation of type I collagen gene in renal resistance vessels and glomeruli in transgenic mice. Role of endothelin. J. Clin. Invest. 101, 2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen T, Verin V, Bochaton‐Piallat M, Popowski Y, Ramaekers F, Debruyne P, Camenzind E, Van Eys G, Gabbiani G (2001) Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation 103, 882. [DOI] [PubMed] [Google Scholar]
- Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ (1995) Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 147, 325. [PMC free article] [PubMed] [Google Scholar]
- Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF (1999) The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc. Res. 44, 232. [DOI] [PubMed] [Google Scholar]
- Denton CP, Zheng B, Shiwen X, Zhang Z, Bou‐Gharios G, Eberspaecher H, Black CM, De Crombrugghe B (2001) Activation of a fibroblast‐specific enhancer of the proalpha2(I) collagen gene in tight‐skin mice. Arthritis Rheum. 44, 712. [DOI] [PubMed] [Google Scholar]
- Deruiter MC, Poelmann RE, Vanmunsteren JC, Mironov V, Markwald RR, Gittenburger‐de Groot AC (1997) Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro . Circ Res. 80, 601. [DOI] [PubMed] [Google Scholar]
- Dettman RW, Denetclaw W, Ordahl CP Jr, Bristow J (1998) Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193, 169. [DOI] [PubMed] [Google Scholar]
- Diaz‐Flores L, Gutierrez R, Varela H, Rancel N, Valladares F (1991) Microvascular pericytes: a review of their morphological and functional characteristics. Histol. Histopathol. 6, 269. [PubMed] [Google Scholar]
- Fauvel‐Lafeve F (1999) Microfibrils from the arterial subendothelium. Int. Rev. Cytol. 188, 1. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Patron C, Radomski MW, Davidge ST (1999) Vascular matrix metalloproteinase‐2 cleaves big endothelin‐1 yielding a novel vasoconstrictor. Circ. Res. 85, 906. [DOI] [PubMed] [Google Scholar]
- Gabbiani G (2003) The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500. [DOI] [PubMed] [Google Scholar]
- Galis ZS (2000) Atheroma morphology and mechanical strength: looks are important, after all – lose the fat. Circ. Res. 86, 1. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Khatri JJ (2002) Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ. Res. 90, 251. [PubMed] [Google Scholar]
- Galis ZS, Muszynski M, Sukhova GK, Simon‐Morrissey E, Libby P (1995) Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Ann. N Y Acad. Sci. 748, 501. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Johnson C, Godin D, Magid R, Shipley JM, Senior RM, Ivan E (2002) Targeted disruption of the matrix metalloproteinase‐9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ. Res. 91, 852–859. [DOI] [PubMed] [Google Scholar]
- Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements JM, Crimmin M, Davidson AH, Drummond AH, Galloway WA, Gilbert R, et al. (1995) Matrix metalloproteinases and processing of pro‐TNF‐alpha. J. Leukoc. Biol. 57, 774. [DOI] [PubMed] [Google Scholar]
- Geary RL, Wong JM, Rossini A, Schwartz SM, Adams LD (2002) Expression profiling identifies 147 genes contributing to a unique primate neointimal smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 22, 2010. [DOI] [PubMed] [Google Scholar]
- George EL, Georges‐Labouesse EN, Patel‐King RS, Rayburn H, Hynes RO (1993) Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 119, 1079. [DOI] [PubMed] [Google Scholar]
- Glukhova M, Koteliansky V, Fondacci C, Marotte F, Rappaport L (1993) Laminin variants and integrin laminin receptors in developing and adult human smooth muscle. Dev. Biol. 157, 437. [DOI] [PubMed] [Google Scholar]
- Halpert I, Sires UI, Roby JD, Potter‐Perigo S, Wight TN, Shapiro SD, Welgus HG, Wickline SA, Parks WC (1996) Matrilysin is expressed by lipid‐laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc. Natl Acad. Sci. USA 93, 9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeneman S, Cleutjens JP, Faber BC, Creemers EE, Van Suylen RJ, Lutgens E, Cleutjens KB, Daemen MJ (2003) The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J. Pathol. 200, 516. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C (1999) Role of PDGF‐B and PDGFR‐beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126, 3047. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Little CD (1999) Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J. Vasc. Res. 36, 2. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Truter S, Bou‐Gharios G, Garrett LA, De Crombrugghe B, Nemoto T, Greenwel P (1998) Activation of Proalpha2 (I) collagen promoter during hepatic fibrogenesis in transgenic mice. Biochem. Biophys. Res. Commun. 250, 606. [DOI] [PubMed] [Google Scholar]
- Jerdan JA, Glaser BM (1986) Retinal microvessel extracellular matrix: an immunofluorescent study. Invest. Ophthalmol. Vis. Sci. 27, 194. [PubMed] [Google Scholar]
- Jones PL, Jones FS, Zhou B, Rabinovitch M (1999) Induction of vascular smooth muscle cell tenascin‐C gene expression by denatured type I collagen is dependent upon a beta3 integrin‐mediated mitogen‐activated protein kinase pathway and a 122‐base pair promoter element. J. Cell Sci. 112, 435. [DOI] [PubMed] [Google Scholar]
- Jussila L, Alitalo K (2002) Vascular growth factors and lymphangiogenesis. Physiol. Rev. 82, 673. [DOI] [PubMed] [Google Scholar]
- Kalluri R (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422. [DOI] [PubMed] [Google Scholar]
- Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN, Virmani R (2002) Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 22, 1642. [DOI] [PubMed] [Google Scholar]
- Krempen K, Grotkopp D, Hall K, Bache A, Gillan A, Rippe RA, Brenner DA, Breindl M (1999) Far upstream regulatory elements enhance position‐independent and uterus‐specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr. 8, 151. [PMC free article] [PubMed] [Google Scholar]
- Lethias C, Labourdette L, Willems R, Comte J, Herbage D (1996) Composition and organization of the extracellular matrix of vein walls: collagen networks. Int. Angiol. 15, 104. [PubMed] [Google Scholar]
- Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT (1998) Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105, 1135. [DOI] [PubMed] [Google Scholar]
- Lijnen HR (2002) Extracellular proteolysis in the development and progression of atherosclerosis. Biochem. Soc. Trans. 30, 163. [DOI] [PubMed] [Google Scholar]
- Lijnen PJ, Petrov VV, Fagard RH (2000) Induction of cardiac fibrosis by transforming growth factor‐beta(1). Mol. Genet. Metab. 71, 418. [DOI] [PubMed] [Google Scholar]
- Liska DJ, Reed MJ, Sage EH, Bornstein P (1994) Cell‐specific expression of alpha 1 (I) collagen‐hGH minigenes in transgenic mice. J. Cell Biol. 125, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohler J, Timpl R, Jaenisch R (1984) Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell 8, 597. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Ziff M (1987) Basement membrane thickening of postcapillary venules and capillaries in rheumatoid synovium. Immunoelectron microscopic and electron microscopic morphometric analysis. Arthritis Rheum. 30, 18. [DOI] [PubMed] [Google Scholar]
- Maurel E, Azema C, Deloly J, Bouissou H (1990) Collagen of the normal and the varicose human saphenous vein: a biochemical study. Clin. Chim. Acta 13, 27. [DOI] [PubMed] [Google Scholar]
- Moreno PR, Purushothaman KR, Fuster V, O'Connor WN (2002) Intimomedial interface damage and adventitial inflammation is increased beneath disrupted atherosclerosis in the aorta: implications for plaque vulnerability. Circulation 105, 2504. [DOI] [PubMed] [Google Scholar]
- Mosse PR, Campbell GR, Wang ZL, Campbell JH (1985) Smooth muscle phenotypic expression in human carotid arteries. I. Comparison of cells from diffuse intimal thickenings adjacent to atheromatous plaques with those of the media. Lab. Invest. 53, 556. [PubMed] [Google Scholar]
- Mosse PR, Campbell GR, Campbell JH (1986) Smooth muscle phenotypic expression in human carotid arteries. II. Atherosclerosis‐free diffuse intimal thickenings compared with the media. Arteriosclerosis 6, 664. [DOI] [PubMed] [Google Scholar]
- Nehls V, Drenckhahn D (1991) Heterogeneity of microvascular pericytes for smooth muscle type alpha actin. J. Cell Biol. 113, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlich AG, Schleicher E (1991) Identification of lymph and blood capillaries by immunohistochemical staining for various basement membrane components. Histochemistry 96, 449. [DOI] [PubMed] [Google Scholar]
- Newby AC, Zaltsman AB (1999) Fibrous cap formation or destruction – the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc. Res. 41, 345. [PubMed] [Google Scholar]
- Niederreither K, D’Souza RN, De Crombrugghe B (1992) Minimal DNA sequences that control the cell lineage‐specific expression of the pro alpha 2(I) collagen promoter in transgenic mice. J.Cell Biol. 119, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya H, Inomata T, Ogihara K (1999) Collagen fiber arrangement in canine hepatic venules. J. Vet. Med. Sci. 61, 21. [DOI] [PubMed] [Google Scholar]
- Nissen R, Cardinale GJ, Udenfriend S (1978) Increased turnover of arterial collagen in hypertensive rats. Proc. Natl Acad. Sci. USA 75, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oemar B, Luscher TF (1997) Connective tissue growth factor, friend or foe? Arterioscler. Thromb. Vasc. Biol. 17, 1483. [DOI] [PubMed] [Google Scholar]
- Pauli BU, Schwartz DE, Thonar EJ, Kuettner KE (1983) Tumor invasion and host extracellular matrix. Cancer Metastasis Rev. 2, 129. [DOI] [PubMed] [Google Scholar]
- Ponticos M, Abraham D, Alexakis C, Lu Q‐L, Black CM, Partridge T, Bou‐Gharios G (2004) Col1a2 enhancer regulates collagen activity during development and in adult tissue repair. Matrix Biol. 22, 619. [DOI] [PubMed] [Google Scholar]
- Rajavashisth TB, Xu XP, Jovinge S, Meisel S, Xu XO, Chai NN, Fishbein MC, Kauls Cercek B, Sharifi B, Shah PK (1999) Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediators. Circulation 99, 3103. [DOI] [PubMed] [Google Scholar]
- Rekhter MD (1999) Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc. Res. 41, 376. [DOI] [PubMed] [Google Scholar]
- Rekhter MD, Hicks GW, Brammer DW, Hallak H, Kindt E, Chen J, Rosebury WS, Anderson MK, Kuipers PJ, Ryan MJ (2000) Hypercholesterolemia causes mechanical weakening of rabbit atheroma: local collagen loss as a prerequisite of plaque rupture. Circ. Res. 86, 101. [DOI] [PubMed] [Google Scholar]
- Rhodin JA (1968) Ultrastructure of mammalian venous capillaries, venules, and small collecting veins. J. Ultrastructure Res. 25, 452. [DOI] [PubMed] [Google Scholar]
- Rich CB, Goud HD, Bashir M, Rosenbloom J, Foster JA (1993) Developmental regulation of aortic elastin gene expression involves disruption of an IGF‐I sensitive repressor complex. Biochem. Biophys. Res. Commun. 196, 1316. [DOI] [PubMed] [Google Scholar]
- Ross R (1999) Atherosclerosis – an inflammatory disease. N. Engl J. Med. 340, 115. [DOI] [PubMed] [Google Scholar]
- Rossert J, Eberspaecher H, De Crombrugghe B (1995) Separate cis‐acting DNA elements of the mouse pro‐alpha 1 (I) collagen promoter direct expression of reporter genes to different type I collagen‐producing cells in transgenic mice. J. Cell Biol. 129, 1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SG, Nozaki Y, Phan SH (2001) Regulation of α‐smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. Int. J. Biochem. Cell Biol. 33, 723. [DOI] [PubMed] [Google Scholar]
- Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P (2001) Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 89, 1111. [DOI] [PubMed] [Google Scholar]
- Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R (2002) Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 8, 403. [DOI] [PubMed] [Google Scholar]
- Schmermund A, Schwartz RS, Adamzik M, Sangiorgi G, Pfeifer EA, Rumberger JA, Burke AP, Farb A, Virmani R (2001) Coronary atherosclerosis in unheralded sudden coronary death under age 50: histo‐pathologic comparison with ‘healthy’ subjects dying out of hospital. Atherosclerosis 155, 499. [DOI] [PubMed] [Google Scholar]
- Schnieke A, Harbers K, Jaenisch R (1983) Embryonic lethal mutation in mice induced by retrovirus insertion into the alpha1 (I) collagen gene. Nature 304, 315. [DOI] [PubMed] [Google Scholar]
- Serini G, Gabbiani G (1999) Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell. Res. 250, 273. [DOI] [PubMed] [Google Scholar]
- Shanahan CM, Weissberg PL (1998) Smooth muscle cell heterogeneity: patterns of gene expression in vascular smooth muscle cells in vitro and in vivo . Arterioscler. Thromb. Vasc. Biol. 18, 333. [DOI] [PubMed] [Google Scholar]
- Sierevogel MJ, Velema E, Van Der Meer FJ, Nijhuis MO, Smeets M, De Kleijn DP, Borst C, Pasterkamp G (2002) Matrix metalloproteinase inhibition reduces adventitial thickening and collagen accumulation following balloon dilation. Cardiovasc. Res. 55, 864. [DOI] [PubMed] [Google Scholar]
- Sims DE (2000) Diversity within pericytes. Clin. Exp. Pharmacol. Physiol. 27, 842. [DOI] [PubMed] [Google Scholar]
- Slack JL, Liska DJ, Bornstein P (1991) An upstream regulatory region mediates high‐level, tissue‐specific expression of the human alpha 1(I) collagen gene in transgenic mice. Mol. Cell Biol. 11, 2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov BP, Mays PK, Khillan JS, Prockop DJ (1993) Tissue‐ and development‐specific expression in transgenic mice of a type I procollagen (COL1A1) minigene construct with 2.3 kb of the promoter region and 2 kb of the 3′‐flanking region. Specificity is independent of the putative regulatory sequences in the first intron. Biochemistry 32, 9242. [DOI] [PubMed] [Google Scholar]
- Stary HC, Blankenhorn DH, Chandler AB, Glagov S, Insull W, Richardson M Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD et al. (1992) A definition of the intima of human arteries and of its atherosclerosis‐prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. 12, 120. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Rosenfeld ME Jr, Schwartz CJ, Wagner WD, Wissler RW (1995) A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 15, 1512. [DOI] [PubMed] [Google Scholar]
- Sterzel RB, Hartner A, Schlotzer‐Schrehardt U, Voit S, Hausknecht B, Doliana R, Colombatti A, Gibson MA, Braghetta P, Bressan GM (2000) Elastic fiber proteins in the glomerular mesangium in vivo and in cell culture. Kidney Int. 58, 1588. [DOI] [PubMed] [Google Scholar]
- Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P (1999) Evidence for increased collagenolysis by interstitial collagenases‐1 and ‐3 in vulnerable human atheromatous plaques. Circulation 99, 2503. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD (1996) Requisite role of Angiopoietin‐1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87, 1171. [DOI] [PubMed] [Google Scholar]
- Thompson RW, Parks WC (1996) Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann. NY Acad. Sci. 800, 157. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349. [DOI] [PubMed] [Google Scholar]
- Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM (2000) Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 20, 1262. [DOI] [PubMed] [Google Scholar]
- Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827. [DOI] [PubMed] [Google Scholar]
- Weiser MC, Belknap JK, Grieshaber SS, Kinsella MG, Majack RA (1996) Developmental regulation of perlecan gene expression in aortic smooth muscle cells. Matrix Biol. 15, 331. [DOI] [PubMed] [Google Scholar]
- Willems IE, Havenith MG, De Mey JG, Daemen MJ (1994) The alpha‐smooth muscle actin‐positive cells in healing human myocardial scars. Am. J. Pathol. 145, 868. [PMC free article] [PubMed] [Google Scholar]
- Williams KJ (2001) Arterial wall chondroitin sulfate proteoglycans: diverse molecules with distinct roles in lipoprotein retention and atherogenesis. Curr. Opin. Lipidol. 12, 477. [DOI] [PubMed] [Google Scholar]
- Witte MH, Bernas MJ, Martin CP, Witte CL (2001) Lymphangiogenesis and lymphangio‐dysplasia: from molecular to clinical lymphology. Microsc. Res. Techn. 55, 122. [DOI] [PubMed] [Google Scholar]
- Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW (1996) Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler. Thromb. Vasc. Biol. 16, 28. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Timpl R, Sasaki T, Chu ML, Ekblom P (1996) Fibulin‐1 and fibulin‐2 expression during organogenesis in the developing mouse embryo. Dev. Dyn. 205, 348. [DOI] [PubMed] [Google Scholar]