

Abstract
The regenerative potential of mesenchymal stromal cells (MSC) holds great promise in using them for treatment of a wide range of debilitating diseases. Several types of culture media and systems have been used for large‐scale expansion of MSCs in vitro; however, the majority of them rely heavily on using foetal bovine serum (FBS)‐supplement for optimal cell proliferation. FBS‐based cultures pose the potential threat of spread of transmissible spongiform encephalopathy and bovine spongiform encephalopathy to MSCs and then to their recipients. A recent trend in cell culture is to change from serum‐use to serum‐free media (SFM). In this context, the current review focuses specifically on employment of various SFM for MSCs and discusses existences of various options with which to substitute FBS. In addition, we analyse MSC population growth kinetic patterns using various SFM for large‐scale production of MSCs.
Introduction
Mesenchymal stromal cells (MSCs) compose an important system of choice in regenerative biopharmaceuticals due to their safety, ability to expand in multiple fold, are hypo‐immunogenic, have tri‐lineage capacity, can repair or regenerate tissues by paracrine or autocrine functions, and help in treatment of a variety of human diseases 1. MSCs can be characterized by a group of surface markers; they are positive for CD105, CD73 and CD90 but do not express CD45, CD34, CD14, CD11b, CD79a, CD19 and HLA‐DR, as set by the International Society for Cellular Therapy guidelines. Generally, MSCs are cultured in foetal bovine serum (FBS)‐containing media to be used in clinical trials, but thus could pose a threat to communication of transmissible spongiform encephalopathy and bovine spongiform encephalopathy to recipient patients. Consequently, there is a need for changing to FBS‐free culture systems for production of clinical grade MSCs, which are completely safe.
Human MSCs (hMSCs) can be infused or injected into the blood stream as a delivery vehicle in allogeneic clinical settings, as they cannot be detected by the host's immune system 2, 3. Large numbers of clinical trials with them are in progress (www.clinicaltrials.gov) and each requires large numbers of cells, in the range of 1–5 million MSCs/kg body weight 4. To meet the huge demand for expanded cells, efficient isolation and expansion processes need to be in place. Cell bank and relevant scalable processes have to be established to cater for market demand subsequent to drug approval. Robust MSC production systems with constant cell yield per batch and consistent quality of cells are an essential element for successful cell therapy clinical trials, even for FBS‐supplemented media for MSCs expansion 5, 6. Increased exploration of disease indications for cell therapy‐based clinical trials demands a large number of doses for multiple indications, which further enhances increased use of FBS for MSC production.
Recent MSC‐based clinical trials did not suffer from the misconception that proof of concept/mechanism of action must be defined or produced before clinical testing 7. Hence, culturing cells for the large numbers of ongoing clinical trials has increased FBS consumption annually in demonstration of safety and efficacy. In 2003, global market for FBS was about 0.5 million litres but had increased to 0.8 million litres by 2008; growth rate is estimated to accelerate by at least 10–15% annually 8. More than a million foetuses of pregnant cows needed to be slaughtered to produce 0.5 million litres of FBS, which involves potential suffering while blood is withdrawn via a needle inserted into the heart, without the use of anaesthesia. In addition, seasonal and continental variations in serum composition lead to lot‐by‐lot variation, which in turn causes changes in morphological, phenotypic and population kinetic changes in MSCs produced. Use of undefined animal serum products in MSC culturing imposes numerous safety issues; 20–50% of commercial FBS is virus positive 9, 10 and internalized xenogeneic antigen during serum‐supplemented culture cannot be eliminated, even after post‐harvest washing procedures of cell therapy products 11, 12. There are ethical, animal welfare and scientific issues for the use of FBS in cell‐based manufacturing systems and we have listed some from available literature (Table 1). Thus, type of cell culture medium used for producing MSCs and safety and efficacy of derived MSCs may be significantly influenced by media chosen and processing steps 13. Due to increasing concerns of safety as well as issues related to consistency and reproducibility, there is increased need to improve stem‐cell bioprocesses to develop robust production process using media free of animal components 12, 14, 15 (Fig. 1).
Table 1.
Disadvantages of FBS‐supplemented media
| S. no. | Disadvantages/potential challenges | References |
|---|---|---|
| 1 | Undefined media components, lot‐to‐lot variation | 16 |
| 2 | High risk of contamination – bacteria, virus and mycoplasma | 9 |
| 3 | High cost of serum | 17 |
| 4 | FBS protein internalized into stem cells and carries risk of transmitting unknown infectious agent | 18 |
| 5 | FBS enhances various non‐MSCs to attach and provide heterogeneous population and proliferate after isolation stage of primordial culture | 19, 20 |
| 5 | FBS‐grown cells raise T‐cell response against transduced MSCs and trigger FBS‐specific immune reactions | 21 |
| 6 | Adventitious agent testing of raw material, intermediate product and final product | 22 |
| 7 | Not all lots of FBS are suitable for isolation and expansion of MSCs and selection of suitable lot of FBS needs rigorous FBS lot validation | 17 |
| 8 | FBS‐supplemented cultures undergo replicative senescence with progressive loss of differentiation capacity and without any cellular transformation characteristics | 23 |
| 9 | Bovine protein attachment to the cells grown in FBS‐containing medium triggers xenogeneic immune response, which affects the viability, safety and efficacy of transplanted MSCs | 24 |
| 10 | FBS‐containing cultures require post‐trypsinization downstream process with extra washing step to remove or decrease FBS content on cells | 19, 25 |
FBS, foetal bovine serum; MSC, mesenchymal stromal cells.
Figure 1.
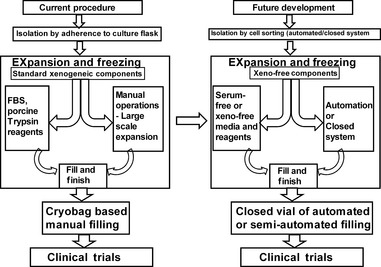
Diagrammatic overview of mesenchymal stromal cell bioprocessing shifting from open system of manual operation to closed controllable system of automation to enhance good manufacturing practices processing and future requirements to enter clinical trails. IMP, investigational medicinal product.
Various serum‐free media (SFM) or humanized media are being studied to develop commercial SFM as an alternative to FBS for culturing of MSCs for clinical applications. Collective reasons for transition to humanized media include safety, efficacy, consistency and reproducibility 9, 10, 11, 12, 13, 18, 26. Furthermore, decisions need to take into account variables such as basal media, glucose concentration in basal media, stable glutamine, Mononuclear cells (MNCs) plating density, MSC expansion seeding density (SD), supplementation of growth factors and cell culture plastic surface quality 27. In this regard, several humanized media have been investigated, including supplementation of autologous human serum (auto‐HS) or allogeneic HS (allo‐HS), human platelet lysates (HPL), thrombin‐activated platelet rich plasma and umbilical cord blood plasma 15, 20, 28, 29. Over the period of this transition, nevertheless, comparison of gold standard FBS supplementation with humanized alternatives should only be done with great care. As most humanized media are of adult origin and different altogether from the eponymous foetal origin FBS, only the best humanized media can be selected for expansion of MSCs for clinical applications.
Many commercially available SFM and disclosed SFM formulations are available with further ones still under development 30, 31, 32, 33, 34, 35. Developing and selecting suitable media, that is compatible with process variables are critical for consistently generating hMSCs that maintain, retain and demonstrate their MSCs characteristics without major deviation in the production process.
Up to now, it has not been possible to develop a universal SFM for all MSCs from various sources. Even when donor MSCs are selected from the same source, they may differ in proliferation kinetics, even when grown in the same culture media. Factors such as donor‐to‐donor variation, age of donor, cell sources/microenvironment of the source, may influence variability in functional characteristics of MSCs such as growth characteristics and differentiation potential 36. Hence, there is a challenge to be able to select suitable media that contribute to efficient isolation, minimization of process fluctuation and retention of multipotency, in long‐term cultures. Thus, we focus from animal component‐free MSC culture media to commercially available SFM or xeno‐free media (XFM), that can be used for cell isolation and expansion procedures for clinical trials or pre‐clinical studies. Isolation and proliferation kinetics of MSCs of various SFM/XFM are compared to gold standard FBS‐supplementation culture. The rapidly evolving field of cell therapy from therapeutic and regulatory points of view, sets new standards for all related aspects.
Classification of culture media
A large number of SFM are commercially available suitable to various cell types, and for various test applications. Use of culture media is increasing exponentially and FBS alternatives are constantly being developed for cell‐based therapies. Alternative culture media, without the use of FBS, confusingly use many different types of terminology synonymous to SFM. Media manufacturers promote such terminologies to appear in many articles to attact more customers. As a result, many terminologies are in use which confuse customers following the standard classification of media described by Jayme et al. 16. Some terminologies include growth factor‐defined media, good manufacturing practices (GMP)‐compliant media, xenogenic‐free medium and humanized medium, and ambiguity extends with titles of articles stating clinical grade production, GMP‐compliant process (and more) used by many authors in their published articles 5, 37, 38, 39, 40, 41. Five classes of medium defined by Jayme et al. 16 include serum‐containing media (SCM), reduced serum media, SFM, protein‐free media (PFM) and chemically defined media (CDM). Usually, SCM contain 10–20% serum, whereas reduced SCM contain 1–5% serum fortified with insulin, transferrin and other nutrients to permit significant reduction in serum. SFM contain a broad range of additive hormones, growth factors, proteins and polyamines, which may themselves be derived from bovine or human sources. Other variants of SFM include animal PFM containing human serum (HS) albumin, human transferrin and human insulin, but animal‐derived lipid, whereas XFM contain HS albumin (HSA), human transferrin, human insulin and CD lipids. Recombinant XFM contain either recombinant or synthetic compounds with CD lipids. The basal component of CDM is protein‐free and well characterized, containing low molecular weight compounds exclusively. A CDM is entirely free of animal derived components and represents the purest form, where all components are well defined and characterized; a few critical components of CDM are recombinant versions or synthetic chemicals of polymers. PFM contain various peptide fragments only (not polypeptides) derived from enzymatic or acid hydrolysis of animal or plant source proteins. On the basis of degree of chemical definition, Brunner et al. have classified media into serum‐free, animal‐derived component‐free or chemically defined media for cell culture applications 42. SFM do not contain serum, but otherwise is chemically undefined, as many or few discrete protein fractions are derived from animal tissue or plant extracts. In contrast, the term ‘CDM’ has been used erroneously instead of ‘SFM’ to capture the commercial market, despite such media containing bovine serum albumin 43, 44. Thus, there is urgent necessity for global harmonization of terminology used in cell culture applications, specially concerning types of media, for better understanding and applicability in the context of regulatory and GMP compliance. Thus to avoid ambiguity, we have attempted to simplify classification of culture media used to grow MSCs, based on whether they are human or animal sourced. As a first category, if the medium is supplemented with HPL, allogeneic or autologous pooled HS, or cord blood serum, it is defined and categorized as humanized culture medium. The second category is SFM; subclasses of SFM are (i) in‐house formulation of SFM, where some or many of the individual components consist of bovine or human sourced items and (ii) commercially available SFM.
Humanized culture media
In the past few years, a number of in vitro studies have been carried out for supplementation of auto‐HS and allo‐HS, HPL for isolation, expansion, differentiation, molecular analysis and evaluation of functional properties of MSCs, derived from various sources including bone marrow (BM), adipose tissue (AT), synovial fluid and Wharton's jelly (WJ). A number of authors has performed assessment of different concentrations of HS including 2%, 5%, 10% and 20% with different basal media (DMEM, a‐DMEM, DMEM‐LG, DMEM‐HG and DMEM‐KO) and compared these with FBS supplementation of 10% or 20% 18, 20, 26, 29, 45, 46, 47, 48, 49, 50, 51, 52, 53. In addition, to enhance efficiency of proliferation, some studies have reported use of basic fibroblast growth factor (bFGF) 54, HSA or insulin/transferrin/selenium supplementation 45. Table 2 summarizes various humanized media used, MSC source and culture characteristics.
Table 2.
Various humanized media showing culture characteristics of MSCs of different sources
| S. no. | Culture media | MSCs source | Isolation of MSCs SD (P0) cells/cm2 | Multipassage SD (cells/cm2) | PD/CPD | PDT | Phenotypic markers | Differentiation potential | Publication |
|---|---|---|---|---|---|---|---|---|---|
| HS | |||||||||
| 1 | DMEM‐F12 + 20% FBS | BM | ND | 5000 | ND | ~84 h | D | ACO | 20 |
| DMEM‐F12 + 20% auto HS | ND | ~53 h | |||||||
| 2 | DMEM‐LG + 10% FBS | BM | ND | 200 | 31.43 ± 3.13 (P7) | 3.72 ± 0.44 (P4, per passage) | D | AO | 29 |
| DMEM‐LG + 10% HS | 14.46 ± 3.46 (P5) | 12.53 ± 6.56 (P4) | |||||||
| 3 | α–MEM + 20% FBS | Synovium | ~166.66; ~1666.6 | 50 | ND | ~140 days | D | C | 50 |
| BM | 1000; 10 000 | ND | ~112 days | ||||||
| α–MEM + 10% auto‐HS | Synovium | ~166.66; ~1666.6 | ND | ~126 days | |||||
| BM | 1000, 10 000 | ND | ~84 days | ||||||
| 4 | α‐MEM + 10% HS | UC | 4000 | 4000 | 7.9 ± 0.02 | ND | D | ND | 45 |
| α‐MEM + 5% HS | 7.6 ± 0.01 | ND | |||||||
| α –MEM + 2% HS + 0.5% ITS + 0.5% HSA | 5.2 ± 0.21 | ND | |||||||
| α‐MEM + 2% HS + 0.5% ITS + 0.5% HSA + 0.5 ng/ml bFGF | 8.1 ± 0.29 | ND | |||||||
| 5 | DMEM‐KO + 10% FBS + 2 ng/ml bFGF | WJ | ND | 5000 | 1.4 × 109 ± 1.1 × 109 | 28.2 ± 2.5 h | D | ACO | 54 |
| DMEM‐KO + 5% alHS + 2 ng/ml bFGF | 1.8 × 108 ± 2.1 × 107 | 35.3 ± 2.4 h | |||||||
| 6 | DMEM‐LG + 10% FBS | AT | ND | 200 | 31.13 + 1.3 (pP8) | ND | D | AO | 48 |
| DMEM‐LG + 10% AB‐HS | 40 + 2.5 (P8) | ND | |||||||
| HPL | |||||||||
| 7 | DMEM‐KO + 10% FBS | BM | ND | 1000 | P1–P5 = 24.03 | 44.52 h | D | ACO | 15 |
| DMEM‐LG + 10% FBS | P1–P5 = 7.76 | 66.37 h | |||||||
| DMEM‐ KO + 10% HPL | P1–P5 = 25.03 | 38.42 h | |||||||
| DMEM‐LG + 10% HPL | P1–P5 = 26.24 | 36.5 h | |||||||
| 8 | BGM + 10% FBS + 1 ng/ml bFGF | BM, UCB |
1 × 105 for BM MNCs; 1 × 106 UCB‐ MNCs |
1000 | ND | 11.2 ± 2.2 days | D | ND | 55 |
| BGM + 10% FBS + 5% HPL | 5.6 ± 0.5 days | ||||||||
| BGM + 10% HPL | 5.8 ± 1.03 days | ||||||||
| BGM + 5% HPL | 3.8 ± 0.7 days | ||||||||
| BGM + 10% HPL + 10% FBS | 9.9 ± 1.4 days | ||||||||
| BGM + 10% FBS | 11.2 ± 2.2 days | ||||||||
| 9 | DMEM‐LG + 1/2.5/10/20% FBS | AT | 100, 10 × 105 | 10000 | 22 ± 4 | 179 ± 19 | D | AC | 56 |
| DMEM‐LG + 1/2.5/10/20% HPL | 43 ± 4 | 94.8 ± 21.7 | |||||||
| 10 | DMEM‐LG + 10% FBS | BM | 2 × 105 | 3000 | ~11.64 | Median 41.4 days | D | AO | 57 |
| DMEM‐LG + 5% HPL | ~27 | Median 27.8 days | |||||||
| 11 | DMEM‐LG + 10% FBS | BM | ND | 200 | 31.43 ± 3.13 (P7) | 3.72 ± 0.44 (P4, per passage) | D | AO | 29 |
| DMEM‐LG + 10% HPL | 52.8 (P8) | 1.9 ± 0.32 (P4) | |||||||
| 12 | DMEM + 10% FBS | BM | ND | 5000 (P5) | ND | 5.0 + 1.0 days | D | ACO | 58 |
| DMEM + 1% HPL | 7.3 days | ||||||||
| DMEM + 2.5% HPL | 3.0 + 0.6 days | ||||||||
| DMEM + 5% HPL | 2.5 + 0.4 days | ||||||||
| DMEM + 10% FBS | AT | ND | 5000 (P5) | ND | 2.9 + 0.4 days | ||||
| DMEM + 5% HPL | ND | 2.0 + 0.2 days | |||||||
| 13 | α‐MEM + 10% FBS | UCB | 1 × 106 | 3 × 105 | 8.53 ± 0.64 | ND | D | ACO | 59 |
| α‐MEM + 10% HPL | 7.62 ± 0.21 | ND | |||||||
| 14 | α‐MEM + 10% HPL | BM | 10 875 | Not done | 2.323 | 165.29 h | D | ND | 60 |
| 15 | Mesencult + 5% HPL | UCB | 160 000 | 4000 | 12.9; 13.3 | 6.5 days | D | AO | 61 |
| 16 | α‐MEM with supplements | BM | 1 × 105 | 1000 (P2) | ND | >20 days | ND | ACO Smooth muscle | 62 |
| BGM + 10% FBS + 1 ng/ml bFGF | 5.6 ± 0.5 days | ||||||||
| BGM + 10% FBS + 5% auto‐HPL | 5.8 ± 1.03 days | ||||||||
| BGM + 10% auto‐HPL | 3.8 ± 0.7 days | ||||||||
| BGM + 10% FBS | 22.4 ± 2.4 days | ||||||||
| 17 | DMEM‐LG + 10% HPL | BM/AT | ND | 1000 (P2–P6) |
Y‐HPL: 10.33 CPD; O‐HPL: 8.99 CPD |
~13–14 days | D | AO | 63 |
| 18 | HPL matrix | BM | ~1.56 × 105 | ND | ~15– 16 CPD | ~37–38 days | D | ACO | 64 |
~, taken approximate values from data/graph or average value; ACO, adipo, chondro, osteo differentiation lineages; Allo‐HS, allogeneic human serum; AT, adipose tissue; Auto‐HS, autologous human serum; BM, bone marrow; CPD, cumulative population doublings; D, defined; HPL, human platelet lysate; MNC, mononuclear cells; MSC, mesenchymal stromal cells; ND, not defined; NM, not mentioned; O, osteogenesis; PD, population doublings; PDT, population doubling time; SD, seeding density; UCB, umbilical cord blood; WJ, Wharton's jelly;
Human serum supplementation
Initial demonstration by Shahdadfar et al. indicated that BM‐MSCs cultured in allo‐HS became growth arrested and died 20. In contrast, Bieback et al. showed that allo‐HS extended proliferation of BM‐MSCs 29; however, population doubling time (PDT) and population doublings (PDs) were lower compared to BM‐MSCs cultured in FBS‐supplemented media. Possible explanations of these results may be differences; first, in use of basal media DMEM‐F12 20 or DMEM‐LG 29, second, SD 5000 cells/cm2 20 and 200 cells/cm2 29, and third, concentration of allo‐HS at 20% 20 or 10% 29. AT‐MSCs cultured in allo‐HS had higher PD 48, and for WJ‐MSCs, PDs were similar to FBS‐supplemented cultures further supplemented with 2 ng/ml bFGF 54. In a further study, synovial‐derived MSC PD was significantly higher than that of BM‐MSCs. PDT was lower in both cultures compared to MSCs supplemented with FBS. This suggests that MSC have source‐dependent variation in PD and PDT. Ten per cent allo‐HS was found to be optimum for proliferation of UC‐MSCs, further enhanced by supplementation with 0.5% insulin/transferrin/selenium, 0.5% HSA, 0.5 ng/ml bFGF with reduced concentration of 2% allo‐HS 45.
Supplementation of auto‐HS has also been investigated for culture of MSCs. Regardless of source of the MSCs and their concentration, auto‐HS caused elevated rate of proliferation compared to those in FBS‐supplemented cultures 20, 48, 49. Auto‐HS‐derived MSCs had donor‐specific growth profiles in terms of PD and PDT, although cumulative cell numbers varied between donor MSCs cultured both in FBS and in auto‐HS, individually 49, 52. Tateishi et al. evaluated potency of auto‐HS and FBS for culturing human synovium MSCs, and bovine synovial MSCs. Human‐derived synovial MSCs had greater potency for proliferation in auto‐HS, whereas bovine‐derived MSCs were more proliferative in FBS. This is an important consideration that MSCs correspondingly mirror species‐specific influence on proliferation 46.
Specificity of HS produces considerable effects on MSC phenotype marker expression; Those derived from synovium, BM and WJ‐derived MSCs are positive for CD13, CD29, CD44, CD54, CD73, CD90, CD105, CD106, CD140b, CD146, CD147, CD166 and HLA‐I, and do not express CD3, CD14, CD19, CD31, CD33, CD34, CD40, CD45, CD80, CD86, CD117, CD144, CD235a and HLA‐DR 29, 45, 46, 50. In contrast, AT‐MSCs cultured in allo‐HS have been shown to have a marked subpopulation of CD45− and CD14− cells at passage 3 (P3). MSCs in FBS‐supplemented cultures at P3 expressed HLA‐II (1.2 ± 0.6), CD45 (11.6 ± 11.9), CD14 (9.5 ± 12.1), and MSCs grown in HS‐supplemented cultures expressed HLA‐II (0.7 ± 0.4), CD45 (61.0 ± 2.1), CD14 (56.2 ± 9.9). Subsequent sequential passaging up to P7 in FBS and HS‐supplemented culture showed a remarkable reduction in CD14 expression from 9.5% and 56.2% to 2.8% and 12.4%, respectively, and in CD45 from 11.6% and 61% to 3% and 5%, respectively. At P7 FBS MSC marker, HLA‐II (0.9 ± 0.7), CD45 (3.0 ± 1.3), CD14 (2.8 ± 3.6) levels were different compared to P7 HS MSC markers HLA‐II (0.1 ± 0.0), CD45 (5.0 ± 4.1), CD14 (12.4 ± 15.1) 48. This clearly signifies that subsequent passaging of MSCs significantly reduced the subpopulations between P3 and P7. However, presence of CD14 and CD45 heterogeneous populations even at P7, clearly do not meet the criteria set by International Society for Cellular Therapy for MSC definition. In addition, synovial MSCs cultured in auto‐HS were dim positive for CD105 and CD106 46, yet in a further study, synovial MSCs were strongly positive 50. These evidence suggest that variation in production processes of HS influence its quality and phenotype marker expression of MSCs.
Genome‐wide array analysis has revealed that MSCs retain stable transcripts and consistent display of normal DNA copy numbers in late passage cultures with auto‐HS 20, 49. Furthermore, no significant chromosomal aberrations have been observed in Comparative genomic hybridization (CGH) analysis 49. Auto‐HS‐derived MSCs tend to maintain an unmethylated state similar to that of uncultured MSCs 49. Ratios of MSCs cultured in FBS and auto‐HS are 20% and 4% in S phase cells respectively. Thus, it is suggested that MSCs grown in FBS rapidly transit through G1 phase, for instance, not yet ready for DNA replication, which may be the reason why MSCs accumulate in S. These MSCs traverse other phases of cell cycle more rapidly 52.
Human platelet lysate
Many studies have evaluated auto‐HPL with different basal growth media – DMEM, α‐DMEM, DMEM‐LG, DMEM‐HG or DMEM‐KO and compared it to with FBS supplementation at 1%, 2.5%, 5%, 10% and 20% 15, 29, 55, 56, 57, 58, 59, 60, 61, 62, 63, 65. Sources of MSCs used for evaluation were BM, AT, umbilical cord blood (UCB) and WJ. Similarly, studies on isolation of MSC with either HPL or FCS resulted in similar CFU‐F frequency; both FBS and HPL supported similar CFU at P0 29, 55, 56, 57, 59, 60, 66 irrespective of source of the MSCs. Differences were observed in HPL‐derived MNCs where colonies appeared larger and were formed of small, densely packed, spindle‐shaped cells.
Many reports have been made clearly demonstrating supplementation of auto‐HPL or allo‐HPL with various basal media result in higher rates of MSC proliferation 15, 29, 55, 56, 57, 58, 59, 60, 61, 65 compared to FBS‐supplemented controls. Ben Azouna et al. have shown that combination of auto‐HPL and FBS with basal media did not result in changed PDT compared to FBS cultures supplemented with 1 ng/ml bFGF 62. However, combination of allo‐HPL and FBS increased PDT of MSCs compared to FBS cultures supplemented with 1 ng/ml bFGF 55. These studies suggested that 5% HPL can be replaced by bFGF, which was further evident with reduced PDT and increased PD compared to bFGF‐supplemented cultures 55, 62. Furthermore, 5% HPL either as auto‐HPL or allo‐HPL can be better than a 10% HPL supplement, which is also evident in their reduced PDT 55, 57, 58. Among basal media, combination of DMEM‐LG with HPL has resulted in reduced PDT compared to others 15, 56, 57. Further evidence suggests that DMEM‐LG with HPL might be support proliferation of of AT‐derived MSCs 58.
In a large‐scale expansion study, using 4 cell factories (Nunc) un‐manipulated (P0) BM MSCs supplemented with 10% HPL, expanded in a single passage, yielded 7.8 ± 1.5 × 108 MSCs in 11–16 days 60. In a further study on large‐scale expansion, carried out at P2 using UCB‐MSCs supplemented with 10% HPL, cell yield was 2.7 ± 1.84 × 108 MSCs with 10% HPL whereas yield was 0.66 ± 0.16 × 108 MSCs with 10% FBS 59. Differences in yield between studies could be due to different sources of MSCs, BM or UCB, differences in seeding densities 30 cells/cm2 and 17 696 ± 2364 cell/cm2, or passage number. Schallmoser et al. 60, carried out isolation and expansion to P1 using pooled allo‐HPL. Considering the above two studies, MSCs can be isolated and expanded in HPL for large‐scale clinical applications of autologous therapy, where total duration of MSC availability to the patients is minimal and further improvements can be made during scale‐up studies for clinical application in autologous settings. While conducting scale‐up studies, it is essential to consider four factors: (i) age of HPL donor – MSCs cultured in HPL from a young person (<25 years) have more cumulative population doublings (CPD) than that from older people (>45 years) 63; (ii) multiple donor pool of allo‐HPL – allo‐HPL pooled from multiple donors has shown significantly higher yield of MSCs than with allo‐HPL from a single donor 66; (iii) virally inactivated HPL 67 that can be processed under GMP conditions 15, 58, 65 to comply with regulations and safety in MSC therapy protocols; (iv) employing HPL matrix to enhance MSC culture expansion 64. HPL matrix has supported MSCs to grow in a multilayer manner at the interface between HPL‐gel and HPL medium; this technique provides non‐enzymatic passaging procedures for culture expansion 64.
Mesenchymal stromal cells cultured in HPL express MSC‐positive markers CD13, CD29, CD44, CD73, CD90, CD105, CD166, HLA‐ABC, and do not express MSC‐negative markers CD14, CD15, CD19, CD33, CD34, CD45, CD80, CD235a, CD117, CD144, HLA‐DR 56, 58, 59, 60, 61, 67. In all studies reported, MSCs cultured in HPL express higher level of positive markers and high geometric mean fluorescence intensity level compared to FBS cultures, irrespective of MSC source. There could be variable marker expression and presence of heterogeneous populations in both HPL‐ and FBS‐supplemented cultures 47. MSCs cultured in HPL have been shown to express fibroblast‐associated protein and CD49d and avb3 (CD51/61). These surface molecules play an important role in cell interactions and homing capabilities. Furthermore, HPL‐cultured MSCs express lower levels of DNAM‐1 ligand poliovirus receptor and Nectin‐2, and NKG2D ligand UPLBP3 68. Consequently, MSCs cultured in HPL have been shown to be resistant to NK cell‐mediated lysis.
Mesenchymal stromal cells expanded in HPL display strong inhibitory effects on T cells and their subsets CD3+, CD3+CD4+, CD3+CD8+ and CD19+ (B cells) 15, 29, 59, 68, although variable effects of inhibitory results have been with CD3‐CD56+ (NK cells) 68, 69. This immunomodulation on alloantigen induced lymphocyte proliferation aided by secretion of anti‐inflammatory cytokines RANTES, IL‐6 and IL‐8 at higher levels, or by reduced secretion of pro‐inflammatory cytokines IFN‐γ, TNF‐α, IL‐10 levels in HPL‐cultured MSCs 55, 68, 69. Further indoleamine 2,3‐dioxygenase (IDO) levels remain low in HPL‐cultured MSCs 61, 68. In contrast, Abdelrazik et al. have shown reduced inhibitory capacity of MSCs cultured in HPL on T‐ and NK‐cell proliferation and function compared to those cultured in SFM, which showed enhanced immunomodulatory properties 68.
CGH array analysis have shown MSCs cultured in HPL did not have any unbalanced chromosomal abnormalities 60, 61. However, MSCs cultured in FBS evaluated at late passage (passage 12) demonstrated cell transformation with clonal aberrations. This included (1:15) translocation found in 1 patient and gain in chromosome 21 with loss of the X chromosome in a another patient. Interestingly, no chromosomal aberrations were observed in the same patients' MSCs cultured in HPL 58. Furthermore, telomerase activity was not detected in MSCs cultured in HPL even at late passages 29, 61. In further work, HPL‐cultured MSCs did not express differentiation markers and were able to differentiate on induction 62. Hence, MSCs cultured in HPL seemed to retain multipotency without losing self‐renewal properties. Differences in methodology of HPL protocols, however, may have contributed to varied differentiation potentials 70.
Serum‐free media
Challenges in use of humanized or FBS‐supplemented culture media have led to progress in development of SFM, but efforts in making GMP‐compliant SFM have not witnessed significant progress over most recent years. SFM not only address challenges to serum‐based formulations but also lead to enhanced consistency in raw materials as well as in the final yield. Furthermore, SFM improve reproducibility of production processes with less probability of culture contamination by bacteria, virus or mycoplasma. SFM for MSCs do not require stringent washes before filling of cryobags and hence contribute to retaining high post‐thaw viability and shelf life 38. Development of GMP‐compliant SFM for culturing MSCs will overcome regulatory hurdles and increase performance of production systems in terms of reproducibility and consistent cell quality.
Disclosed in‐house serum‐free formulations
In‐house serum‐free formulations for MSCs have several advantages over commercially available SFM. These formulations help to design and optimize nutritional requirements specific to the in‐house donor MSCs. These disclosed formulations further contribute to simplification of post‐harvest bioprocessing with better access to trouble‐shooting and control over the processes. In addition, this also enables higher cell yield and higher potency of donor‐specific MSCs with increased control over physiological culture conditions. It increases robustness, consistency of processes and are free from vendor‐related cost fluctuations, market withdrawal and non‐availability. In‐house preparation of SFM can be significantly cheaper and lead to better control over complete media stability, as they are prepared and used afresh for each occasion, in production settings. Moreover, such systems do not require storage of bulk media in cold rooms and reduce repeat quality control testing, storage space and cost. Additional time required for repeated preparation of in‐house SFM for each production batch can be compensated by the many other advantages. Initial time investment to optimize cell culture medium may result in higher productivity and reduced cost, with better biological function and process control 71. It enhances simultaneous reshuffling of multiple medium components in developing seed media, production and feeding media depending on growth and nutritional requirements 72. At present, there are several serum‐free formulations published that allow culture of MSCs in SFM and several media have been developed and optimized to support in‐house donor MSC expansion requirements 11, 32, 33, 34, 35, 37, 73, 74, 75, 76, 77. A concise list of all disclosed SFM formulations for MSCs is tabulated in Table S1.
The first disclosed serum‐free formulation was developed in 1995 by Lennon et al. for rat BM‐MSCs (RDMF) using insulin, BSA, PDGF‐β and bFGF, in 60% DMEM‐LG and 40% MCBD‐201 basal medium 74. Chemically defined RDMF SFM was tested and cells grown in subsequent passages on pre‐isolated MSCs, for culture age of day 10; cells were large and spread‐out and with irregular projections. Only an osteochondral assay was performed, in porous ceramic cubes, to display of culture characteristics. In contrast, Gronthos et al. isolated STRO‐1‐positive BM precursor cells using magnetic‐activated cell sorting; cells were further grown in SFM containing α‐MEM, insulin, transferrin, BSA, and human LDL, and cultured on fibronectin‐coated plates in the absence of serum 78. The group extensively studied colony growth characteristics and contributions of EGF, PDGF, IGF‐1 and bFGF to expansion. Marshak et al. in 79 patented CD medium for BM MSCs expanded from P1 to P3 using CDM for proliferating pre‐isolated MSCs 79. Rayes et al. in 2002 isolated multipotent adult progenitor cells from human BM, by using micro magnetic beads 76. Cells were subcultured at SD of 2000 (2K) and 8K in fibronectin‐coated culture vessel for more than 20 PDs; low serum formulation with 2% FBS gave equivalent results to 20% FBS controls. Identical formulation as SFM was used by Sorensen et al. in 2007 as one of 4 culture media with no serum. Isolation and expansion were also studied in various SFM 73. Further heterogeneous populations of MSCs were selected by plastic adherence, which resulted in less than 70% of cells expressing positive markers, although it's possible there was other non‐MSC cell contamination. However, these cells had low PDT compared to the other 3 tested culture media. In 2006, Lui et al. used the statistical method of fractional factorial design and combined it with a steepest ascent approach to optimize multicomponent media for cord blood MSC expansion (CB‐MSCs) 80. This was the first published article on factorial design of statistical methods of media optimization for MSC and was performed using pre‐isolated CB‐MSCs. Optimized SFM supported cell proliferation under high SD with average PDs per passage of 1.07 and PDT of 74.1 h. On the other hand, Wu et al. in 79 designed a CD‐SFM for population growth of rabbit BM MSCs, which resulted in 50‐fold increase in cell number in 10 days 77. Parker et al. in 2010 optimized serum‐free formulation using previously isolated MSCs from adipose tissue (ASC) subcultured for 2 passages at SD of 2 K/cm2. Growth kinetics, phenotype marker profile and differentiation potential were studied. Rajala et al., Hudson et al. and Mimura et al. disclosed formulations that supported cell expansion for only a single or two passages at higher SD and with low growth rate 32, 37, 75. Table 3 summarizes various disclosed SFM used, MSC source and culture characteristic.
Table 3.
Cultural characteristics of serum‐free formulations for MSCs
| Year | Media group | MSCs – Source | P0 seeding density/cm2 | Multipassage SD/cm2 | PDS | PDT (h) | Phenotypic | Differentiation | Publication |
|---|---|---|---|---|---|---|---|---|---|
| 1995 | RDMF | rat BM‐MSCs | I.S | 7K (P1) | ND | − | − | O | 74 |
| 1999 | patent – CDM | BM‐MSCs | I.S | P2–P3 (2.5–3K) | 15 CPD | − | + | O | (P#5 908 782)79 |
| 2002 | Reyes | BM‐MSCs | I.S/2.85 mn | 2–8K | 20 | 28.2 | + | Endothelial | 73, 76 |
| 2007 | OSFM | CB‐MSC | I.S | 10K (P1–P9) | 9.71 CPDs/9 passage (1.07 PDs/passage) | 74.1 | − | ACO | 80 |
| 2007 | AR8 SFM | ASCs | I.S | 2K, P1–P5 | 1.77 (P1–P3) | 138 | + | ACO | 11 |
| 2009 | CD SFM | rabbit BM‐MSCs | NM | NM | ~5.64 PDP | ~42.52 | − | AO | 77 |
| 2010 | mTeSR | BM‐MSCs | – | 2.5K (P2–P3) | ~4.1 (one passage) | ~82 | + | ACO | 32 |
| 2010 | XF –RegES | ASCs | I.S (Allo‐HS) | 4.5K (one P) | ND | − | + | ACO | 75 |
| 2010 | PPRF MSC6 | BM‐MSCs | 1.5 L (13–14 PDs) | 5K | 62 ± 4 – CPD (P1–P10) (PDP‐6.2) | 21–26 | + | ACO | 33, 34, 35 |
| 2010 | K‐M | P/S | NM | 7 days – | P yields 1.2 mn and S yields 0.5 mn in 7 days of time | − | ACO | 81 | |
| 2011 | DhESF10 | BM‐MSCs | N/App (immortalized hMSC line used) | 10K | ND | − | + | AO | 37 |
| 2013 | DMEM/SF/FGF/EGF | Endometrial MSCs | ISF | 2K (P0–P3) | 6.4 PD per passage | 28.4 ± 12.4 | + | AOM | 82 |
− +, is satisfactory or mentioned; –, is not done or not mentioned; ~, approximate value taken from data/graph or average value; ACO, adipo, chondro, osteo differentiation lineages; IS, isolated the MSCs at P0 in serum‐supplemented cultures; K = 1000 means 7K = 7000; NM, not mentioned; O, osteogenesis; P/S, Human periodontal ligament stem cells (PDLSCs), human stem cells from exfoliated deciduous teeth (SHEDs) not mentioned the phenotype marker expression level.
Several studies using in‐house SFM have shown lower expression of positive markers, for example 88.2% CD105 37, almost 65–68% of all three markers CD90, CD73 and CD13 76, 75% CD105 75. Table S2 shows phenotype marker expression profile of MSCs during culture optimization processes of various disclosed SFM. Media supporting attachment, self‐renewal and growth, during either isolation by plastic adherence or expansion of pure populations, and subsequently passaged cells under SFM affect phenotype marker expression levels. Yet, these changes in phenotype marker expression have been due to attempts at modifying existing xeno‐free human embryonic stem‐cell medium such as RegES and mTeSR (Stem Cell Technologies, St. Katharinen, Germany, http://www.stemcell.com) for culturing hMSCs 37, 75. mTeSR media were used to grow and expand a specific genetically immortalized hMSC line. Changes of slow growth, lower positive expression of marker phenotype may be due to sudden shift from serum to serum‐free conditions for cells previously grown in serum‐positive conditions. Hence, it is necessary to check the phenotype marker profile of primordial harvested cultures, when newer SFM are developed for isolation and expansion of MSCs. In 2010, Tarle et al. developed a SFM, K‐M, for expansion of human periodontal ligament stem cells (PDLSCs) and human stem cells from exfoliated deciduous teeth (SHEDs) 81. Results of comparison of K‐M media with other 4 media showed equivalent proliferation with that of Lonza Therapeak and K‐M media, whereas in PDLSCs and SHEDs, proliferation rate was higher than of cells cultured in FBS‐supplemented media. It should be noted that culturing cells for short periods of time, 7 days or one or two passages, is not sufficient to declare that a SFM is robust, as stem cells themselves have proliferative capacity in basal culture media. Multipassage expansion of MSCs has been successfully performed from isolation to P10 showing strength of optimized media of Pharmaceutical Production Research Facility (PPRF). Recently, Jung et al. published a series of studies on PPRF msc6 media and developed serum‐free formulation that supports isolation and expansion of MSCs 33, 34, 35. Cultures were fully characterized in terms of PD, morphology, phenotype and multipotency. Recently, PPRF msc6 were the first successfully disclosed defined media, showing superior isolation and cell expansion profiles, over controls in FBS culture media. Population doubling per passage (PDP) of PPRF msc6 was 6.2 from P1 onwards thus competing with commercial SFM/XFM. Jung et al. initiated a comparative study involving other commercially available media such as Mesencult‐XF (MSX; Stem Cell Technologies), StemPro MSC‐XF (Invitrogen, Life Technologies, Grand Island, NY, USA), MSCGM‐CD (Lonza) along with PPRF msc6 and FBS‐supplemented media as controls. None of the commercially available media supported better cell population growth or proliferation than PPRF msc6 33. Recently, Rajaraman et al. showed PDP of 6.4 using endometrial MSCs grown on fibronectin‐coated surfaces of DMEM/SF/FGF/EGF formulations and have derived comparable results with those of Lonza Therapeak on fibronectin‐coated surfaces 82. Probable reasons for higher PDP in DMEM/SF/FGF/EGF formulations might be using naive MSCs of endometrial origin and also using lower SD (2000 cells per cm2 compared to 5000 cells per cm2) of PPRF media. These significantly higher PDPs are uncommon even for gold standard FBS‐supplemented cultures with high SD 15. Further, careful, thorough and in‐depth molecular (proteomics and genomics) and cell‐based immune assays are needed for selecting appropriate media for clinical scale production of MSCs. PPRF msc6 medium can be taken as a base line to further develop suitable in‐house media for donor MSCs.
Commercially available serum‐free formulations
Commercially available CD‐SFM support higher cell population growth rates and are most commonly used by many customers. They have proven safety and regulatory compliance. Many stem cell manufacturers use commercially available CD‐SFM in process development without modifications. However, demand for making target‐specific MSCs and with special in‐process steps requires the industry to develop customized media as well. An advanced statistical tool, Design of Experiments combined with high throughput screening has been used to develop a feeding regime and appropriate nutrients, and is more relevant for CD‐SFM in further reducing development cycle time 83, 84, 85, 86. These new approaches for cell‐based manufacturing require support to develop production media, and commercially available CD‐SFM form a baseline for customizing culture media for large‐scale application. Commercially pre‐formulated CD‐SFM reduce and eliminate qualification costs for each material or ingredient, thereby reducing expense in corrective and preventive actions throughout a component's life cycle. Other advantages of CD‐SFM include consistency and high‐quality media with support from vendors and initiative for process analytical technology, and increase operational excellence. Commercially pre‐formulated CD‐SFM reduce and eliminate mixing tanks, pH, osmolality adjustments, raw material quality control testing and documentation; improve inventory management with reduced number of material components; decrease media preparation space suites and manpower and reduce technical training and documentation. Moreover, commercially pre‐formulated CD‐SFM overcomes the issues and risk posed by in‐house SFM preparation. These issues are lot‐to‐lot variability in raw materials, challenges in consistency of media preparation, mixing, and media hold stability studies 87.
Ability to support efficient isolation, multipassage expansion with maintaining multipotency, and acceptable phenotype characteristics has been the main factor of choice for selection between several commercially available SFM that have been introduced for expansion of MSCs. Table 4 summarizes various commercially available appropriate SFM.
Table 4.
Various commercially available serum‐free media
| S. no. | Serum‐free media/product name | Cat no. | Company | Media type | References cited |
|---|---|---|---|---|---|
| 1 | BD Mosaic™ hMSC serum‐free medium (kit) | 355701 | BD Biosciences | CD, SF | 25 |
| 2 | CellGro™ | 24803–0500 | CellGenix | SF, PE | 38 |
| 3 | HEScGRO | SCM020 | Merck Millipore | CD, SF, ACF | 88 |
| 4 | Mesenchymal stem cell growth medium DXF | C‐28019 | PromoCell | CD, SF, XF | Not cited |
| 5 | MesenCult®‐XF (culture kit) | 5429 | Stem Cell Technologies | CD, SF, ACF | 82, 89, 90, 91, 92, 93, 94, 95 |
| 6 | MesenGro | ZRD‐MGro‐500 | StemRD | CDM | 38 |
| 7 | MSC Qualified PLus™ | PLS2 | Compass Biomedical | XF | Not cited |
| 8 | MSC‐Gro™ (SF, complete) | SC00B3 | Vitro Biopharma | SF, CG | 38 |
| 9 | MSCGS‐ACF (MSC supplements) | 7572 | ScienCell Research | SF | 38 |
| 10 | mTeSR | 5850 | Stem Cell Technologies | SF | 32 |
| 11 | PRIME‐XV™ MSC Expansion SFM | 31000 | Irvine Scientific | SF | Not cited |
| 12 | RS‐Novo™ and GEM‐Novo | N/App | Kerry Bio‐Sciences | CD, SF, ACF | Not cited |
| 13 | SPE‐IV | SPE‐IV‐EBM/500 | Abecell‐Bio | SF | 38 |
| 14 | Stemline MSC expansion medium | S1569 | Sigma Alderich | CD, SF, AF | 96, 97 |
| 15 | StemPro® MSC SFM (media kit) | A10332‐01 | Life Technologies | CD, SF, | 30, 81, 98, 99 |
| 16 | StemPro® MSC SFM‐XF (media kit) | A10675‐01 | Life Technologies | CD, SF, AF | 31, 82, 100, 101, 102 |
| 17 | StemXVivo™ | CCM014 | R&D Systems, Inc. | SFM | 38 |
| 18 | STK2 | N/App | Two Cells Co., Ltd. | CD, SF, AF | 103, 104 |
| 19 | TheraPEAK™ MSCGM‐CD™ (Bullet kit) | 190632 | Lonza | CD, SF, AF | 33, 82, 103, 104 |
| 20 | Ultrasor G (lyophilized) | 15950‐017 | Pall Biosepra | SF | 105 |
AF, animal component‐free; Cat no., catalogue number; CD, chemically defined; CG, clinical grade (especially for MSC clinical trials); PE, preclinical ex vivo use only; PF, protein‐free; SF, serum‐free; XF, xeno‐free.
However, a number of other novel commercially available SFM have arrived on the market with claims of increased efficiency and productivity as well as new clinical grade materials. Some offer many advantages such as enhanced cell proliferation, higher capacity for multilineage differentiation and preservation of better stemness than FBS‐based cultured MSCs, apart from regulatory and clinical grade benefits. These new products of SFM media and supplements need to support and attain higher expansion and culture densities equivalent to or higher than traditional FBS cultures.
Commercial MSCGM medium (MSCGM Bullet Kit™; CellSystems, St. Katharinen, Germany, http://www.cellsystems.de), was initially used by Bieback et al. and soon after, MesenCult (Stem Cell Technologies) was commercially available as growth‐promoting medium for isolation of MSCs from umbilical cord blood 106. These two known MSC‐specific commercially available SCM were tested for isolation and propagation of MSCs; a DMEM‐based MSCGM supported cell isolation, whereas McCoys‐based MesenCult was used successfully in only isolating MSCs from BM 106. This demonstrates that choice of basal medium is equally important and should have synergetic relationship with MSC isolation. Stemline media – commercially available serum supplement media known to promote MSC growth was also used for ex vivo expansion of UC‐MSCs 96.
As per our knowledge, the first commercially available serum‐free formulations for culturing of MSCs were developed by using a serum substitute Ultroser G, by Meuleman et al. 105. Isolation and expansion of BM MSCs using Ultroser G were performed at the lowest possible lower density of 86 000 at P0 and 1000 cells per cm2 at P1 and showed superior isolation and expansion over those of SCM controls. MSCs isolated in both media were phenotypically and morphologically similar, preserved multipotency and might be the first optimal SFM for BM‐MSCs. In contrast, Ultraser G did not support cell population growth beyond two passages. On the other hand, Battula et al. also used commercially available serum replacement of Knockout Serum replacement (KO‐SR – Invitrogen) to DMEM‐KO for isolating and expanding MSCs from BM and non‐amniotic placenta, in serum‐free conditions with bFGF supplementation 107. These studies demonstrated satisfactory proliferation of MSCs on gelatin‐coated plates while using SFM, and showed increased expression of SSEA‐4, frizzled‐9 (FZD‐9), Oct 4 and nestin, by using KO‐SR and DMEM‐KO, as they are ESC media. P0 SD was 0.13 million per cm2 but mutlipassage expansion and SD used were not mentioned.
StemPro MSC‐SFM from Invitrogen (Gibco, Life Technologies) represents MSC‐specific commercially available media and a series of articles have been published on isolation and expansion of MSCs 30, 98, 99, 108. Probably, Stempro is the first commercially available SFM and Ng et al. demonstrated isolation and multipassage (P0–P9) of MSCs at SD of 10 000 cells per cm2 using this medium 108. However, when the donor pool was used at high SD, it led to increased osteogenic lineage differentiation compared to serum controls. Although comparison was made between serum and serum‐free MSC cultures and extensive transcriptional profiling was made to discover important signalling pathways in differentiation, discrepancy in SD with serum (3000 cells per cm2) and SFM grown (10 000 cells per cm2) were not considered while arriving at the conclusion.
In 2009, Lindroos et al., Agata et al. and Ishikawa et al. further demonstrated the use of other commercial SFM for isolation, population growth and expansion of MSCs more efficiently than SCM 98, 100, 104. Agata et al. made use of the findings of Ng et al. StemPro MSC‐SFM (SP‐SFM) propensity for differentiation into osteogenic lineages, and used them efficiently in bone tissue engineering with BM‐MSCs. BM‐MSC isolation efficiency and in vivo bone‐forming abilities were higher in 4 donor tests using SP‐SFM compared to SCM. Serum‐free expanded BM‐MSCs had remarkable bone formation – 25.1% compared to serum‐expanded BM‐MSCs 21.1%; however, this difference was not statistically significant. Early passage BM‐MSCs grown in SFM might be beneficial for bone tissue engineering. Lindroos et al. evaluated two versions of StemProMSC SFM xeno‐free (with and without lipid supplement) with adipose stem cells and compared it to FBS‐supplementation, as well as HS‐supplemented media, as controls (ASCs) 100. ASC were able to expand to 5000 cells per cm2 in SP SFM‐XF and attained the higher CPD of 7.9 (more than in other 3 media) and demonstrated tri‐lineage differentiation potential. Lindroos et al. observed higher expression of CD166 in SP SFM, whereas Agata et al. reported moderate levels of CD166 in SP SFM‐XF. The difference in the CD 166 level could be attributed to different variants of StemPro, although MSC sources were also were not identical. Similarly, Ishikawa et al. confirmed significantly higher hMSC proliferation in STK2 medium compared to serum‐supplemented medium of DMEM and MSCGM (Lonza). SFM expanded MSCs were tested for bone‐forming ability with hydroxyl‐apatite where the lowest inhibitory effect was in STK2 medium. These points once more suggest that type of expansion medium may influence MSC characteristics 109.
Table 5 provides a qualitative summary of population growth profile trends of MSCs in various commercially available SFM. Gradual increase in publication from 2009 to 2012 concerning serum‐free culturing of MSCs has led to widespread implementation of SFM culturing for commercialization and to meet regulatory standards.
Table 5.
Culture characteristics of commercially available serum‐free media
| S.no. | Year | References | Media group | MSCs source | P0 seeding density/cm2 | Media sub group tested | Multipassage and seeding density/cm2 | CPDS | PDP | PDT (h) | Phenotype | Differentiation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2013 | 25 | BD‐SFM | BM‐MSCs | IS – P3 | KO‐FBS | P4–P7 – 1K | 19.6 | 4.9 | 43.3 h | D | ACO |
| BD‐SFM‐1K | P4–P8 – 1K | 21 | 4.2 | 45.6 h | D | ACO | ||||||
| BD‐SFM‐3K | P4–P8 – 3K | 13.5 | 2.7 | 42.9 h | ND | ND | ||||||
| 2 | 2013 | 110 | BD‐SFM | BM‐MSCs | IS – P1 | MSCGM (lonza) | P2–P4 – 4–5K | ~4 | ND | ND | D | ACO |
| BD Mosaic | P2–P4 – 4–5K | ~11 | ND | ND | D | ACO | ||||||
| MSX | P2–P4 – 4–5K | ~10.5 | ND | ND | D | ACO | ||||||
| SP‐SFM | P2–P4 – 4–5K | ~8 | ND | ND | D | ACO | ||||||
| 3 | 2010 | 30 | Stempro MSC SFM | BM‐MSCs | ISF – 0.26 mn | Stempro SFM | P1–P5 – 10K | 3.33 | 0.666 | 46.3 ± 7 | D | ACO |
| ISF – 0.266 mn | Stempro SFM | P6–P13 – 10K | 20 | 2.5 | 46.3 ± 8 | ND | ND | |||||
| IS – 0.26 mn | SCM | P6–P13 – 3K | ~16.5 | 2.0625 | 46.3 + ‐8 | D | ND | |||||
| 4 | 2010 | 99 | Stempro SFM | UCMSC | 1K | GM‐FBS | P1–P5 – 10K | ~11.6 | 2.32 | ND | ND | AEO |
| SP SFM + 2%HS | P1–P5 – 10K | ~15.8 | 3.16 | ND | ND | AEO | ||||||
| Mesencult ACF | P1–P5 – 10K | ~16.6 | 3.32 | ND | ND | AEO | ||||||
| 5 | 2008 | 108 | Stempro SFM | BM‐MSCs | IS | SP SFM + bFGF + TGF.b, + PDGF + insulin | (pooled donor) P5–P9 – 10K | ~16.3 | 3.26 | ND | ND | ACO |
| DMEM + FBS | (pooled) P5–P9 – 3K | ~12.28 | 2.45 | ND | ND | ACO | ||||||
| 6 | 2009 | 98 | Stempro SFM | BM‐MSCs | ISF | SP SFM | P1–P3 = 5–10K | ND | ND | ND | D | O |
| IS | A‐MEM. FBS | P1–P3 –5–10K | ND | ND | ND | D | ND | |||||
| 7 | 2013 | 53 | Stempro SFM‐XF | ASCs | ND | FBS | P1–P5 – 2.5K | ~5.5 | ~1.1 | ND | D | ACO |
| ND | HS | P1–P5 – 2.5K | ~6 | ~1.2 | ND | D | ACO | |||||
| ND | XF/SF CM | P1–P5 – 2.5K | ~12.7 | ~2.55 | ND | D | ACO | |||||
| ND | XF/SF CS | P1–P5 – 2.5K | ~11 | ~2.2 | ND | D | ACO | |||||
| 8 | 2012 | 31 | Stempro SFM‐XF | BM‐MSCs | IS &SF | SP‐XF | P0–P9 = 5–10K | 18 | 1.8 | 48‐72 | D | ACO |
| SCM | P0–P9 – 5–10K | ND | ND | 96 | ND | ND | ||||||
| ASCs | IS (P4) | SP‐XF | P3–P9 – 3K | 16.6 | 1.66 | 48‐72 | D | ND | ||||
| SCM | P3–P9 – 3K | ND | ND | ~96 h | ND | ND | ||||||
| 9 | 2009 | 100 | Stempro SFM‐XF | ASCs | IS | Control‐FBS | P4–P8 – 5K | ~1.9 | ~0.38 | 23‐25 | D | ACO |
| Iso in HS | Control‐HS | P4–P8 – 5K | ~2.1 | ~0.42 | 23‐25 | D | ACO | |||||
| SP‐XF | P4–P8 – 5K | ~7.9 | ~1.58 | 29‐48 | D | ND | ||||||
| SP‐XF‐LM | P4–P8 – 5K | ~7.5 | ~1.5 | 29‐48 | ND | ACO | ||||||
| 10 | 2012 | 102 | SP SFM‐XF with 5% o2. | ASCs | Iso in FBS | SP‐XF 5%02 | P3–P9 – 2K | 21.5 | 3.07 | 57.6 h | D | ACO |
| A‐MEM‐5%02 | P3–P9 – 2K | 16.5 | 2.35 | 74.4 | D | ACO | ||||||
| A‐MEM‐20%02 | P3–P9 – 2K | 13.4 | 1.91 | 86.4 | D | ACO | ||||||
| 11 | 2012 | 91 | MSX | BM‐MSCs | ISF – 6L | MSX | P0&P1–P4 1.5K | 23.3 | 5.82 | ND | D | C |
| IS – 6L | FBS | P0&P1–P4 1.5K | 14.6 | 3.65 | ND | |||||||
| 12 | 2012 | 90 | MSX | L‐MSCs | P0.Y = 76K | DF12.FBS | P0–P3 – 1.92 mn | ND | ND | ND | ND | ACO |
| P0.Y = 7K | 2%B‐27 medium | ND | ||||||||||
| P0.Y = 6K | DK‐SFM | ND | ||||||||||
| P0.Y = 0.34 mn | MSX | P0–P3 – 2.94 mn | ||||||||||
| 13 | 2010 | 103 | STK2 medium a | hMSCs | ND | MSCGM | 6K/cm2 | ~15.6 | ND | ~76.8 h | ND | ND |
| STK2 | 6K/cm2 | ~23.2 | ND | ~51.6 h | ND | ND | ||||||
| 14 | 2012 | 97 | Stemline | WJ‐MSCs bovine | ND | Stemline Expansion media | P1–P60 – 40K/cm2 | ND | ND | ND | D | ACO |
| 15 | 2010 | 81 | Stempro MSC SFM and TP | PDLSCs and SHEDS | ND | FBS‐M | Cultured for 7 days in 4 different media | PDLSCs provide 1.2 mn whereas SHEDS yields 5L in 7 days of time. Cells seems to be best grown in TP and KM< media. | ND | AC0 | ||
| DF‐ SDM | ND | |||||||||||
| DF‐ (K‐M) | ACO | |||||||||||
| SP‐SFM | ND | |||||||||||
| TP | ND | |||||||||||
~, approx value calculated from graphs or average value; 1K = 1000 cells means 10K is 10 000 cells; ACO, adipogenesis, chondrogenesis, osteogenesis; AEO, adipogenesis, endothelial cells, osteogenesis; CM, coating matrix kit (coating free supplements); CS, cell start; D, defined; DF, disclosed formulations; FBS, foetal bovine serum; HS, human serum; IS, isolation of MSCs in serum conditions; ISF, isolated in serum free conditions; Iso, isolation; mn, millions; MSX, mesencult‐XF; ND, not defined; P, passage means P1 is passage 1; SF, serum‐free; TP, Therapeak (Lonza). XF, xeno‐free; Y, yield; (tabulated as per media group).
In 2010, Chase et al. showed sequential passaging of MSCs from P0 to P5 and P6 to P13 with two independent experiments using SP MSC SFM supplemented with bFGF, TGF‐β and PDGF‐BB at a SD of 10 000 cells per cm2 30. In addition, they grew P6–P13 in serum‐containing medium for comparison. PDP of 4 donor‐pooled pre‐isolated MSCs (grown in SCM from P0 to P5) from P6 to P13 revealed 2.5 PDP, whereas individual donor MSCs isolated and passaged up to P5 achieved only 0.66 PDP. This suggests that SP MSC SFM, even after supplementing with 3 growth factors at high SD, had significantly less PDP from P1 to P5 in individual donor MSCs. However, Hartmann et al. reported optimal growth of 5‐fold increase in yield, after supplementing with 2% HS at a high SD in SP MSC SFM. In comparison, MesenCult animal component‐free succeeded in being the superior medium, achieving PDP of 3.32 for UC‐MSCs grown up to P5 in complete serum‐free conditions 99. Tarle et al. performed a proliferation assay comparing 4 different SFM media with that of serum‐supplemented culture, and found that SP MSC SFM did not support population growth of either of the two stem‐cell lines they used (PDLSCs and SHEDs) 81; these cells seem to be grow best in Therapeak (Lonza) and K‐M media. Serum‐supplemented culture of MSCGM (Lonza) in STK2 was used at 6000 cells per cm2 SD; CPDs of 15.6 and 23.25, respectively, were attained 103. Further microarray analysis of MSCs grown in STK2 medium revealed significantly higher gene expression levels compared to MSCGM‐derived MSCs.
Historically, MSCs have been grown on treated plastic surfaces as adherent cultures in suitable culture media – typically called the 2‐D cell culture process. Interestingly, several researchers have tried to grow MSCs as 3D cultures using micro‐carriers, suspension cultures, and scaffolds, in bioreactors. In 2011, Santos et al. cultured BM MSCs and ASC in SP MSC SFM‐XF to test feasibility of micro‐carrier (micro‐beads) cultures and witnessed only an 18‐fold increase in cell yield after 14 days 101. Rajaraman et al. also observed the lower yield of 2.2 PDP on fibronectin‐coated micro‐beads (3D culture) suggesting greater expansion of 6.4 PDP in 2D compared to 3‐D culturing 82. Consistently uniform seeding of cells and their harvest are very difficult to optimize in 3‐D cultures to achieve higher yield for every production batch. Hence, only 2‐D expansion of MSCs was taken for consideration of comparison in Table 5.
Miwa et al. have reported complete xeno‐free isolation and expansion of BM‐MSCs using MSX in comparison with serum‐supplemented culture of MSCs at SD of 0.6 million per cm2, of MNCs at P1, and 1500 MSCs per cm2 from P2 onwards 91. CPDs were 22–23 in MSX and 13–14 in serum supplement cultures; PDP are been presented in Table 5. PDP data clearly indicate that MSX medium is one of the most suitable media for cell expansion and has the potential to replace serum‐based medium for large‐scale cell population growth. Strength of MSX for its growth and expansion properties has further been indicated in its multiple usage by different groups and also as it supports stem cells from various sources. The work of Yu et al. on successfully establishing feeder‐free reprogramming conditions using MSX and StemSpan with bFGF and N2B27 supplements 95, further strengthens the above conclusion. However, basic safety and efficacy studies are needed on MSX‐derived MSC to prove their suitability for clinical trials.
In 2012, MSX was shown to support population growth of limbal MSCs and UC‐MSCs 89, 90, 94. Bray et al. demonstrated that MSX was able to maintain the MSC phenotype more efficiently than serum‐supplemented media 89. These findings led to work on corneolimbal tissue engineering from Bombax mori silk fibroin and further indicated that MSX causes retention of MSC‐specific characteristics compared to FBS‐supplemented media. Bray et al. evaluated a method for culturing limbal MSCs by using MSX, and demonstrated significantly higher yield in MSC isolation compared to two disclosed SFM and FBS‐supplemented media 90. However, elevated expression of CD141 raises concerns over its usage and needs further investigation before being translated into a clinical protocol. Wagey et al. developed an outline process for optimal isolation, enumeration and expansion of hMSCs in xeno‐free conditions 93. Simultaneously, two independent groups published articles 31, 102 on SP MSC‐XF optimization to increase cell yield by reducing SD, and oxygen content to 5%. Chase et al. demonstrated ASC expansion from P3 to P9 at SD of 3000 cells per cm2, and witnessed PDP of around 1.66 and 1.8 in BM MSCs cultured at SD of 10 000 cells per cm2 31. This indicates that further basic studies are required for optimization of MSC in serum‐free expansions.
A number of modifications can be introduced in standard procedures for culturing in SFM to further enhance proliferation capabilities. Similar studies using fibrin coating or exposure to hypoxia indicate an opportunity for further improvement 102, 111. Although Chase at al. demonstrated superior proliferation levels in hypoxic conditions, they observed only 79.21% CD29, 84.30% CD73 and 83.36% CD105‐positive marker profiles of immune‐phenotype in SP MSC SFM. However, integration of xeno‐free and hypoxic conditions supports long‐term culturing of ASCs, although leading to lower expression of CD29, CD73 and CD105, needs to be further studied before clinical application. Recently, Cardoso et al. reported successful culturing of bovine WJ‐derived MSCs under serum‐free conditions up to 60 consecutive passages using stemline MSC expansion medium 97. This finding has not been considered for comparative analysis as the source is bovine; isolation and expansion culture conditions were not mentioned and growth profiles have not been stated. These cells have been grown in 3‐D culture at initial SD of 40 000 cells per cm2 and differentiation ability, phenotype markers, telomerase activity and karyotyping were carried out.
Our group has recently published an article on comparison of 5 different commercially available SFM on their ability to support population growth and expansion of pre‐isolated MSCs 25. The five different SFM are SP MSC SFM, SP MSC SFM‐XF, MSX, BD‐SFM and MSCGM‐CD (Thera‐peak, Lonza), but our initial experiments resulted in higher yield only in MSX and BD‐SFM along with controls. MSX was not selected for further testing due to higher HLA‐DR expression; newer BD‐SFM media were studied in depth at lower SD of 1000 cells per cm2 in large‐scale cultures along with controls. PDP of 4.2 in BD‐SFM compared to PDP of 4.9 in controls qualifies it as one of the alternative commercially available SFM alongside SP‐SFM as well as MSX. Furthermore, very importantly, our studies revealed that BD‐SFM supports even completely FBS‐free isolation and expansion of BM‐MSCs and resulted in comparable cell yield to that of MSX (Gottipamula S, Ashwin KM, Muttigi MS, Suresh K, Kolkundkar U, Seetharam RN, unpublished data). In an equivalent type of study, Crapnell et al. compared their newly developed BD Mosaic SFM – developed by using a bioinformatics driven approach (BD discovery platform) – with 4 different commercially available SFM 110. A similar result was obtained, suggesting efficient population growth in BD‐SFM, while maintaining MSC morphological, phonotypical and functional characteristics. Although MSX is a generally suitable medium for isolation of MSCs from many sources, Rajaraman et al. were unable to isolate endometrial MSCs using it; however, they found that Therapeak (Lonza) supported their population growth and expansion when using fibronectin‐coated culture plates 82. These data repeatedly indicate that screening available SFM is most important to identify the most suitable SFM for MSCs, as donor age and type of MSC source (adipose, BM, WJ endometrial, UC, amniotic fluid and more) affect suitability of SFM. Collectively, the data indicate that isolation and expansion of MSCs from various sources can be performed with currently available SFM, but extensive screening and feasibility studies are needed for phenotype and functional characterization of MSCs. In addition, improvements in commercially available media can be achieved by the end‐user by altering supplement or substrate concentration for optimization, as per the donor MSC growth requirements, as well as for keeping standard controls. Related efforts should be made though by the customer, to retain an alternative vendor for supply of SFM as substitute, in case of an on‐demand product hike, SFM manufacturing crisis or non‐compliance SFM.
It has been generally observed that phenotype marker expression level of ASCs changes when grown in serum‐free culture conditions; Table S3 provides a concise report of phenotype characterization of MSCs cultured in various commercially available SFM. Surprisingly, HLA‐ABC were reported by Lindroos et al. to not be expressed by ASC when cultured in SP MSC SFM‐XF, whereas in the same medium, Chase et al. and Yang et al. independently observed 95.1 and 99.37% HLA‐ABC, respectively 31, 100, 102. There is difference in expression of phenotype marker profile of MSCs based on type of their derivation source. Identical observations were clearly demonstrated in W8B2 phenotype positive marker of BM‐MSCs and non‐expression by PL‐MSCs when grown in KO‐SR serum‐free formulations 107. Prominent expression levels of CD105 were observed in BM‐MSCs in SP MSC SFM compared to serum‐supplemented culture 98.
In summary, over the last decade, the literature clearly suggests that there have been a number of studies on humanized media standardization based on user requirements. In all these investigations, various methods were employed for preparation of HS and HPL. Furthermore, there have been comparative experiments that differentiate between media, based on culture characteristics. MSCs cultured in humanized media showed immunomodulatory properties, home to the target tissue, have stable transcripts, exhibit normal DNA copy number without any chromosomal aberrations and have acceptable phenotypic markers. Thus, currently available information indicates appropriate usage of HS and HPL for large‐scale expansion of MSCs for therapeutic applications. At present, five clinical trials are ongoing using HPL‐derived MSCs (http://clinicaltrials.gov/).
Increased safety and regulatory concerns regarding risk of potential adventitious agent introduction in cell therapy products through SCM use, need to be addressed and defined for better quality assurance and reproducibility of processes. Development of technologies, methodologies and strategies towards production of clinical grade xeno‐free MSCs includes a number of components, as outlined in Fig. 2. Various SFM may be used to eliminate constituents of animal origin without compromising functional and biological properties of MSCs. These various SFM formulations have been subjected screening, integrated manufacturing processing and implementation of strategic process control, to evaluate retention of MSC characteristics, multipotency, and to minimize the contamination risk of adventitious agent and lot‐to‐lot variation in production. Further research is required to make donor MSC‐specific media and processes to address challenges to suit media to clinical as well as therapeutic manufacturing of cell‐based products.
Figure 2.
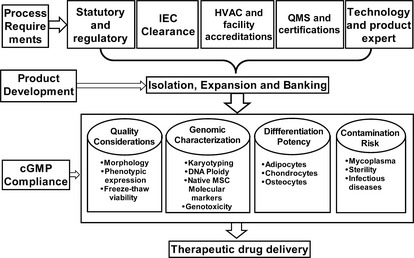
Outline of critical components for the derivation of clinical grade human mesenchymal stromal cells. IEC, institutional ethical committee; HVAC, heating, ventilation, air‐conditioning; QMS, quality management system; cGMP, current good manufacturing practice. [Correction added on 14 October 2013, after first online publication: The correct version of Figure 2 has now been included.]
Conflicts of interest and disclosure
The authors have no conflict of interests, apart from the above.
Supporting information
Table S1. Various disclosed serum free media formulation for MSCs.
Table S2. Phenotypic marker expression of MSCs frown in disclosed SFM.
Table S3. Phenotypic characterizations of MSCs in various commercially available SFM.
Acknowledgements
This work was fully funded by Stempeutics Research Pvt. Ltd, Manipal, India.
References
- 1. Prockop DJ (2007) “Stemness” does not explain the repair of many tissues by mesenchymal stem/multipotent stromal cells (MSCs). Clin. Pharmacol. Ther. 82, 241–243. [DOI] [PubMed] [Google Scholar]
- 2. Le Blanc K (2003) Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy 5, 485–489. [DOI] [PubMed] [Google Scholar]
- 3. Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O (2003) Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 57, 11–20. [DOI] [PubMed] [Google Scholar]
- 4. Subbanna PK (2007) Mesenchymal stem cells for treating GVHD: in‐vivo fate and optimal dose. Med. Hypotheses 69, 469–470. [DOI] [PubMed] [Google Scholar]
- 5. Sensebé L, Bourin P (2009) Mesenchymal stem cells for therapeutic purposes. Transplantation 87, S49–S53. [DOI] [PubMed] [Google Scholar]
- 6. Tarte K, Gaillard J, Lataillade JJ, Fouillard L, Becker M, Mossafa H et al (2010) Clinical‐grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood 115, 1549–1553. [DOI] [PubMed] [Google Scholar]
- 7. Phinney DG, Sensebe L (2013) Mesenchymal stromal cells: misconceptions and evolving concepts. Cytotherapy 15, 140–145. [DOI] [PubMed] [Google Scholar]
- 8. Gstraunthaler G (2003) Alternatives to the use of fetal bovine serum: serum‐free cell culture. ALTEX 20, 275–281. [PubMed] [Google Scholar]
- 9. Levings RL, Wessman SJ (1991) Bovine viral diarrhea virus contamination of nutrient serum, cell cultures and viral vaccines. Dev. Biol. Stand. 75, 177–181. [PubMed] [Google Scholar]
- 10. Wessman SJ, Levings RL (1999) Benefits and risks due to animal serum used in cell culture production. Dev. Biol. Stand. 99, 3–8. [PubMed] [Google Scholar]
- 11. Parker AM, Shang H, Khurgel M, Katz AJ (2007) Low serum and serum‐free culture of multipotential human adipose stem cells. Cytotherapy 9, 637–646. [DOI] [PubMed] [Google Scholar]
- 12. Spees JL, Gregory CA, Singh H, Tucker HA, Peister A, Lynch PJ et al (2004) Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol. Ther. 9, 747–756. [DOI] [PubMed] [Google Scholar]
- 13. Battiwalla M, Hematti P (2009) Mesenchymal stem cells in hematopoietic stem cell transplantation. Cytotherapy 11, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schallmoser K, Bartmann C, Rohde E, Reinisch A, Kashofer K, Stadelmeyer E et al (2007) Human platelet lysate can replace fetal bovine serum for clinical‐scale expansion of functional mesenchymal stromal cells. Transfusion 47, 1436–1446. [DOI] [PubMed] [Google Scholar]
- 15. Gottipamula S, Sharma A, Krishnamurthy S, Majumdar AS, Seetharam RN (2012) Human platelet lysate is an alternative to fetal bovine serum for large‐scale expansion of bone marrow‐derived mesenchymal stromal cells. Biotechnol. Lett. 34, 1367–1374. [DOI] [PubMed] [Google Scholar]
- 16. Jayme DW, Smith SR (2000) Media formulation options and manufacturing process controls to safeguard against introduction of animal origin contaminants in animal cell culture. Cytotechnology 33, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Valk J, Mellor D, Brands R, Fischer R, Gruber F, Gstraunthaler G et al (2004) The humane collection of fetal bovine serum and possibilities for serum‐free cell and tissue culture. Toxicol. In Vitro 18, 1–12. [DOI] [PubMed] [Google Scholar]
- 18. Dimarakis I, Levicar N (2006) Cell culture medium composition and translational adult bone marrow‐derived stem cell research. Stem Cells 24, 1407–1408. [DOI] [PubMed] [Google Scholar]
- 19. Tekkatte C, Gunasingh GP, Cherian KM, Sankaranarayanan K (2011) “Humanized” stem cell culture techniques: the animal serum controversy. Stem Cells Int. 2011, 504723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shahdadfar A, Fronsdal K, Haug T, Reinholt FP, Brinchmann JE (2005) In vitro expansion of human mesenchymal stem cells: choice of serum is a determinant of cell proliferation, differentiation, gene expression, and transcriptome stability. Stem Cells 23, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 21. Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY et al (2002) Isolated allogeneic bone marrow‐derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 99, 8932–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Erickson GA, Bolin SR, Landgraf JG (1991) Viral contamination of fetal bovine serum used for tissue culture: risks and concerns. Dev. Biol. Stand. 75, 173–175. [PubMed] [Google Scholar]
- 23. Bieback K, Hecker A, Schlechter T, Hofmann I, Brousos N, Redmer T et al (2012) Replicative aging and differentiation potential of human adipose tissue‐derived mesenchymal stromal cells expanded in pooled human or fetal bovine serum. Cytotherapy 14, 570–583. [DOI] [PubMed] [Google Scholar]
- 24. Heiskanen A, Satomaa T, Tiitinen S, Laitinen A, Mannelin S, Impola U et al (2007) N‐glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells 25, 197–202. [DOI] [PubMed] [Google Scholar]
- 25. Gottipamula S, Muttigi MS, Chaansa S, Ashwin KM, Priya N, Kolkundkar U et al (2013) Large‐scale expansion of pre‐isolated bone marrow mesenchymal stromal cells in serum‐free conditions. J. Tissue Eng. Regen. Med. doi: 10.1002/term.1713. (PMID:23495227) [DOI] [PubMed] [Google Scholar]
- 26. Berger MG, Veyrat‐Masson R, Rapatel C, Descamps S, Chassagne J, Boiret‐Dupre N (2006) Cell culture medium composition and translational adult bone marrow‐derived stem cell research. Stem Cells 24, 2888–2890. [DOI] [PubMed] [Google Scholar]
- 27. Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M (2006) Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells 24, 462–471. [DOI] [PubMed] [Google Scholar]
- 28. Baba K, Yamazaki Y, Ishiguro M, Kumazawa K, Aoyagi K, Ikemoto S et al (2013) Osteogenic potential of human umbilical cord‐derived mesenchymal stromal cells cultured with umbilical cord blood‐derived fibrin: a preliminary study. J. Craniomaxillofac. Surg. doi: 10.1016/j.jcms.2013.01.025. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 29. Bieback K, Hecker A, Kocaomer A, Lannert H, Schallmoser K, Strunk D et al (2009) Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells 27, 2331–2341. [DOI] [PubMed] [Google Scholar]
- 30. Chase LG, Lakshmipathy U, Solchaga LA, Rao MS, Vemuri MC (2010) A novel serum‐free medium for the expansion of human mesenchymal stem cells. Stem Cell Res. Ther. 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chase LG, Yang S, Zachar V, Yang Z, Lakshmipathy U, Bradford J et al (2012) Development and characterization of a clinically compliant xeno‐free culture medium in good manufacturing practice for human multipotent mesenchymal stem cells. Stem Cells Transl. Med. 1, 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hudson JE, Mills RJ, Frith JE, Brooke G, Jaramillo‐Ferrada P, Wolvetang EJ et al (2011) A defined medium and substrate for expansion of human mesenchymal stromal cell progenitors that enriches for osteo‐ and chondrogenic precursors. Stem Cells Dev. 20, 77–87. [DOI] [PubMed] [Google Scholar]
- 33. Jung S, Panchalingam KM, Rosenberg L, Behie LA (2012) Ex vivo expansion of human mesenchymal stem cells in defined serum‐free media. Stem Cells Int. 2012, 123030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung S, Sen A, Rosenberg L, Behie LA (2010) Identification of growth and attachment factors for the serum‐free isolation and expansion of human mesenchymal stromal cells. Cytotherapy 12, 637–657. [DOI] [PubMed] [Google Scholar]
- 35. Jung S, Sen A, Rosenberg L, Behie LA (2011) Human mesenchymal stem cell culture: rapid and efficient isolation and expansion in a defined serum‐free medium. J. Tissue Eng. Regen. Med. 6, 391–403. [DOI] [PubMed] [Google Scholar]
- 36. Jaiswal N, Haynesworth SE, Caplan AI, Bruder SP (1997) Osteogenic differentiation of purified, culture‐expanded human mesenchymal stem cells in vitro. J. Cell. Biochem. 64, 295–312. [PubMed] [Google Scholar]
- 37. Mimura S, Kimura N, Hirata M, Tateyama D, Hayashida M, Umezawa A et al (2011) Growth factor‐defined culture medium for human mesenchymal stem cells. Int. J. Dev. Biol. 55, 181–187. [DOI] [PubMed] [Google Scholar]
- 38. Kinzebach S, Bieback K (2012) Expansion of mesenchymal stem/stromal cells under xenogenic‐free culture conditions. Adv. Biochem. Eng. Biotechnol. 129, 33–57. [DOI] [PubMed] [Google Scholar]
- 39. De Luca A, Lamura L, Gallo M, Maffia V, Normanno N (2012) Mesenchymal stem cell‐derived interleukin‐6 and vascular endothelial growth factor promote breast cancer cell migration. J. Cell. Biochem. 113, 3363–3370. [DOI] [PubMed] [Google Scholar]
- 40. Jung J, Moon N, Ahn JY, Oh EJ, Kim M, Cho CS et al (2009) Mesenchymal stromal cells expanded in human allogenic cord blood serum display higher self‐renewal and enhanced osteogenic potential. Stem Cells Dev. 18, 559–571. [DOI] [PubMed] [Google Scholar]
- 41. Bieback K, Schallmoser K, Kluter H, Strunk D (2008) Clinical protocols for the isolation and expansion of mesenchymal stromal cells. Transfus. Med. Hemother. 35, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brunner D, Frank J, Appl H, Schoffl H, Pfaller W, Gstraunthaler G (2010) Serum‐free cell culture: the serum‐free media interactive online database. ALTEX 27, 53–62. [DOI] [PubMed] [Google Scholar]
- 43. Yao S, Chen S, Clark J, Hao E, Beattie GM, Hayek A et al (2006) Long‐term self‐renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc. Natl. Acad. Sci. USA 103, 6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW (2008) NS21: re‐defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 171, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hatlapatka T, Moretti P, Lavrentieva A, Hass R, Marquardt N, Jacobs R et al (2011) Optimization of culture conditions for the expansion of umbilical cord‐derived mesenchymal stem or stromal cell‐like cells using xeno‐free culture conditions. Tissue Eng. Part C Methods 17, 485–493. [DOI] [PubMed] [Google Scholar]
- 46. Tateishi K, Ando W, Higuchi C, Hart DA, Hashimoto J, Nakata K et al (2008) Comparison of human serum with fetal bovine serum for expansion and differentiation of human synovial MSC: potential feasibility for clinical applications. Cell Transplant. 17, 549–557. [DOI] [PubMed] [Google Scholar]
- 47. Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M (2006) Cell culture medium composition and translational adult bone marrow‐derived stem cell research. Stem Cells 24, 1409–1410. [DOI] [PubMed] [Google Scholar]
- 48. Kocaoemer A, Kern S, Kluter H, Bieback K (2007) Human AB serum and thrombin‐activated platelet‐rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 25, 1270–1278. [DOI] [PubMed] [Google Scholar]
- 49. Dahl JA, Duggal S, Coulston N, Millar D, Melki J, Shahdadfar A et al (2008) Genetic and epigenetic instability of human bone marrow mesenchymal stem cells expanded in autologous serum or fetal bovine serum. Int. J. Dev. Biol. 52, 1033–1042. [DOI] [PubMed] [Google Scholar]
- 50. Nimura A, Muneta T, Koga H, Mochizuki T, Suzuki K, Makino H et al (2008) Increased proliferation of human synovial mesenchymal stem cells with autologous human serum: comparisons with bone marrow mesenchymal stem cells and with fetal bovine serum. Arthritis Rheum. 58, 501–510. [DOI] [PubMed] [Google Scholar]
- 51. Nimura A, Muneta T, Otabe K, Koga H, Ju YJ, Mochizuki T et al (2011) Analysis of human synovial and bone marrow mesenchymal stem cells in relation to heat‐inactivation of autologous and fetal bovine serums. BMC Musculoskeletal Disord. 11, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Duggal S, Brinchmann JE (2011) Importance of serum source for the in vitro replicative senescence of human bone marrow derived mesenchymal stem cells. J. Cell. Physiol. 226, 2908–2915. [DOI] [PubMed] [Google Scholar]
- 53. Patrikoski M, Juntunen M, Boucher S, Campbell A, Vemuri MC, Mannerstrom B et al (2013) Development of fully defined xeno‐free culture system for the preparation and propagation of cell therapy‐compliant human adipose stem cells. Stem Cell Res. Ther. 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Venugopal P, Balasubramanian S, Majumdar AS, Ta M (2011) Isolation, characterization, and gene expression analysis of Wharton's jelly‐derived mesenchymal stem cells under xeno‐free culture conditions. Stem Cells Cloning 4, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jenhani F, Durand V, Ben Azouna N, Thallet S, Ben Othmen T, Bejaoui M et al (2011) Human cytokine expression profile in various conditioned media for in vitro expansion bone marrow and umbilical cord blood immunophenotyped mesenchymal stem cells. Transpl. Proc. 43, 639–643. [DOI] [PubMed] [Google Scholar]
- 56. Cholewa D, Stiehl T, Schellenberg A, Bokermann G, Joussen S, Koch C et al (2011) Expansion of adipose mesenchymal stromal cells is affected by human platelet lysate and plating density. Cell Transplant. 20, 1409–1422. [DOI] [PubMed] [Google Scholar]
- 57. Capelli C, Domenghini M, Borleri G, Bellavita P, Poma R, Carobbio A et al (2007) Human platelet lysate allows expansion and clinical grade production of mesenchymal stromal cells from small samples of bone marrow aspirates or marrow filter washouts. Bone Marrow Transplant. 40, 785–791. [DOI] [PubMed] [Google Scholar]
- 58. Crespo‐Diaz R, Behfar A, Butler GW, Padley DJ, Sarr MG, Bartunek J et al (2011) Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant. 20, 797–811. [DOI] [PubMed] [Google Scholar]
- 59. Reinisch A, Bartmann C, Rohde E, Schallmoser K, Bjelic‐Radisic V, Lanzer G et al (2007) Humanized system to propagate cord blood‐derived multipotent mesenchymal stromal cells for clinical application. Regen. Med. 2, 371–382. [DOI] [PubMed] [Google Scholar]
- 60. Schallmoser K, Rohde E, Reinisch A, Bartmann C, Thaler D, Drexler C et al (2008) Rapid large‐scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng. Part C Methods 14, 185–196. [DOI] [PubMed] [Google Scholar]
- 61. Avanzini MA, Bernardo ME, Cometa AM, Perotti C, Zaffaroni N, Novara F et al (2009) Generation of mesenchymal stromal cells in the presence of platelet lysate: a phenotypic and functional comparison of umbilical cord blood‐ and bone marrow‐derived progenitors. Haematologica 94, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ben Azouna N, Jenhani F, Regaya Z, Berraeis L, Ben Othman T, Ducrocq E et al (2012) Phenotypical and functional characteristics of mesenchymal stem cells from bone marrow: comparison of culture using different media supplemented with human platelet lysate or fetal bovine serum. Stem Cell Res. Ther. 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lohmann M, Walenda G, Hemeda H, Joussen S, Drescher W, Jockenhoevel S et al (2012) Donor age of human platelet lysate affects proliferation and differentiation of mesenchymal stem cells. PLoS One 7, e37839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walenda G, Hemeda H, Schneider RK, Merkel R, Hoffmann B, Wagner W (2012) Human platelet lysate gel provides a novel three dimensional‐matrix for enhanced culture expansion of mesenchymal stromal cells. Tissue Eng. Part C Methods 18, 924–934. [DOI] [PubMed] [Google Scholar]
- 65. Warnke PH, Humpe A, Strunk D, Stephens S, Warnke F, Wiltfang J et al (2013) A clinically‐feasible protocol for using human platelet lysate and mesenchymal stem cells in regenerative therapies. J. Craniomaxillofac. Surg. 41, 153–161. [DOI] [PubMed] [Google Scholar]
- 66. Horn P, Bokermann G, Cholewa D, Bork S, Walenda T, Koch C et al (2010) Impact of individual platelet lysates on isolation and growth of human mesenchymal stromal cells. Cytotherapy 12, 888–898. [DOI] [PubMed] [Google Scholar]
- 67. Shih DT, Chen JC, Chen WY, Kuo YP, Su CY, Burnouf T (2010) Expansion of adipose tissue mesenchymal stromal progenitors in serum‐free medium supplemented with virally inactivated allogeneic human platelet lysate. Transfusion 51, 770–778. [DOI] [PubMed] [Google Scholar]
- 68. Abdelrazik H, Spaggiari GM, Chiossone L, Moretta L (2011) Mesenchymal stem cells expanded in human platelet lysate display a decreased inhibitory capacity on T‐ and NK‐cell proliferation and function. Eur. J. Immunol. 41, 3281–3290. [DOI] [PubMed] [Google Scholar]
- 69. Flemming A, Schallmoser K, Strunk D, Stolk M, Volk HD, Seifert M (2011) Immunomodulative efficacy of bone marrow‐derived mesenchymal stem cells cultured in human platelet lysate. J. Clin. Immunol. 31, 1143–1156. [DOI] [PubMed] [Google Scholar]
- 70. Kazemnejad S, Allameh A, Gharehbaghian A, Soleimani M, Amirizadeh N, Jazayeri M (2008) Efficient replacing of fetal bovine serum with human platelet releasate during propagation and differentiation of human bone‐marrow‐derived mesenchymal stem cells to functional hepatocyte‐like cells. Vox Sang. 95, 149–158. [DOI] [PubMed] [Google Scholar]
- 71. Jayme DW, Blackman KE (1985) Culture media for propagation of mammalian cells, viruses, and other biologicals. Adv. Biotechnol. Process 5, 1–30. [PubMed] [Google Scholar]
- 72. Jordan M, Voisard D, Berthoud A, Tercier L, Kleuser B, Baer G et al (2013) Cell culture medium improvement by rigorous shuffling of components using media blending. Cytotechnology 65, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Haack‐Sorensen M, Friis T, Bindslev L, Mortensen S, Johnsen HE, Kastrup J (2008) Comparison of different culture conditions for human mesenchymal stromal cells for clinical stem cell therapy. Scand. J. Clin. Lab. Invest. 68, 192–203. [DOI] [PubMed] [Google Scholar]
- 74. Lennon DP, Haynesworth SE, Young RG, Dennis JE, Caplan AI (1995) A chemically defined medium supports in vitro proliferation and maintains the osteochondral potential of rat marrow‐derived mesenchymal stem cells. Exp. Cell Res. 219, 211–222. [DOI] [PubMed] [Google Scholar]
- 75. Rajala K, Lindroos B, Hussein SM, Lappalainen RS, Pekkanen‐Mattila M, Inzunza J et al (2010) A defined and xeno‐free culture method enabling the establishment of clinical‐grade human embryonic, induced pluripotent and adipose stem cells. PLoS One 5, e10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM (2002) Origin of endothelial progenitors in human postnatal bone marrow. J. Clin. Invest. 109, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu W, Zhou Y, Tan W (2009) In vitro culture of bone marrow‐derived mesenchymal stem cells in a chemically‐defined serum‐free medium. Sheng Wu Gong Cheng Xue Bao 25, 121–128. [PubMed] [Google Scholar]
- 78. Gronthos S, Simmons PJ (1995) The growth factor requirements of STRO‐1‐positive human bone marrow stromal precursors under serum‐deprived conditions in vitro. Blood 85, 929–940. [PubMed] [Google Scholar]
- 79. Marshak DR, Holecek JJ (1999) Chemically defined medium for human mesenchymal stem cells. Google Patents United States Patent 5, 908, 782. [Google Scholar]
- 80. Liu C‐H, Wu M‐L, Hwang S‐M (2007) Optimization of serum free medium for cord blood mesenchymal stem cells. Biochem. Eng. J. 33, 1–9. [Google Scholar]
- 81. Tarle SA, Shi S, Kaigler D (2010) Development of a serum‐free system to expand dental‐derived stem cells: PDLSCs and SHEDs. J. Cell. Physiol. 226, 66–73. [DOI] [PubMed] [Google Scholar]
- 82. Rajaraman G, White J, Tan KS, Ulrich D, Rosamilia A, Werkmeister J et al (2013) Optimization and scale‐up culture of human endometrial multipotent mesenchymal stromal cells: potential for clinical application. Tissue Eng. Part C Methods 19, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim EJ, Kim NS, Lee GM (1998) Development of a serum‐free medium for the production of humanized antibody from Chinese hamster ovary cells using a statistical design. In Vitro Cell. Dev. Biol. Anim. 34, 757–761. [DOI] [PubMed] [Google Scholar]
- 84. Kim EJ, Kim NS, Lee GM (1999) Development of a serum‐free medium for dihydrofolate reductase‐deficient Chinese hamster ovary cells (DG44) using a statistical design: beneficial effect of weaning of cells. In Vitro Cell. Dev. Biol. Anim. 35, 178–182. [DOI] [PubMed] [Google Scholar]
- 85. Lee GM, Kim EJ, Kim NS, Yoon SK, Ahn YH, Song JY (1999) Development of a serum‐free medium for the production of erythropoietin by suspension culture of recombinant Chinese hamster ovary cells using a statistical design. J. Biotechnol. 69, 85–93. [DOI] [PubMed] [Google Scholar]
- 86. Castro PM, Hayter PM, Ison AP, Bull AT (1992) Application of a statistical design to the optimization of culture medium for recombinant interferon‐gamma production by Chinese hamster ovary cells. Appl. Microbiol. Biotechnol. 38, 84–90. [DOI] [PubMed] [Google Scholar]
- 87. Li F, Vijayasankaran N, Shen AY, Kiss R, Amanullah A (2010) Cell culture processes for monoclonal antibody production. MAbs. 2, 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Meng G, Liu S, Rancourt DE (2012) Synergistic effect of medium, matrix, and exogenous factors on the adhesion and growth of human pluripotent stem cells under defined, xeno‐free conditions. Stem Cells Dev. 21, 2036–2048. [DOI] [PubMed] [Google Scholar]
- 89. Bray LJ, George KA, Hutmacher DW, Chirila TV, Harkin DG (2012) A dual‐layer silk fibroin scaffold for reconstructing the human corneal limbus. Biomaterials 33, 3529–3538. [DOI] [PubMed] [Google Scholar]
- 90. Bray LJ, Heazlewood CF, Atkinson K, Hutmacher DW, Harkin DG (2012) Evaluation of methods for cultivating limbal mesenchymal stromal cells. Cytotherapy 14, 936–947. [DOI] [PubMed] [Google Scholar]
- 91. Miwa H, Hashimoto Y, Tensho K, Wakitani S, Takagi M (2011) Xeno‐free proliferation of human bone marrow mesenchymal stem cells. Cytotechnology 64, 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pal R, Hanwate M, Jan M, Totey S (2009) Phenotypic and functional comparison of optimum culture conditions for upscaling of bone marrow‐derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 3, 163–174. [DOI] [PubMed] [Google Scholar]
- 93. Wagey R, Short B (2012) Isolation, enumeration, and expansion of human mesenchymal stem cells in culture. Methods Mol. Biol. 946, 315–334. [DOI] [PubMed] [Google Scholar]
- 94. Yang T, Chen GH, Xue SL, Qiao M, Liu HW, Tian H et al (2012) Comparison of the biological characteristics of serum‐free and fetal bovine serum‐contained medium cultured umbilical cord‐derived mesenchymal stem cells. Zhonghua Xue Ye Xue Za Zhi 33, 715–719. [DOI] [PubMed] [Google Scholar]
- 95. Yu J, Chau KF, Vodyanik MA, Jiang J, Jiang Y (2011) Efficient feeder‐free episomal reprogramming with small molecules. PLoS One 6, e17557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McNiece I, Harrington J, Turney J, Kellner J, Shpall EJ (2004) Ex vivo expansion of cord blood mononuclear cells on mesenchymal stem cells. Cytotherapy 6, 311–317. [DOI] [PubMed] [Google Scholar]
- 97. Cardoso TC, Ferrari HF, Garcia AF, Novais JB, Silva‐Frade C, Ferrarezi MC et al (2012) Isolation and characterization of Wharton's jelly‐derived multipotent mesenchymal stromal cells obtained from bovine umbilical cord and maintained in a defined serum‐free three‐dimensional system. BMC Biotechnol. 12, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Agata H, Watanabe N, Ishii Y, Kubo N, Ohshima S, Yamazaki M et al (2009) Feasibility and efficacy of bone tissue engineering using human bone marrow stromal cells cultivated in serum‐free conditions. Biochem. Biophys. Res. Commun. 382, 353–358. [DOI] [PubMed] [Google Scholar]
- 99. Hartmann I, Hollweck T, Haffner S, Krebs M, Meiser B, Reichart B et al (2010) Umbilical cord tissue‐derived mesenchymal stem cells grow best under GMP‐compliant culture conditions and maintain their phenotypic and functional properties. J. Immunol. Methods 363, 80–89. [DOI] [PubMed] [Google Scholar]
- 100. Lindroos B, Boucher S, Chase L, Kuokkanen H, Huhtala H, Haataja R et al (2009) Serum‐free, xeno‐free culture media maintain the proliferation rate and multipotentiality of adipose stem cells in vitro. Cytotherapy 11, 958–972. [DOI] [PubMed] [Google Scholar]
- 101. Santos F, Andrade PZ, Abecasis MM, Gimble JM, Chase LG, Campbell AM et al (2011) Toward a clinical‐grade expansion of mesenchymal stem cells from human sources: a microcarrier‐based culture system under xeno‐free conditions. Tissue Eng. Part C Methods 17, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yang S, Pilgaard L, Chase LG, Boucher S, Vemuri MC, Fink T et al (2012) Defined xenogeneic‐free and hypoxic environment provides superior conditions for long‐term expansion of human adipose‐derived stem cells. Tissue Eng. Part C Methods 18, 593–602. [DOI] [PubMed] [Google Scholar]
- 103. Sawada R, Yamada T, Tsuchiya T, Matsuoka A (2010) Microarray analysis of the effects of serum‐free medium on gene expression changes in human mesenchymal stem cells during the in vitro culture. Yakugaku Zasshi 130, 1387–1393. [DOI] [PubMed] [Google Scholar]
- 104. Ishikawa I, Sawada R, Kato Y, Tsuji K, Shao J, Yamada T et al (2009) Effectivity of the novel serum‐free medium STK2 for proliferating human mesenchymal stem cells. Yakugaku Zasshi 129, 381–384. [DOI] [PubMed] [Google Scholar]
- 105. Meuleman N, Tondreau T, Delforge A, Dejeneffe M, Massy M, Libertalis M et al (2006) Human marrow mesenchymal stem cell culture: serum‐free medium allows better expansion than classical alpha‐MEM medium. Eur. J. Haematol. 76, 309–316. [DOI] [PubMed] [Google Scholar]
- 106. Bieback K, Kern S, Kluter H, Eichler H (2004) Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 22, 625–634. [DOI] [PubMed] [Google Scholar]
- 107. Battula VL, Bareiss PM, Treml S, Conrad S, Albert I, Hojak S et al (2007) Human placenta and bone marrow derived MSC cultured in serum‐free, b‐FGF‐containing medium express cell surface frizzled‐9 and SSEA‐4 and give rise to multilineage differentiation. Differentiation 75, 279–291. [DOI] [PubMed] [Google Scholar]
- 108. Ng F, Boucher S, Koh S, Sastry KS, Chase L, Lakshmipathy U et al (2008) PDGF, TGF‐beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 112, 295–307. [DOI] [PubMed] [Google Scholar]
- 109. Apel A, Groth A, Schlesinger S, Bruns H, Schemmer P, Buchler MW et al (2009) Suitability of human mesenchymal stem cells for gene therapy depends on the expansion medium. Exp. Cell Res. 315, 498–507. [DOI] [PubMed] [Google Scholar]
- 110. Crapnell K, Blaesius R, Hastings A, Lennon DP, Caplan AI, Bruder SP (2013) Growth, differentiation capacity, and function of mesenchymal stem cells expanded in serum‐free medium developed via combinatorial screening. Exp. Cell Res. 319, 1409–1418. [DOI] [PubMed] [Google Scholar]
- 111. Fink T, Rasmussen JG, Lund P, Pilgaard L, Soballe K, Zachar V (2011) Isolation and expansion of adipose‐derived stem cells for tissue engineering. Front. Biosci. (Elite Ed.) 3, 256–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Various disclosed serum free media formulation for MSCs.
Table S2. Phenotypic marker expression of MSCs frown in disclosed SFM.
Table S3. Phenotypic characterizations of MSCs in various commercially available SFM.