

Abstract
Cystathionine β-synthase (CBS) is a key regulator of sulfur amino acid metabolism, taking homocysteine from the methionine cycle to the biosynthesis of cysteine via the trans-sulfuration pathway. CBS is also a predominant source of H2S biogenesis. Roles for CBS have been reported for neuronal death pursuant to cerebral ischemia, promoting ovarian tumor growth, and maintaining drug-resistant phenotype by controlling redox behavior and regulating mitochondrial bioenergetics. The trans-sulfuration pathway is well-conserved in eukaryotes, but the analogous enzymes have different enzymatic behavior in different organisms. CBSs from the higher organisms contain a heme in an N-terminal domain. Though the presence of the heme, whose functions in CBSs have yet to be elucidated, is biochemically interesting, it hampers UV–vis absorption spectroscopy investigations of pyridoxal 5′-phosphate (PLP) species. CBS from Saccharomyces cerevisiae (yCBS) naturally lacks the heme-containing N-terminal domain, which makes it an ideal model for spectroscopic studies of the enzymological reaction catalyzed and allows structural studies of the basic yCBS catalytic core (yCBS-cc). Here we present the crystal structure of yCBS-cc, solved to 1.5 Å. Crystal structures of yCBS-cc in complex with enzymatic reaction intermediates have been captured, providing a structural basis for residues involved in catalysis. Finally, the structure of the yCBScc cofactor complex generated by incubation with an inhibitor shows apparent off-pathway chemistry not normally seen with CBS.
Graphical Abstract
l-Methionine is an essential sulfur-containing amino acid for mammals. Its metabolism comprises two intersecting metabolic pathways: the synthesis pathway for methionine via the S-adenosylmethionine (SAM) cycle and the trans-sulfuration pathway leading to glutathione synthesis. Both pathways compete for homocysteine, a central intermediate that is generated from methionine and adenosine triphosphate (ATP).1 In the trans-sulfuration pathway, believed to be the sole route for cysteine biosynthesis in vertebrates, homocysteine is converted to cysteine. Cystathionine β-synthase (CBS, EC 4.2.1.22) is the key enzyme that regulates the flux of homocysteine through this pathway.2,3 Two trans-sulfuration pathways are known: the forward and reverse pathways. The forward pathway is present in organisms such as bacteria,4 plants,5 and yeast6 and involves the transfer of the thiol group from cysteine to homocysteine on the way to methionine. The reverse pathway is present in many organisms, including humans, and involves the transfer of the thiol group from homocysteine to cysteine.7
CBS catalyzes the first of two pyridoxal 5′-phosphate (PLP)-dependent reactions that convert the essential amino acid l-methionine to l-cysteine by the condensation of l-homo-cysteine and l-serine to yield (L,L)-cystathionine, which is then converted to l-cysteine by the action of cystathionine γ-lyase (Figure 1).2,8 Homocysteine is toxic in mammals, and mutations in the gene encoding the human form of the enzyme (hCBS) are the major causes of homocystinuria.9–12 Deficiency in the activity of the enzyme leads to accumulation of l-homocysteine, a risk factor for cardiovascular, Alzheimer’s, and other human diseases.13–15
Figure 1.

Reactions catalyzed by CBS.8 (A) Canonical reaction of CBS. (B) CBS reactions that generate or utilize H2S.
In addition, CBS is the predominant source of H2S biogenesis in the brain, where increased levels are associated with neuronal death pursuant to cerebral ischemia (vide infra).16–18 Recently, it has been shown that CBS silencing can inhibit ovarian tumor growth and sensitize ovarian cancer cells to cisplatin.19
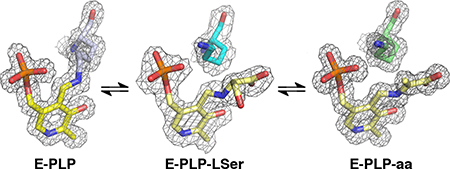
The synthesis of cystathionine starts when the internal aldimine between the enzyme active-site lysine and pyridoxal 5′-phosphate (E-PLP) is displaced by the incoming α-amino acid l-serine (Figure 2). The serine external aldimine adduct (E-PLP-l-ser) forms an aminoacrylate intermediate (E-PLP-aa) that reacts with the incoming second substrate, such as l-homocysteine, to form the (L,L)-cystathionine external aldimine, which is then displaced by the active-site lysine to regenerate the active enzyme. The enzyme can also operate in the reverse direction, in which the PLP form of the enzyme reacts with cystathionine directly and can convert to the serine external aldimine or the native internal aldimine.20 This reverse reaction with cystathionine has also been reported as far as the aminoacrylate intermediate.21–23 The enzyme does not readily perform a reaction often seen for PLP-dependent enzymes, the further reaction of the external aldimine to form a ketamine followed by β-elimination and release of an α-keto product and the pyridoxamine phosphate form of the enzyme (E-PMP). The rate of pyruvate formation from serine in the absence of homocysteine has been reported to be less than 2% of the tritium exchange rate of the α-proton of serine.24
Figure 2.

Proposed enzymatic mechanism for the yCBS reaction with absorption maxima.20 Possible quinonoid intermediates between E-serine (E-PLP-l-ser) and E-aminoacrylate (E-PLP-aa) or between E-aminoacrylate and E-cystathionine are not shown. The growing consensus among those working on fold-type-II PLP-dependent enzymes is that the ring nitrogen does not undergo protonation during the catalytic cycle in these enzymes.25 However, at pH 6.5 the expectation is that the ring nitrogen is protonated as shown.
The eukaryotic CBS has several domains, depending on the organism from which it is derived.26 Thus, the human (hCBS) and Drosophila melanogaster (dCBS) enzymes have three domains, while the enzyme from yeast (yCBS) has only two. Of the three domains, two are regulatory and one is catalytic. The catalytic domain contains the active site and is flanked by an N-terminal heme-binding domain (hCBS and dCBS) and a C-terminal Bateman module27,28 consisting of two CBS domains (hCBS, dCBS, yCBS).29 Sequence comparisons of CBSs exhibit extensive homology between yeast and parts of hCBS and dCBS, particularly in the catalytic core domains, which contain the PLP-cofactor binding sites (the sequence identities in this domain for yCBS/hCBS and yCBS/dCBS are 52%; Figure S130). The main differences for the overall protein come from the presence of an N-terminal extension on hCBS and dCBS that binds a heme (residues 1–71 and 1–41, respectively), which is absent in yCBS. The C-terminal domains (sequence identities: yCBS/hCBS < 40%, yCBS/dCBS < 50%), composed of two tandem CBS domains, are common to all CBSs although their functions seem to be different. Three-dimensional structures of a C-terminally truncated form of hCBS containing the heme-binding and catalytic core domains,31,32 a full-length hCBS construct missing an internal loop (hCBSΔ516–525),33–35 and full-length dCBS36 and two structures from bacterial CBSs (Lactobacillus plantarum37 and Bacillus anthracis38) that have only the catalytic domain have been reported. In all cases, the catalytic domain belongs to fold-type-II of the PLP-dependent enzymes.
The differences in activity among the CBSs are related to the influences of the regulatory domains, making direct comparisons among enzymes from different species difficult. It has been suggested that the heme-binding domain serves a regulatory function, possibly as a redox sensor.39 Truncation of this domain in the human protein produces an enzyme that is still active, albeit less so (19%) than the wild type.40 The C-terminal Bateman module also has a regulatory function. In the absence of allosteric regulators, truncation of this domain activates the truncated enzyme over the full-length enzyme by ~5-fold for the human enzyme40 and ~2-fold for the yeast enzyme.20 In addition, deletion of the Bateman module from hCBS and yCBS leads to a change in oligomerization from a tetrameric form to a dimeric form of the enzyme.41,42 In contrast, dCBS is dimeric in the presence or absence of this domain.36
SAM can act as an allosteric regulator of the enzyme. In the absence of SAM, specific activities of 106, 1227, and 1245 units/mg of protein for the reaction forming cystathionine from serine and homocysteine have been reported for the hCBS, dCBS, and yCBS enzymes, respectively. In the presence of SAM, these activities are reported to be 424, 1279, and 1260 units/mg of protein, respectively.42,43 The same trend was observed in a later study not only for the cystathionine synthesis reaction but also for other reactions catalyzed by CBS, such as H2S-generating reactions or H2S-utilizing reactions.26 The effect on hCBS is striking, especially in comparison with dCBS and yCBS, where SAM has very little or no effect. The C-terminal domain in the human full-length enzyme has an autoinhibitory effect that is alleviated by truncation of the domain or by binding of SAM.44 A structural interpretation of these results for hCBS focuses on different conformations of the Bateman module relative to the catalytic domain. The interaction of SAM with the Bateman module leads to conformational changes whereby autoinhibition of this regulatory region is eliminated, making the active site more accessible to substrate.26,33–35,45,46 The Drosophila enzyme is constitutively active, does not bind SAM, and is insoluble when C-terminally truncated. Yeast CBS is activated by C-terminal truncation but does not bind to either SAM or ATP.26
The physiological relevance of the CBS reaction derives from its importance in homeostasis of homocysteine, a toxic substance in eukaryotes.47 Several alternate reactions have been described that utilize cysteine in either β-elimination or β-replacement reactions to yield H2S.8 The importance of H2S stems from its involvement as a factor in modulating cell function in the vasculature and in the cerebrum (for a recent review, see ref 48) Although there are two other enzymatic sources of H2S (cystathionine γ-lyase and 3-mercaptopyruvate sulfotransferase), CBS serves as the principal source of H2S biogenesis in the brain.18,49–51 Numerous experiments linking cysteine, CBS, and H2S levels support the notion that CBS is the key enzyme modulating levels of cysteine-derived H2S in the cerebral cortex and that H2S mediates ischemic damage pursuant to stroke.52–57
Given the evidence that implicates elevated levels of H2S in ischemic neuronal damage, an inhibitor of the enzyme could be an important therapeutic agent for treatment of stroke. A unique inhibitor of the enzyme [(L,L)-2,7-bis(hydrazino)-1,8-octanedioic acid] has been designed that shows promise as a modulator against the deleterious effects of stroke.18 The compound has been shown to inhibit purified CBS and to reduce H2S production in a neuroblastoma model study designed to simulate ischemic conditions, thereby reducing cell death. In a rat transient middle cerebral artery occlusion (tMCAO) model for ischemia, the inhibitor reduces infarct volume by ~83% or 66% relative to control when the inhibitor is injected 30 min prior to or 60 min after the onset of tMCAO, respectively.18 Gaining additional insight into how this compound and other CBS inhibitors interact with the enzyme is an essential early step toward the development of more druglike compounds that effectively inhibit CBS.
In order to gain further understanding of the mechanism of this enzyme and to understand how regulation and inhibition occur, the yCBS catalytic core (yCBS-cc) was studied, and intermediates in the reaction were trapped crystallographically. Spectroscopic studies of the catalytic domains in the human and Drosophila enzymes are hampered by interference by the heme-binding domains, which absorb in the same spectral region. Thus, the yeast enzyme, which consists only of the core catalytic domain and the CBS domain, provides a model system from which to study the basal condensation reaction without regulation by the heme and CBS domains and to study inhibition mechanisms that are related to the PLP-dependent active site without interference from a regulatory domain.
Although CBSs from various sources display different kinetic properties and regulation, they all catalyze the same overall reactions. Therefore, the kinetic mechanisms are expected to be the same, and information from one enzyme can be transferred to another. Kinetic studies of the yeast enzyme showed that hydrolysis of the external aldimine of cystathionine is the rate-determining step in the reaction leading to cystathionine.32,58 Consequently, it was possible to trap an intermediate along the reaction path. To that end, we have determined the structures of the catalytic domain and those of two intermediates: the external aldimine formed between PLP and serine and that formed between PLP and the aminoacrylate intermediate in the reaction. Intermediates have also been trapped for dCBS,36 but one of them is not the same as for yCBS. The structure of the enzyme soaked with the hydrazine-based inhibitor suggests that the compound is an “inactivator” in that it converts the enzyme into the pyridoxamine form, which is inactive as a producer of H2S.
MATERIALS AND METHODS
Cloning, Expression, and Purification of Recombinant yCBS Proteins
yCBS DNA was cloned from the yeast genome using primers for pYPT200 in the forward and reverse directions. The full-length and catalytic-core yCBS constructs were designed using reported methods with some modifications.42 The gene was amplified using the following primers: for full-length yCBS (residues 1–508), the forward primer was 5′-ggccagCATATGatgactaaatctgagcagcaagc, and the reverse primer was 5′-ccgtgCTCGAGtcatgctaagtagctcag; for yCBS-cc (residues 1–353), the same forward primer was used with a different reverse primer, 5′-ccgtgCTCGAGtcacagctttgaagagtc. The PCR products were digested with NdeI and XhoI (New England Biolabs) and ligated into a pET-28(+) vector (Novagen) containing an N-terminal His tag.
All of the yCBS constructs were transformed into Escherichia coli expression strain BL21(DE3). Cells were grown overnight at 37 °C in 5 mL of LB broth containing 50 μg/mL kanamycin. The overnight culture was inoculated into 1 L of LB broth containing 50 μg/mL kanamycin at 37 °C with shaking at 200 rpm. Once the culture reached an optical density at 600 nm (OD600) of approximately 0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 500 μM. Fresh kanamycin or ampicillin was added, and the culture was grown overnight at 30 °C at 150 rpm. Cells were pelleted and resuspended in lysis buffer containing 1 mM PLP, 1 mM dithiothreitol (DTT), and one complete EDTA-free Protease Inhibitor Cocktail Tablet (Roche) in 50 mM Tris buffer (pH 8.0). Cells were sonicated for 20 s on and 40 s off for a total of 4 min of sonication time. This suspension was centrifuged at 10 000 rpm for 1 h, and the supernatant was collected and filtered to remove any cell debris.
The enzyme was purified using first a nickel agarose column (Ni-NTA Agarose, Qiagen). Ni-NTA resin was washed and equilibrated with lysis buffer containing 10 mM imidazole. The filtered supernatant was passed through the column and then washed with 50 mL of wash buffer #1 (20 mM imidazole, 1 mM DTT, 1 mM PLP, and 50 mM Tris pH 8.0) followed by 50 mL of wash buffer #2 (50 mM imidazole, 1 mM DTT, 1 mM PLP, and 50 mM Tris pH 8.0). The yCBS constructs were eluted with 25 mL of elution buffer (250 mM imidazole, 1 mM DTT, 1 mM PLP, and 50 mM Tris pH 8.0).
Next, the eluent was passed through a Mono Q column that was equilibrated with buffer A (1 mM DTT and 50 mM Tris pH 8.0). The protein was eluted using a gradient of buffer B (1 M NaCl, 1 mM DTT, and 50 mM Tris pH 8.0). The yCBS constructs eluted at around 150 mM NaCl. Final protein fractions were collected and flash-frozen in liquid nitrogen for storage at −80 °C.
Crystallization
Both full-length and catalytic-core yCBSs were set up for crystallization. Only the catalytic core succeeded. The yCBS-cc was crystallized in hanging drops containing 10 mg/mL protein, 30% PEG 400, 100 mM calcium acetate, and 100 mM Tris pH 8.0 at 4 °C against a well solution containing 40% PEG 400 and 50 mM Tris pH 8.0. Crystals were yellow, indicating the presence of the PLP-derived internal aldimine. Crystals were cryoprotected with 40% PEG 400/50 mM Tris pH 8.0 and flash-frozen in liquid nitrogen. The substrate l-serine was cocrystallized with yCBS-cc at pH 6.5 and 8.0 with a concentration ratio of 2:1. At pH 8.0, yellow crystals appeared overnight. At pH 6.5, orange crystals, indicative of the PLP–aminoacrylate external aldimine at the active site, appeared overnight. Crystals were harvested, soaked in appropriate cryoprotectant, and flash-frozen in liquid nitrogen for data collection. The hydrazine inhibitor at 500 μM was soaked into crystals for 1 h at room temperature and pH 8. The color of the crystal changed from yellow to colorless. Crystals were harvested, soaked in appropriate cryoprotectant, and flash-frozen in liquid nitrogen for data collection.
X-ray Diffraction Data Collection, Phasing, and Refinement
Data sets and refinement statistics are listed in Table S1. Diffraction data for substrate-free yCBS-cc, yCBS-cc with the substrate l-serine at pH 6.5 and 8.0, and yCBS-cc with hydrazine inhibitor were collected using synchrotron radiation at 100 K at APS SBC-CAT, beamline 19-ID-D, using an ADSC Quantum 315 detector. The diffraction data were processed using the program package HKL2000.59 The structure of the yCBS-cc construct was solved by molecular replacement using a monomer of the catalytic domain of hCBS (PDB ID 1JBQ31) as a search model. All of the structures were refined using the program package PHENIX60 with model building in Coot.61 In later rounds of refinement, the electron density in yCBS-cc cocrystallized with l-serine at pH 8.0 was modeled and refined with either the PLP–l-ser adduct or the PLP–aa adduct in the active site. Only the PLP–l-ser adduct fit the electron density and was amenable to refinement (Figure S2). In this structure, there was weak electron density that might suggest a potential alternate conformation for K53 as the geminal diamine. However, because it was so weak, it was not modeled. The electron density in the yCBS-cc/hydrazine inhibitor complex indicated that the enzyme had turned over into the PMP form, and the model was built accordingly (Figure S2). Model quality was assessed throughout using MolProbity.62 During the last stage of this process, models were submitted to the online PDB_REDO server,63 followed by a final round of refinement using REFMAC.64 All of the models included residues 4–348 of the protein and the relevant PLP species.
All structural figures were created using MacPyMOL (PyMOL version 1.8.2.3 Enhanced for Mac OS X: The PyMOL Molecular Graphics System, Schrödinger, LLC, New York). Schematics were generated with ChemDraw (ChemDraw Professional, version 16.0.1.4, PerkinElmer Informatics, Inc., Cambridge, MA). Distances for key interactions between atoms are reported in Table S2. Sequence comparisons and structural alignments were done using the Dali server.30 Atomic coordinates and structure factors for these crystal structures have been deposited in the Protein Data Bank (www.rcsb.org65) with accession codes 6C2H, 6C2Q, 6C2Z, and 6C4P.
Kinetic Assays
The activities of the full-length and catalytic-core yCBS were determined using the lead(II) sulfide-forming assay.45 l-cysteine was used as the substrate, and β-mercaptoethanol was excluded to avoid disruption of the protein oligomeric state. The assays were conducted in 10 mM phosphate buffer (pH 8.0) and 0.4 mM lead(II) acetate, and the progress of the reaction was followed spectroscopically at 300 nm. A standard curve for the generation of PbS was generated using NaSH at concentrations ranging from 10 to 100 μM. A standard curve was generated using constant enzyme and homocysteine (5 mM) concentrations, varying the l-cysteine concentration from 10 to 100 mM. The results displayed Michaelis–Menten-type kinetics and agreed with published results (Table S3).45 The reaction of the enzyme with the hydrazine compound was followed by incubating the enzyme at 50 μM with 50 to 500 μM compound in Tris pH 8.0 at room temperature and recording the UV–vis absorption spectrum from 250 to 400 nm over time (Figure S3).
Oligomerization and Stability/Ligand Binding
Analytical Gel Filtration
A high molecular weight (HMW) gel filtration calibration kit (GE Healthcare) was used for protein molecular weight determination. Protein samples of yCBS-cc in 150 mM NaCl/50 mM Tris pH 8.0 at 2 mg/mL were passed through a Superdex 200 5/15 GL column (GE) on an FPLC system (ÄKTA FPLC, GE), and their elution volumes and partition coefficients were determined.
Native Polyacrylamide Gel Electrophoresis
Protein samples were run through 10% PAGE separation gels at 4 °C. Some samples were incubated with 1 mM SAM or 1 mM ATP for 30 min at room temperature prior to the native PAGE analysis. In all cases, yCBS-cc appeared as a dimer in solution.
ThermoFluor Stability Assay
Protein samples were exchanged into 100 μM HEPES buffer (pH 8.0). Solutions containing protein at 5 μM and 1X Sypro Orange (Invitrogen) alone or with added small molecules (1 mM) were mixed and added to a 96-well RT-PCR plate. The change in fluorescence was monitored over the temperature range from 25 to 95 °C at a heating rate of 0.3 °C/min in an RT-PCR machine (StepOnePlus, Applied Biosystems). Data were normalized with appropriate buffer-only controls. The full-length protein seems to have two unfolding transitions, one starting at about 30 °C and the other at 58 °C (Table S4 and Figure S4).
Circular Dichroism
Protein samples were buffer-exchanged into 1× PBS (pH 7.4). Protein samples at 20 μM were scanned from 260 to 200 nm using a J-810 CD spectrometer (Jasco). Measurements were taken in triplicate at 20 °C. CD melting curves in the presence of inhibitors were obtained from protein samples that were preincubated at room temperature for 10 min. yCBS unfolding was monitored at 222 nm from 25 to 72 °C. The curves for full-length and catalytic-core yCBS displayed unfolding transitions at 57.5 and 59.2 °C, respectively.
RESULTS AND DISCUSSION
Biochemical and Structural Characterization of yCBS
The C-terminally truncated form of the yeast enzyme has the same CD spectrum (not shown) as the full-length enzyme, indicating that it is folded. The UV–vis spectrum of the full-length enzyme (not shown) is identical to that reported previously, with a major peak at 412 nm characteristic of the internal aldimine with PLP and a shoulder at around 320 nm whose interpretation is not clear, possibly a different protonation state of the cofactor.26 The spectrum of yCBS-cc (Figure S3A) shows the peak at 412 nm but not the shoulder at 320 nm. Since the only difference between these two proteins is the loss of the CBS domain, the shoulder in the full-length enzyme may be due to interactions between the catalytic core and the CBS domain.
The stabilities of the protein in the full-length and truncated forms are very similar (Table S4) but are lower than that of full-length hCBS. Two methods were used to determine the unfolding temperatures of the proteins: CD spectroscopy and ThermoFluor assay. The TM values obtained varied slightly between measurements, but the differences observed for the full-length form versus the catalytic core are consistent (Table S4). The differences observed may be due to the slightly different protein concentrations used (20 μM for CD and 5 μM for ThermoFluor) and the possibility that oligomers are more stable at higher concentrations. The TMs of the full-length protein and catalytic core indicate that the CBS domain contributes only very little to the thermal stabilization of the enzyme, at best stabilizing it by 1–2 °C. An early weak unfolding transition for the full-length form is observed starting around 30–35 °C (Figure S4). This transition can be attributed to the CBS domain since it is not observed in the absence of that domain. Addition of SAM has a negligible effect on TM, indicating that SAM, if it binds at all, does not stabilize the protein. However, the early transition seen in the full-length protein is no longer observed in the presence of SAM.
Structure of the Catalytic Core
The structure of yCBS-cc is a homodimer consisting of two monomers related by crystallographic symmetry that form a tight dimer (Figure 3). The dimer interface buries 2235 Å2 of surface area, representing 15% of the monomeric surface area and involving 61 residues from each monomer. Gel filtration and native gel data all agree with a dimer in solution for both full-length yCBS and yCBS-cc. The N-terminus of each monomer extends into the other monomer, forming interactions that contribute to the dimeric interface. The overall fold resembles that of the tryptophan synthase (β-subunit) family of fold-type-II PLP-dependent enzymes and is similar to the catalytic cores of hCBS (PDB ID 4COO,35 root-mean-square deviation (rmsd) = 1.2 Å) and dCBS (PDB ID 3PC2,36 rmsd = 1.6 Å) (Figure S6). There are some minor differences due to small insertions or deletions in yCBS relative to hCBS and dCBS, but they are in loops on the surface (residues 74, 181–185, and 304–306; asterisks in Figure 3). All of these insertions, which are far removed from the active site and the dimer interface, are not expected to contribute to differences in enzymatic activity or structural integrity of the enzyme.
Figure 3.

The yCBS catalytic core (residues 1–353) is a head-to-tail homodimer. One monomer is colored light blue, and the other monomer is colored according to secondary structure, in which α-helices are red, β-strands are cyan, and loop regions are green. PLP and the covalently linked K53 are shown in ball-and-stick with carbon atoms colored yellow. Asterisks and magenta-colored ribbons indicate locations of sequence insertions of yCBS residues (residues 74, 181–185, and 304–306; Figure S1) relative to the hCBS and dCBS protein sequences. There is one cis-proline in the structure, at Q229-P230, which forms a tight turn between a strand and the following short α-helix. All of the structures reported here show this same cis-proline configuration. A comparison of the overall structures of all of the yCBS-cc structures reported here is shown in Figure S5. No major rearrangements are observed.
Binding of SAM has a significant effect on the human enzyme but practically none on the Drosophila or yeast enzymes. Both the full-length and Δ516–525 truncated hCBS enzymes are significantly activated by binding of SAM.44 The effect is ascribed to a conformational change of the Bateman module relative to the catalytic domain from an inactivated conformation to an activated one in which SAM is involved in a domain–domain interaction that helps to stabilize the catalytic dimer.34,35 The full-length dCBS is observed only in the conformation that corresponds to the activated state of hCBS, with or without SAM.36 The C-terminus of the catalytic domain of the yeast enzyme is ordered and extends in a direction that is different from that of the C-terminal CBS domains in hCBS and dCBS. It is not known how significant that observation could be, except that yCBS is activated only slightly (approximately 2-fold) by the presence of the Bateman module but is not activated by SAM, and the linker orientation indicates that the domains may not interact at all (Figure S6).
The effect of the absence of the N-terminal heme-binding domain on stability cannot be determined directly. Comparisons of the unfolding transitions of yCBS and hCBS indicate that this domain may contribute to stabilization of the protein. However, since the overall sequences are not the same, the differences in stability may not be due to the N-terminal domain. The N-terminal domains in hCBS (70 residues) and dCBS (40 residues) are sequence extensions that are absent in yCBS. The heme that is part of this domain is cradled by approximately 15 residues within this extension and the surface of the catalytic domain. The corresponding surface of yCBS where the heme would interact would not accommodate a heme in the same position. The remaining residues of the extension are involved in contacts with the surface of the catalytic domain.
The yCBS-cc monomer contains two structurally conserved salt bridges that are also seen in the structures of other members of the fold-type-II family of PLP-dependent enzymes. These are E174/K42 and E44/R55 (E239/K108 and E110/R121 in hCBS; E208/K77 and E79/R90 in dCBS). These salt bridges occur on the si side of the PLP cofactor and seem to have a structural stabilization role. The structural importance of E44/R55 is underscored by the proximity of R55 to K53 (the active-site lysine; vide infra) and by the fact that both salt bridges have limited or no solvent accessibility.
The Active Site
All of the residues that interact with the cofactor are contributed from the monomer in which the active site is found (Figure 4). The cofactor forms a Schiff base with K53 and makes numerous hydrogen-bonding interactions with the surrounding residues, many of which are conserved among PLP-dependent enzymes or are indicative of the type of reaction catalyzed. N84 interacts with the phenolic oxygen, a type of interaction seen in all fold-type-II PLP-dependent enzymes (either N or Q) and frequently in other fold types as well. The phosphate is held in place at the center of loop 196–200, interacting mainly with backbone amides. The pyridine nitrogen interacts with S289, an interaction also found in tryptophan synthase. A serine in this position is also seen for Oacetylserine sulfhydrylase (OASS), an interaction thought to disfavor the formation of a quinonoid intermediate. This intermediate has not been observed by rapid-scanning spectroscopy in either OASS or yCBS.66 It has, however, been observed in tryptophan synthase, where the serine that interacts with the pyridoxal nitrogen also interacts with an adjacent serine, which is thought to help stabilize such a quinonoid intermediate.67 Interestingly, several PLP-dependent enzymes, including OASS, tryptophan synthase,68 threonine deaminase,69 and CBS, all of which go through an aminoacrylate intermediate as part of the reaction catalyzed, have a serine interacting with the pyridine nitrogen.
Figure 4.

The yCBS catalytic core active site. (A) PLP is held in the active site as an internal aldimine formed by a Schiff base linkage with K53. The Fobs – Fcalc electron density contoured at 3σ from a simulated annealing omit map is shown in green. An acetate ion has been modeled into the active site and is held in place by interactions with residues 81–84. The carboxylate groups of the E-PLP-l-ser and E-PLP-aa intermediates are similarly coordinated (Figure 5). Atoms are colored according to element as follows: gray, carbon; blue, nitrogen; red, oxygen; orange, phosphorus. The carbon atoms of PLP and acetate are colored yellow. (B) Schematic diagram of the yCBS-cc active site. Hydrogens are shown for side-chain and backbone amide groups only because their locations can be defined chemically, whereas those of side-chain hydroxyl groups, especially those involved in hydrogen bonds, cannot. Distances for key interactions between atoms are reported in Table S2.
A salt bridge with a structural role that is important for the integrity of the active site is E244/K327. Residue 244 is part of a loop from residues 244–246 that covers the re face of the pyridine ring of PLP and is positioned by this salt bridge interaction. The interaction becomes important at the external aldimine stage of the reaction, when the cofactor is no longer tethered to the protein and can easily diffuse out of the active site. The loop (244-EGI-246) and K327 are conserved in other CBS enzymes. In dCBS, binding of substrate induces another loop containing S116 (yCBS S82) to shift toward the PLP cofactor (Figure S7). This change, along with movement of other loops in the region, effectively close access to the PLP site. In contrast, all of these loops in the yCBS-cc structures presented here adopt a “closed” conformation. The aminoacrylate external aldimine forms of both yCBS-cc (PDB ID 6C2Z) and dCBS (PDB ID 3PC336) also adopt a “closed” conformation with respect to these loops, as do the internal aldimine forms of hCBS (PDB IDs 4L3V33 and 4COO35). However, structures of both truncated forms of hCBS (PDB IDs 1JBQ31 and 1M5432) and of the SAM-activated full-length hCBS (PDB ID 4PCU34) adopt an “open” loop conformation similar to that seen in the internal aldimine dCBS structure. It is possible that these loops also adopt a different configuration in full-length yCBS.
There is an acetate ion in the active site of yCBS-cc, presumably derived from the crystallization mother liquor (Figure 4A). An acetate ion is also observed in the structure of hCBS (PDB ID 4COO35), but it is oriented differently than observed in yCBS-cc. The orientation of the anion in yCBS-cc is supported by the types of interactions made with protein residues and favors hydrogen bonding with backbone and sidechain atoms over waters. Anions derived from crystallization media have been observed in the active sites of structures of other PLP-dependent enzymes, for instance, acetate in alanine racemase,70 sulfate in aspartate aminotransferase,71 and sulfate in the CBS from Lactobacillus.37 In all cases, the anion occupies the position that the carboxylate of a ligand does in structures of intermediate or substrate analogues. The structures of yCBS-cc intermediates show the same pattern of interactions (vide infra) for such a carboxylate group.
Structures of yCBS Intermediates
The mechanism of the CBS canonical reaction is shown in Figure 2. All of the intermediates have signature UV absorption maxima, although some of these signals overlap. In an attempt to characterize the steps in this mechanism, some of these intermediates have been trapped crystallographically at different pH values.
yCBS PLP–l-Serine External Aldimine
The first step in the CBS-catalyzed reaction is the replacement of the internal aldimine by the substrate, forming the external aldimine. The structure of this complex was obtained by cocrystallization of yCBS-cc with l-serine at pH 8.0. The electron density for this adduct is clear and was interpreted as the external aldimine with l-serine (Figures 5A,B and S2C,D). In the refined structure, the α-carbon of the serine moiety is not planar, indicating that the next step of the reaction has not yet occurred. A similar intermediate was trapped in dCBS but was interpreted as a carbanionic intermediate36 based on planarity at the α-carbon of the l-serine adduct. Attempts to model the electron density observed for the yeast adduct as a carbanion could not be refined as a planar moiety and always reverted to a model where the α-carbon has sp3 geometry. The differences between the Drosophila structure and this one may be due to differences in pH at which the intermediate was crystallized (pH 7.0 for dCBS vs pH 8.0 for yCBS-cc). The pyridine ring of the cofactor is tilted away from the position of the internal aldimine by about 11° (Figure S8) and away from the active-site lysine and toward S82. The active-site lysine, which has been released from the cofactor, interacts with the phosphate of the PLP moiety and forms a hydrogen bond to a water molecule that is also seen in the internal aldimine structure (Figure 4). Although the Lys side chain points away from the intermediate, the active configuration where Lys can carry out acid–base chemistry is readily accessible by simple conformational rearrangement of the side chain. A water molecule is seen in all of the structures reported here and is held in place by hydrogen bonds with one oxygen of the phosphate, the amide oxygen of N163, and the ring nitrogen of H167. The orientation of the amide side chain of N163 is interpreted in terms of an interaction between the amide nitrogen and the backbone carbonyl of Q157.
Figure 5.

yCBS-cc active site intermediates. (A) The PLP–l-serine intermediate active site at pH 8.0. K53 is shifted away from PLP, and the pyridine ring is tilted away from its position in the apoenzyme by about 11° (Figure S8). The Fobs – Fcalc electron density contoured in green at 3σ from a simulated annealing omit map supports the external aldimine intermediate modeled into the active site. (B) The hydroxyl group of S82 is within hydrogen-bonding distance of the β-OH leaving group of the l-serine intermediate. The carboxylate group of the intermediate makes similar interactions as the acetate noted in the internal aldimine yCBS-cc active site (Figure 4). (C) The PLP–l-aminoacrylate intermediate active site at pH 6.5. K53 is shifted away from PLP. The carboxylate group of the intermediate makes similar interactions as the acetate noted in the internal aldimine yCBS-cc active site (Figure 4). (D) A water in the active site indicates the expected position of the attacking sulfur on the substrate. This water is also noted in the internal aldimine active site (Figure 4) but is absent in the E-PLP-l-ser intermediate active site in (A), where it is displaced by the β-OH group of the serine moiety, and in the PMP complex active site (Figure 7), where lower-resolution electron density does not support placing it there. Atoms are colored according to element as follows: cyan or green, carbon; blue, nitrogen; red, oxygen; orange, phosphorus. The carbon atoms of PLP are colored yellow.
yCBS PLP–Aminoacrylate Intermediate
The rate-determining step for yCBS, as it is for hCBS and dCBS, is the conversion of the aminoacrylate intermediate to the product, suggesting that this intermediate should be kinetically accessible. This external aldimine intermediate between the enzyme and serine has been reported for the dCBS36 and Lactobacillus plantarum enzymes.37 For yCBS, this intermediate was trapped at pH 6.5. The electron density for this intermediate is unambiguous. The structure (Figure 5C,D) shows a planar arrangement from the nitrogen of the pyridine ring through the carboxylate group of the acrylate moiety. The pyridine ring of the cofactor is again tilted away from the position of the internal aldimine, and the active-site lysine makes the same interactions as it does in the serine adduct structure.
The remarkable feature of this intermediate is the position of the carboxylate group, which corresponds perfectly with the position of the acetate ion in the active site of the substrate-free enzyme. Superposition of the aminoacrylate model with that of the E-PLP-l-ser adduct emphasizes the nonplanarity of the serine α-carbon. The structure and conformation of E-PLP-aa match those reported for dCBS, making the same inter-actions.36
A water molecule is positioned in the active site of the aminoacrylate intermediate, held in place by hydrogen bonds between the side-chain OHs of S82 and Y248 and the backbone NH of G245. The position of this water molecule coincides with the expected position of an attacking sulfur atom in the addition reaction of homocysteine to the acrylate to form cystathionine or the addition of H2S to form cysteine. A water molecule is also observed in the same position in the aminoacrylate dCBS structure, indicating that it is potentially conserved.36 This same water is observed in the yCBS internal aldimine complex structure but not in that of native dCBS.36
Inhibition of yCBS and yCBS-cc
Two types of inhibitors were tested with the enzyme: D-cycloserine, a mechanism-based inactivator of some PLP-dependent enzymes that can undergo a transamination reaction to give a dead-end complex, and an inhibitor designed by the Berkowitz laboratory18 that is based on structural and mechanistic considerations of the catalyzed reaction.
To test the potential of cycloserine as an inhibitor of yCBS, the inactivator was mixed with the protein, and the course of inactivation was followed spectrophotometrically. The resulting complex was tested for stability using CD and ThermoFluor assays (Table S4). The absorption spectrum does not support the formation of an adduct. However, in the presence of 1 mM cycloserine, the full-length and catalytic-core enzymes are destabilized by 12 and 14.5 °C, respectively. Attempts to obtain a crystal structure of the complex with cycloserine failed.
The hydrazine inhibitor was expected to give an adduct similar to that observed for BioA of Mycobacterium tuberculosis.72 The affinity of the hydrazine inhibitor was reported to be Ki = 48 ± 2 μM versus homocysteine as the substrate,18 indicating that full inactivation is to be expected. Reaction of the hydrazine inhibitor with the enzyme did not change the CD spectra for either yCBS or yCBS-cc, indicating that the overall structures of the proteins were not altered by interaction with the inhibitor. Destabilization of the protein has been observed in other PLP-dependent enzyme studies using hydrazine compounds.72 In the case of yCBS, the presence of the inhibitor destabilized both the full-length and catalytic-core enzymes by about 6 °C. A possible explanation for the decrease in thermal stability when the enzyme is treated with cycloserine or the hydrazine compound is that the PLP cofactor is no longer covalently attached to the protein, thereby removing the stabilization that such a covalent interaction can provide.
The nature of the product of the reaction between the hydrazine compound and the enzyme was studied in two ways: the change in the absorption spectrum of the enzyme upon incubation with the compound and the crystal structure of the adduct. The absorption spectrum of the compound reacting with free PLP had been reported previously18 and was used to interpret the absorption spectra of the compound reacting with yCBS and yCBS-cc. The reaction of the compound with free PLP at pH 8.0 showed a loss of the peak at 390 nm (free PLP) and the formation of two peaks at 305 and 329 nm, which were interpreted as the rearrangement of the hydrazine adduct to a hydrazone adduct (Figure 6).18 The reaction of the compound with yCBS or yCBS-cc at pH 8.0 gave rise to peaks at 308 and 336 nm with the disappearance of the internal aldimine peak at 412 nm. The slight differences in the new absorption peaks compared with those for free PLP and the enzyme can be attributed to the effect of the protein environment. The initial interpretation of these spectra was that a hydrazone adduct had formed (Figure 6). The time course of the reaction was determined spectroscopically at pH 8.0 and room temperature (Figure S3). Under these conditions, the rate of the reaction was concentration-dependent, and the reaction was almost complete within 1 h at 0.5 mM added compound. Therefore, it should be possible to capture the expected hydrazone product in the crystal by soaking for an hour.
Figure 6.

Potential mechanisms for tautomerization and subsequent fragmentation of the PLP hydrazone expected to form from the designed hydrazine inhibitor and the internal aldimine form of the CBS enzyme. The crystallographically observed reduced form of the cofactor (PMP), under redox-neutral conditions, suggests that the initial PLP hydrazone may tautomerize to the azo form, which itself could undergo N–N cleavage following a second tautomerization and decarboxylation (pathway A) or via hydration and disproportionation (pathway B), at least at sufficient levels to result in significant active-site occupancy by the PMP form of the cofactor. Although the pyridine nitrogen appears as protonated in this figure, there is some question about its state of protonation (Figure 2).
Structure of the Inhibited Enzyme
The structure of the hydrazine inhibitor bound to yCBS-cc yielded a surprise relative to the expected outcome but not inconsistent with biochemical observations such as loss of color and loss of thermal stability. The interpretation of hydrazine inhibition in vivo was based on the expectation that the inhibitor can form an essentially irreversible dead-end complex featuring a tightly bound PLP–inhibitor hydrazone. The structure shows that the enzyme did indeed turn over into a dead-end complex, but not the expected one (Figures 6 and 7). The electron density in the active site for this structure can only be interpreted as the PMP form of the enzyme on the basis of interactions that the PLP species makes. On the basis of the above spectroscopic data, the expected product of the reaction between the enzyme and inhibitor is the hydrazone, which forms within minutes but disappears with consequent formation of PMP, rather than the initially interpreted PLP–hydrazone complex.18 Therefore, over sufficient time, the enzyme–inhibitor adduct has followed a mechanistic path that yields the pyridoxamine form of the cofactor bound in the active site. An attempt to capture the putative adduct at lower pH failed. Crystals of yCBS-cc soaked with the hydrazine compound at pH 6.5 gave the structure of the native enzyme.
Figure 7.

PMP active site at pH 8.0. (A) Active site of yCBS-cc after soaking with hydrazine. Fobs – Fcalc electron density contoured at 3σ from a simulated annealing omit map is shown in green. K53 is shifted away from PMP. Acetate does not appear to be present in this active site; a water molecule is bound instead (WA). (B) K53 is shifted away from its internal aldimine position and interacts with a water molecule that is held in place by surrounding residues. This latter water is also present in the other active-site structures reported here (Figures 4 and 5) and interacts with K53 in all of the external aldimine intermediate structures (Figure 5). Atoms are colored according to element as follows: magenta, carbon; blue, nitrogen; red, oxygen; orange, phosphorus. The carbon atoms of PLP are colored yellow.
There is certainly precedent for hydrazine-based inhibition of PLP enzymes, including the clinically widely used drug for Parkinson’s disease, carbidopa, a 3′,4′-dihydroxyphenylalanine (DOPA) decarboxylase inhibitor,73,74 and hydrazino analogues of γ-aminobutyric acid (GABA) that are known to inhibit GABA transaminase.75 However, in these cases, the inhibitors are thought to act by forming stable PLP–hydrazone adducts in the active site. Indeed, for carbidopa, this enzyme-bound PLP hydrazone has been observed crystallographically.76 There are also a number of inhibitors of oxidase-type enzymes that bear a hydrazine moiety.77–81 At least in one such case, there is crystallographic evidence for tautomerization from the hydrazone form to the azo form of the inhibitor.82 This is seen in the active site of the enzyme bovine serum amine oxidase that utilizes a topaquinone (2′,4′5′-trihydroxyphenylalanine) cofactor. Interestingly, the favored azo tautomer in the bovine enzyme appears to be the opposite of the favored hydrazone tautomer in the corresponding E. coli amine oxidase active site. On the basis of the notion that such a hydrazone–azo tautomerization equilibration can obtain in an enzyme active site, we illustrate in Figure 6 two pathways, both emanating from this azo tautomer (highlighted in the center of the figure), that could give rise to the observed PMP-bound form of the cofactor here. As pertains to pathway A, it should be noted that the generation of a nitrile via decarboxylation and C==N–X cleavage has been postulated for related intermediates in both enzymatic83 and nonenzymatic systems.84 The possibility of such mechanisms, at least over extended times, will be the subject of future studies.
CONCLUSION
The intermediates that have been trapped in the yeast CBS catalytic core in this study provide added structures from which the mechanistic pathway of the reaction can be described. The E-PLP-l-ser structure for yCBS-cc reported here is for the first step, formation of the external aldimine with serine bound. Elimination of the hydroxyl from the serine side chain to form the aminoacrylate intermediate, E-PLP-aa (Figure 2), could then proceed through the intermediate described for dCBS and interpreted as a carbanionic intermediate.36
We have also shown that yCBS can undergo off-pathway reactions with the appropriate reagents. Cystathionine β-synthase does not normally go through the PLP/external aldimine/PMP cycle and its reverse, exhibited by many other PLP-dependent enzymes. Arguably, it only goes as far as the aminoacrylate external aldimine followed by addition of the incoming nucleophile to the acrylate moiety. Release of product and re-formation of the internal aldimine form of the enzyme bypasses the PMP form of the enzyme that would be derived from hydrolysis of the external aldimine in the standard ping-pong mechanism. The implication of the structure of a PMP enzyme when treated with hydrazine inhibitor is that the reverse reaction to release the hydrazine adduct and re-form the PLP form of the enzyme is slow and that, given time, alternative pathways are possible, including tautomerization and subsequent inhibitor–cofactor fragmentation, leading to the inactive PMP form of the enzyme. Although many PLP-dependent enzymes are highly specific for a particular type of reaction, they are known to undergo side reactions. For instance, alanine racemase can undergo a transamination half-reaction when exposed to cycloserine.85 The observation of off-pathway chemistry opens up new possibilities for inhibitor design and modification in the quest to develop potent and effective small-molecule inhibitors of CBS with potential applications in the protection against neuronal damage pursuant to ischemic stroke.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Jacqueline Naffin-Olivos, Junfeng Shi, and CeFeng Liu and thank Edith Miles and Barry Snider for helpful discussions during the course of this project. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source using beamline 19-ID. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02–06CH11357.
Funding
This work was supported by funds from National Institute of Health Grant GM32415 to G.A.P. and D.R., by Grant-in-Aid 16GRNT313400012 from the American Heart Association to D.B.B., and by NIH RR016544 for facilities support.
ABBREVIATIONS
- CBS
cystathionine β-synthase
- PLP
pyridoxal 5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- yCBS
cystathionine β-synthase from Saccharomyces cerevisiae
- yCBS-cc
catalytic core of cystathionine β-synthase from Saccharomyces cerevisiae consisting of residues 1–353 and an N-terminal His6 tag
- hCBS
cystathionine β-synthase from Homo sapiens
- dCBS
cystathionine β-synthase from Drosophila melanogaster
- DOPA
3′,4′-dihydroxyphenylalanine
- GABA
γ-aminobutyric acid
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00092.
yCBS kinetic parameters, unfolding transitions, crystallographic data collection and refinement statistics, distances of interactions in the active site, structure-based sequence alignment, and ThermoFluor assay results (PDF)
REFERENCES
- (1).Finkelstein JD, and Martin JJ (1984) Methionine metabolism in mammals. Distribution of homocysteine between competing pathways. J. Biol. Chem 259, 9508–9513. [PubMed] [Google Scholar]
- (2).Banerjee R, Evande R, Kabil Ö, Ojha S, and Taoka S (2003) Reaction mechanism and regulation of cystathionine β-synthase. Biochim. Biophys. Acta, Proteins Proteomics 1647, 30–35. [DOI] [PubMed] [Google Scholar]
- (3).Banerjee R, and Zou C (2005) Redox regulation and reaction mechanism of human cystathionine-β-synthase: a PLP-dependent hemesensor protein. Arch. Biochem. Biophys 433, 144–156. [DOI] [PubMed] [Google Scholar]
- (4).Auger S, Danchin A, and Martin-Verstraete I (2002) Global Expression Profile of Bacillus subtilis Grown in the Presence of Sulfate or Methionine. J. Bacteriol 184, 5179–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Macnicol PK, Datko AH, Giovanelli J, and Mudd SH (1981) Homocysteine Biosynthesis in Green Plants: Physiological Importance of the Transsulfuration Pathway in Lemna paucicostata. Plant Physiol. 68, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cherest H, Thomas D, and Surdin-Kerjan Y (1993) Cysteine biosynthesis in Saccharomyces cerevisiae occurs through the transsulfuration pathway which has been built up by enzyme recruitment. J. Bacteriol 175, 5366–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Thomas D, and Surdin-Kerjan Y (1997) Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev 61, 503–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Singh S, Padovani D, Leslie RA, Chiku T, and Banerjee R (2009) Relative Contributions of Cystathionine β-Synthase and γ-Cystathionase to H2S Biogenesis via Alternative Trans-sulfuration Reactions. J. Biol. Chem 284, 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Miles EW, and Kraus JP (2004) Cystathionine β-Synthase: Structure, Function, Regulation, and Location of Homocystinuria-causing Mutations. J. Biol. Chem 279, 29871–29874. [DOI] [PubMed] [Google Scholar]
- (10).Jhee K-H, and Kruger WD (2005) The Role of Cystathionine β-Synthase in Homocysteine Metabolism. Antioxid. Redox Signaling 7, 813–822. [DOI] [PubMed] [Google Scholar]
- (11).Sanchez-Ruiz JM, Lopez-Lacomba JL, Cortijo M, and Mateo PL (1988) Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry 27, 1648–1652. [DOI] [PubMed] [Google Scholar]
- (12).Kraus JP, Janosǐk M, Kožich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LAJ, Blom H, Boers GHJ, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, and Gaustadnes M (1999) Cystathionine β-synthase mutations in homocystinuria. Hum. Mutat 13, 362–375. [DOI] [PubMed] [Google Scholar]
- (13).Refsum H, Ueland PM, Nygård O, and Vollset SE (1998) Homocysteine and cardiovascular disease. Annu. Rev. Med 49, 31–62. [DOI] [PubMed] [Google Scholar]
- (14).Mills JL, McPartlin JM, Kirke PN, Lee YJ, Conley MR, Weir DG, and Scott JM (1995) Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 345, 149–151. [DOI] [PubMed] [Google Scholar]
- (15).Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, and Ueland PM (1998) Folate, Vitamin B12, and Serum Total Homocysteine Levels in Confirmed Alzheimer Disease. Arch. Neurol 55, 1449–1455. [DOI] [PubMed] [Google Scholar]
- (16).Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, and Kimura H (2005) Cystathionine β-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 19, 1854–1856. [DOI] [PubMed] [Google Scholar]
- (17).Lee M, Schwab C, Yu S, McGeer E, and McGeer PL (2009) Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 30, 1523–1534. [DOI] [PubMed] [Google Scholar]
- (18).McCune CD, Chan SJ, Beio ML, Shen W, Chung WJ, Szczesniak LM, Chai C, Koh SQ, Wong PT-H, and Berkowitz DB (2016) “Zipped Synthesis” by Cross-Metathesis Provides a Cystathionine β-Synthase Inhibitor That Attenuates Cellular H2S Levels and Reduces Neuronal Infarction in a Rat Ischemic Stroke Model. ACS Cent. Sci 2, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal E, Weaver AL, Visscher DW, Cliby W, Sood AK, Bhattacharya R, and Mukherjee P (2013) Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS One 8, e79167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jhee K-H, McPhie P, and Miles EW (2000) Yeast Cystathionine β-Synthase Is a Pyridoxal Phosphate Enzyme but, Unlike the Human Enzyme, Is Not a Heme Protein. J. Biol. Chem 275, 11541–11544. [DOI] [PubMed] [Google Scholar]
- (21).Brown FC, and Gordon PH (1971) Cystathionine synthase from rat liver: partial purification and properties. Can. J. Biochem 49, 484–491. [DOI] [PubMed] [Google Scholar]
- (22).Taoka S, and Banerjee R (2002) Stopped-flow Kinetic Analysis of the Reaction Catalyzed by the Full-length Yeast Cystathionine β-Synthase. J. Biol. Chem 277, 22421–22425. [DOI] [PubMed] [Google Scholar]
- (23).Aitken SM, and Kirsch JF (2003) Kinetics of the yeast cystathionine beta-synthase forward and reverse reactions: continuous assays and the equilibrium constant for the reaction. Biochemistry 42, 571–578. [DOI] [PubMed] [Google Scholar]
- (24).Borcsok E, and Abeles RH (1982) Mechanism of action of cystathionine synthase. Arch. Biochem. Biophys 213, 695–707. [DOI] [PubMed] [Google Scholar]
- (25).Caulkins BG, Young RP, Kudla RA, Yang C, Bittbauer TJ, Bastin B, Hilario E, Fan L, Marsella MJ, Dunn MF, and Mueller LJ (2016) NMR Crystallography of a Carbanionic Intermediate in Tryptophan Synthase: Chemical Structure, Tautomerization, and Reaction Specificity. J. Am. Chem. Soc 138, 15214–15226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Majtan T, Pey AL, Fernández R, Fernández JA, Martínez-Cruz LA, and Kraus JP (2014) Domain Organization, Catalysis and Regulation of Eukaryotic Cystathionine Beta-Synthases. PLoS One 9, e105290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Baykov AA, Tuominen HK, and Lahti R (2011) The CBS Domain: A Protein Module with an Emerging Prominent Role in Regulation. ACS Chem. Biol 6, 1156–1163. [DOI] [PubMed] [Google Scholar]
- (28).Ereño-Orbea J, Oyenarte I, and Martínez-Cruz LA (2013) CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys 540, 70–81. [DOI] [PubMed] [Google Scholar]
- (29).Bateman A (1997) The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci 22, 12–13. [DOI] [PubMed] [Google Scholar]
- (30).Holm L, and Rosenström P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Meier M, Janosik M, Kery V, Kraus JP, and Burkhard P (2001) Structure of human cystathionine β-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 20, 3910–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Taoka S, Lepore BW, Kabil Ö, Ojha S, Ringe D, and Banerjee R (2002) Human Cystathionine β-Synthase Is a Heme Sensor Protein. Evidence That the Redox Sensor Is Heme and Not the Vicinal Cysteines in the CXXC Motif Seen in the Crystal Structure of the Truncated Enzyme. Biochemistry 41, 10454–10461. [DOI] [PubMed] [Google Scholar]
- (33).Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, and Martínez-Cruz LA (2013) Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proc. Natl. Acad. Sci. U. S. A. 110, E3790–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, and Martínez-Cruz LA (2014) Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proc. Natl. Acad. Sci. U. S. A 111, E3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McCorvie TJ, Kopec J, Hyung S-J, Fitzpatrick F, Feng X, Termine D, Strain-Damerell C, Vollmar M, Fleming J, Janz JM, Bulawa C, and Yue WW (2014) Inter-domain Communication of Human Cystathionine β-Synthase: Structural Basis of S-Adenodyl-l-Methionine Activation. J. Biol. Chem 289, 36018–36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Koutmos M, Kabil O, Smith JL, and Banerjee R (2010) Structural basis for substrate activation and regulation by cystathionine beta-synthase (CBS) domains in cystathionine β-synthase. Proc. Natl. Acad. Sci. U. S. A 107, 20958–20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Matoba Y, Yoshida T, Izuhara-Kihara H, Noda M, and Sugiyama M (2017) Crystallographic and mutational analyses of cystathionine β-synthase in the H2S-synthetic gene cluster in Lactobacillus plantarum. Protein Sci. Publ. Protein Soc 26, 763–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Devi S, Abdul Rehman SA, Tarique KF, and Gourinath S (2017) Structural characterization and functional analysis of cystathionine β-synthase: an enzyme involved in the reverse transsulfuration pathway of Bacillus anthracis. FEBS J. 284, 3862–3880. [DOI] [PubMed] [Google Scholar]
- (39).Vicente JB, Colaço HG, Mendes MIS, Sarti P, Leandro P, and Giuffrè A (2014) NO · Binds Human Cystathionine β-Synthase Quickly and Tightly. J. Biol. Chem 289, 8579–8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Oliveriusová J, Kery V, Maclean KN, and Kraus JP (2002) Deletion Mutagenesis of Human Cystathionine β-Synthase: Impact on Activity, Oligomeric Staus, and S-Adendosylmethionine regulation. J. Biol. Chem 277, 48386–48394. [DOI] [PubMed] [Google Scholar]
- (41).Kery V, Poneleit L, Meyer JD, Manning MC, and Kraus JP (1999) Binding of Pyridoxal 5′-Phosphate to the Heme Protein Human Cystathionine β-Synthase. Biochemistry 38, 2716–2724. [DOI] [PubMed] [Google Scholar]
- (42).Jhee K-H, McPhie P, and Miles EW (2000) Domain Architecture of the Heme-Independent Yeast Cystathionine β-Synthase Provides Insights into Mechanisms of Catalysis and Regulation. Biochemistry 39, 10548–10556. [DOI] [PubMed] [Google Scholar]
- (43).Su Y, Majtan T, Freeman KM, Linck R, Ponter S, Kraus JP, and Burstyn JN (2013) Comparative study of enzyme activity and heme reactivity in Drosophila melanogaster and Homo sapiens cystathionine β-synthases. Biochemistry 52, 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Taoka S, Widjaja L, and Banerjee R (1999) Assignment of Enzymatic Functions to Specific Regions of the PLP-Dependent Heme Protein Cystathionine β-Synthase. Biochemistry 38, 13155–13161. [DOI] [PubMed] [Google Scholar]
- (45).Janošík M, Kery V, Gaustadnes M, Maclean KN, and Kraus JP (2001) Regulation of Human Cystathionine β-Synthase by S-Adenosyl-l-methionine: Evidence for Two Catalytically Active Conformations Involving an Autoinhibitory Domain in the C-Terminal Region. Biochemistry 40, 10625–10633. [DOI] [PubMed] [Google Scholar]
- (46).Pey AL, Majtan T, Sanchez-Ruiz JM, and Kraus JP (2013) Human cystathionine β-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): complex regulation of CBS activity and stability by SAM. Biochem. J 449, 109–121. [DOI] [PubMed] [Google Scholar]
- (47).Perła-Kaján J, Twardowski T, and Jakubowski H (2007) Mechanisms of homocysteine toxicity in humans. Amino Acids 32, 561–572. [DOI] [PubMed] [Google Scholar]
- (48).Kimura H (2015) Signaling Molecules: Hydrogen Sulfide and Polysulfide. Antioxid. Redox Signaling 22, 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Erickson PF, Maxwell IH, Su LJ, Baumann M, and Glode LM (1990) Sequence of cDNA for rat cystathionine γ-lyase and comparison of deduced amino acid sequence with related Escherichia coli enzymes. Biochem. J 269, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Swaroop M, Bradley K, Ohura T, Tahara T, Roper MD, Rosenberg LE, and Kraus JP (1992) Rat cystathionine β-synthase. Gene organization and alternative splicing. J. Biol. Chem 267, 11455–11461. [PubMed] [Google Scholar]
- (51).Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, and Kimura H (2009) 3-Mercaptopyruvate sulfur-transferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signaling 11, 703–714. [DOI] [PubMed] [Google Scholar]
- (52).Whiteman M, Li L, Rose P, Tan C-H, Parkinson DB, and Moore PK (2010) The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox Signaling 12, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Abe K, and Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kimura H (2000) Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun 267, 129–133. [DOI] [PubMed] [Google Scholar]
- (55).Wong PTH, Qu K, Chimon GN, Seah ABH, Chang HM, Wong MC, Ng YK, Rumpel H, Halliwell B, and Chen CPLH (2006) High plasma cyst(e)ine level may indicate poor clinical outcome in patients with acute stroke: possible involvement of hydrogen sulfide. J. Neuropathol. Exp. Neurol 65, 109–115. [DOI] [PubMed] [Google Scholar]
- (56).Chan SJ, Chai C, Lim TW, Yamamoto M, Lo EH, Lai MKP, and Wong PTH (2015) Cystathionine β-synthase inhibition is a potential therapeutic approach to treatment of ischemic injury. ASN Neuro 7 (2), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhao H, Chan S-J, Ng Y-K, and Wong PT-H (2013) Brain 3-Mercaptopyruvate Sulfurtransferase (3MST): Cellular Localization and Downregulation after Acute Stroke. PLoS One 8, e67322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Jhee KH, Niks D, McPhie P, Dunn MF, and Miles EW (2001) The reaction of yeast cystathionine beta-synthase is rate-limited by the conversion of aminoacrylate to cystathionine. Biochemistry 40, 10873–10880. [DOI] [PubMed] [Google Scholar]
- (59).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (60).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Joosten RP, Long F, Murshudov GN, and Perrakis A (2014) The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Williams RAM, Westrop GD, and Coombs GH (2009) Two pathways for cysteine biosynthesis in Leishmania major. Biochem. J 420, 451–462. [DOI] [PubMed] [Google Scholar]
- (67).Pan P, Woehl E, and Dunn MF (1997) Protein architecture, dynamics and allostery in tryptophan synthase channeling. Trends Biochem. Sci 22, 22–27. [DOI] [PubMed] [Google Scholar]
- (68).Hyde CC, Ahmed SA, Padlan EA, Miles EW, and Davies DR (1988) Three-dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem 263, 17857–17871. [PubMed] [Google Scholar]
- (69).Gallagher DT, Gilliland GL, Xiao G, Zondlo J, Fisher KE, Chinchilla D, and Eisenstein E (1998) Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 6, 465–475. [DOI] [PubMed] [Google Scholar]
- (70).Shaw JP, Petsko GA, and Ringe D (1997) Determination of the Structure of Alanine Racemase from Bacillus stearothermophilus at 1.9-Å Resolution. Biochemistry 36, 1329–1342. [DOI] [PubMed] [Google Scholar]
- (71).Almo SC, Smith DL, Danishefsky AT, and Ringe D (1994) The structural basis for the altered substrate specificity of the R292D active site mutant of aspartate aminotransferase from E. coli. Protein Eng., Des. Sel 7, 405–412. [DOI] [PubMed] [Google Scholar]
- (72).Dai R, Wilson DJ, Geders TW, Aldrich CC, and Finzel BC (2014) Inhibition of Mycobacterium tuberculosis Transaminase BioA by Aryl Hydrazines and Hydrazides. ChemBioChem 15, 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Mittur A, Gupta S, and Modi NB (2017) Pharmacokinetics of Rytary, An Extended-Release Capsule Formulation of Carbidopa-Levodopa. Clin. Pharmacokinet 56, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Whitfield AC, Moore BT, and Daniels RN (2014) Classics in Chemical Neuroscience: Levodopa. ACS Chem. Neurosci 5, 1192–1197. [DOI] [PubMed] [Google Scholar]
- (75).Lightcap ES, and Silverman RB (1996) Slow-binding inhibition of γ-aminobutyric acid aminotransferase by hydrazine analogs. J. Med. Chem 39, 686–94. [DOI] [PubMed] [Google Scholar]
- (76).Burkhard P, Dominici P, Borri-Voltattorni C, Jansonius JN, and Malashkevich VN (2001) Structural insight into Parkinson’s disease treatment from drug-inhibited DOPA decarboxylase. Nat. Struct. Biol 8, 963–967. [DOI] [PubMed] [Google Scholar]
- (77).Binda C, Wang J, Li M, Hubalek F, Mattevi A, and Edmondson DE (2008) Structural and Mechanistic Studies of Arylalkylhydrazine Inhibition of Human Monoamine Oxidases A and B. Biochemistry 47, 5616–5625. [DOI] [PubMed] [Google Scholar]
- (78).Burke AA, Severson ES, Mool S, Solares Bucaro MJ, Greenaway FT, and Jakobsche CE (2017) Comparing hydrazine-derived reactive groups as inhibitors of quinone-dependent amine oxidases. J. Enzyme Inhib. Med. Chem 32, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Carradori S, D’Ascenzio M, De Monte C, Secci D, and Yanez M (2013) Synthesis and Selective Human Monoamine Oxidase B Inhibition of Heterocyclic Hybrids Based on Hydrazine and Thiazole Scaffolds. Arch. Pharm 346, 17–22. [DOI] [PubMed] [Google Scholar]
- (80).Dar A, Khan KM, Ateeq HS, Khan S, Rahat S, Perveen S, and Supuran CT (2005) Inhibition of monoamine oxidase-A activity in rat brain by synthetic hydrazines: Structure–activity relationship (SAR). J. Enzyme Inhib. Med. Chem 20, 269–274. [DOI] [PubMed] [Google Scholar]
- (81).Fung S-PS, Wang H, Tomek P, Squire CJ, Flanagan JU, Palmer BD, Bridewell DJA, Tijono SM, Jamie JF, and Ching L-M (2013) Discovery and characterization of hydrazines as inhibitors of the immune suppressive enzyme, indoleamine 2,3-dioxygenase 1 (IDO1). Bioorg. Med. Chem 21, 7595–7603. [DOI] [PubMed] [Google Scholar]
- (82).De Matteis G, Agostinelli E, Mondovi B, and Morpurgo L (1999) The metal function in the reactions of bovine serum amine oxidase with substrates and hydrazine inhibitors. JBIC, J. Biol. Inorg. Chem 4, 348–353. [DOI] [PubMed] [Google Scholar]
- (83).Kato Y, Tsuda T, and Asano Y (2007) Purification and partial characterization of N-hydroxy-l-phenylalanine decarboxylase/oxidase from Bacillus sp. strain OxB-1, an enzyme involved in aldoxime biosynthesis in the “aldoxime–nitrile pathway”. Biochim. Biophys. Acta, Proteins Proteomics 1774, 856–865. [DOI] [PubMed] [Google Scholar]
- (84).Acheson RM, Hunt PG, Littlewood DM, Murrer BA, and Rosenberg HE (1978) The synthesis, reactions, and spectra of 1-acetoxy-, 1-hydroxy-, and 1-methoxy-indoles. J. Chem. Soc., Perkin Trans 1 0, 1117–1125. [Google Scholar]
- (85).Fenn TD, Stamper GF, Morollo AA, and Ringe D (2003) A Side Reaction of Alanine Racemase: Transamination of Cycloserine. Biochemistry 42, 5775–5783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.